
Genetic and Antigenic Characteristics of Highly Pathogenic Avian Influenza A(H5N8) Viruses Circulating in Domestic Poultry in Egypt, 2017–2021

 ,
,  , , ,
, , , 

Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection and Virus Isolation
2.2. Sequencing and Sequence Analysis
2.3. Antigenic Analysis
3. Results
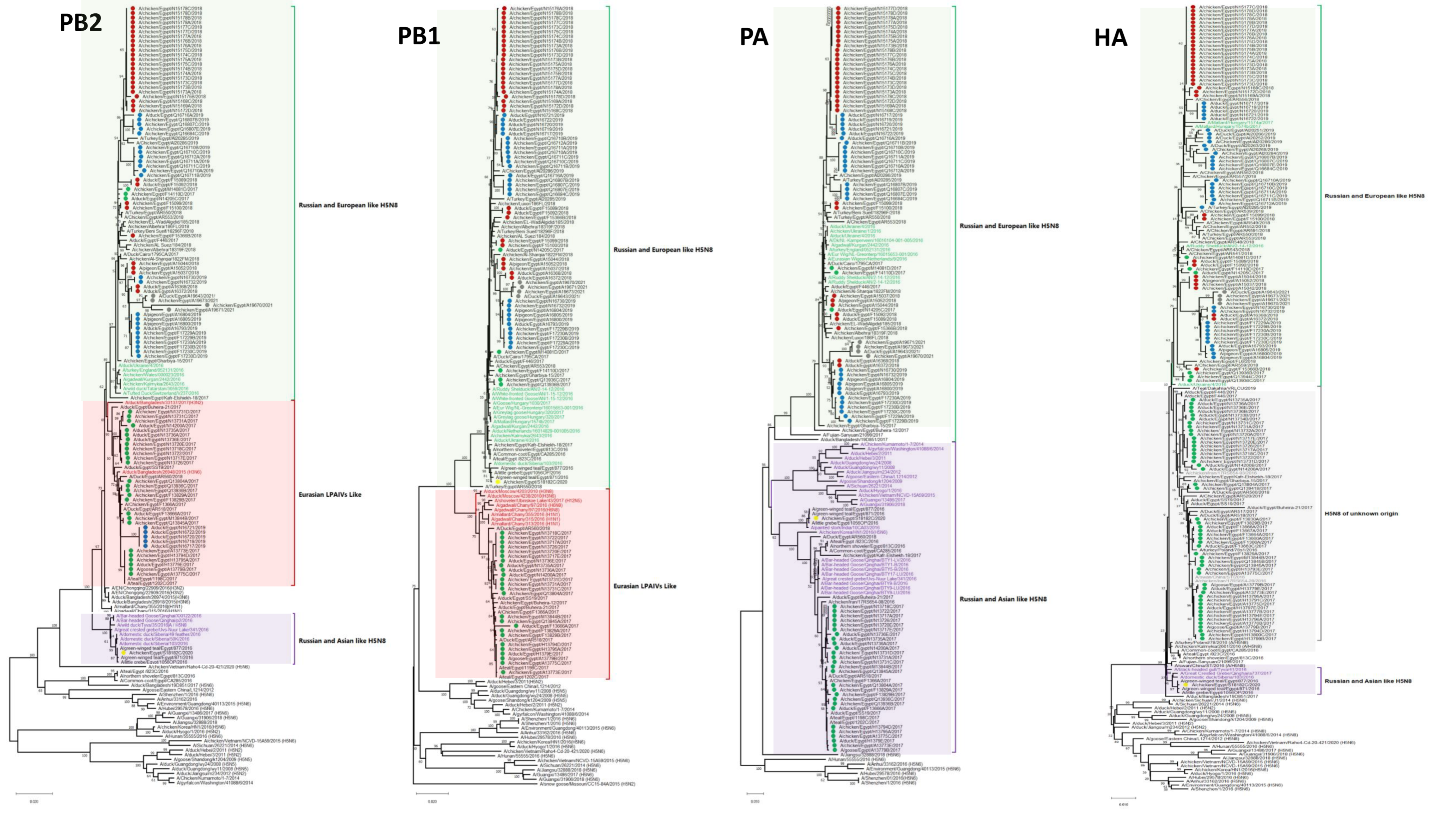
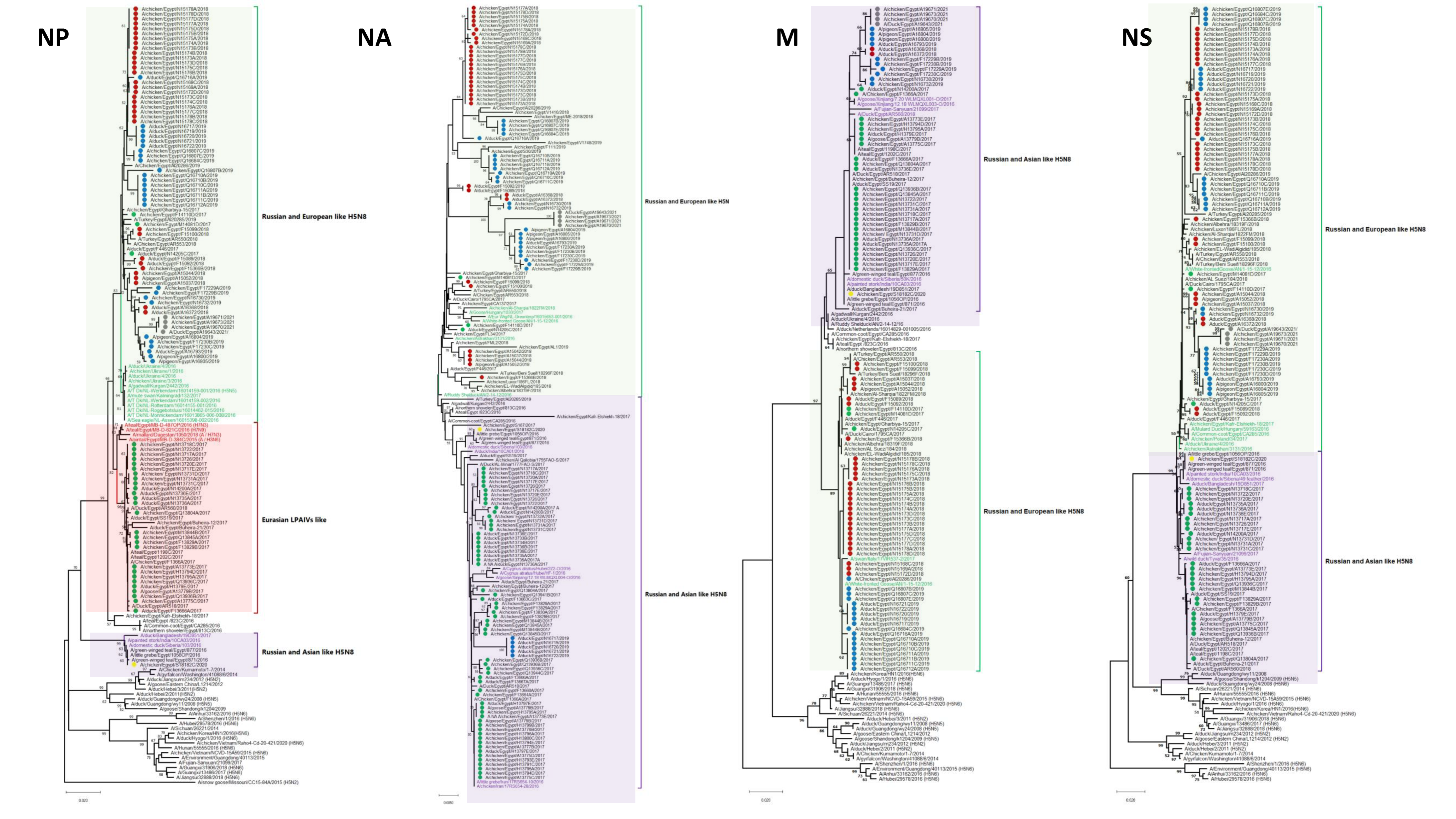
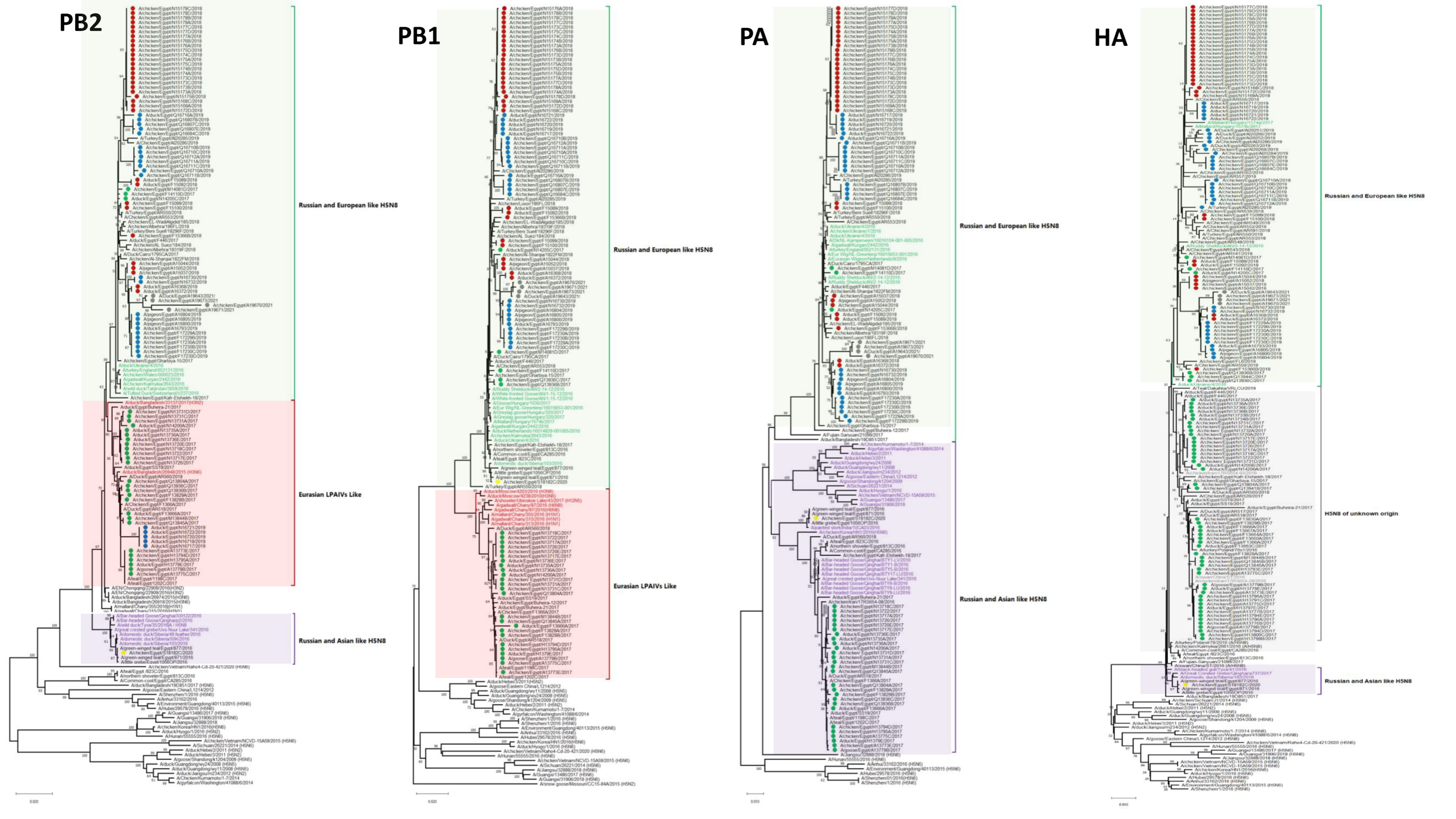
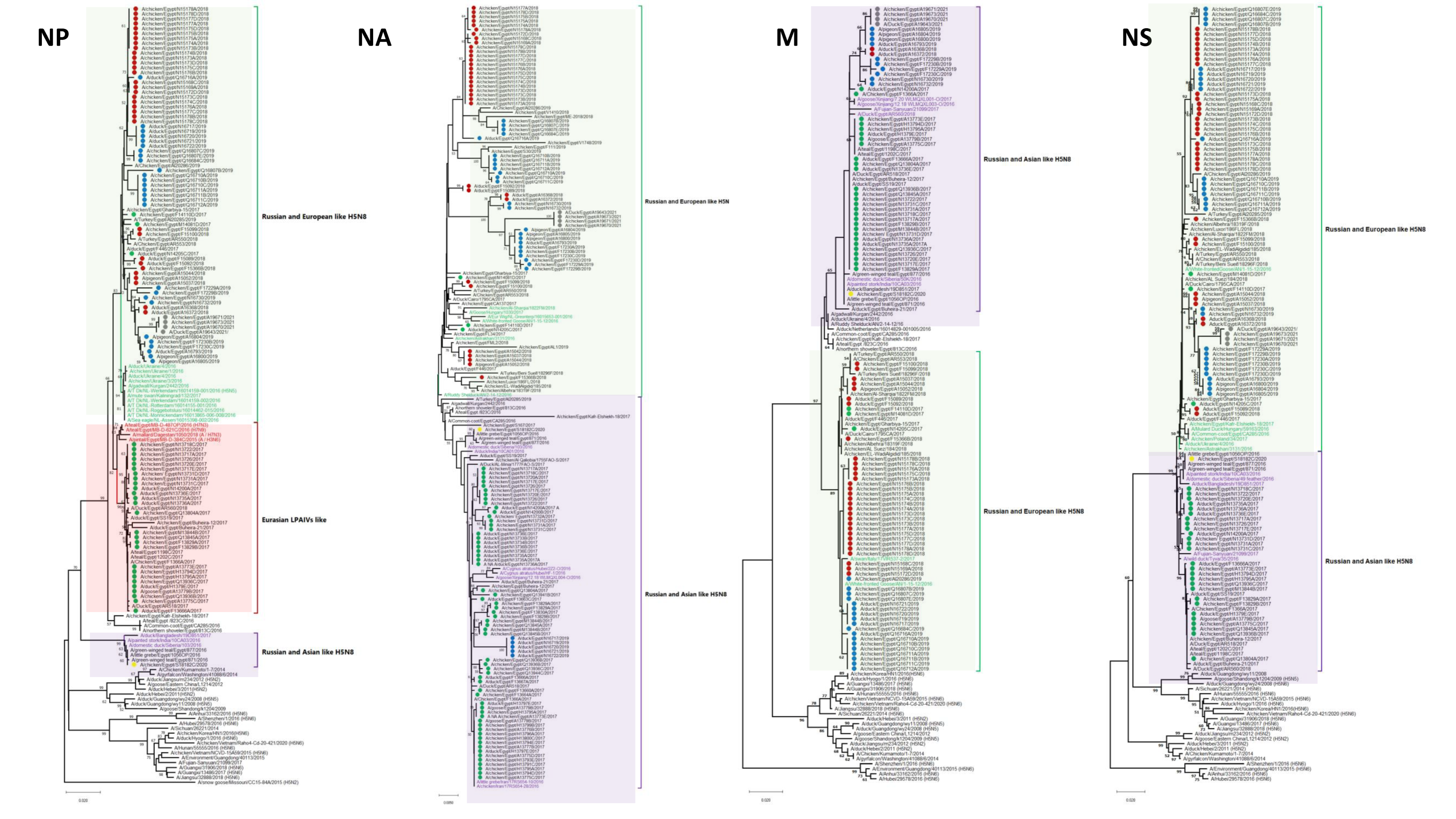
3.1. Phylogenetic Analysis and Sequence Similarity
3.2. Genetic Characterization
3.2.1. Internal Segments
3.2.2. HA
3.2.3. NA
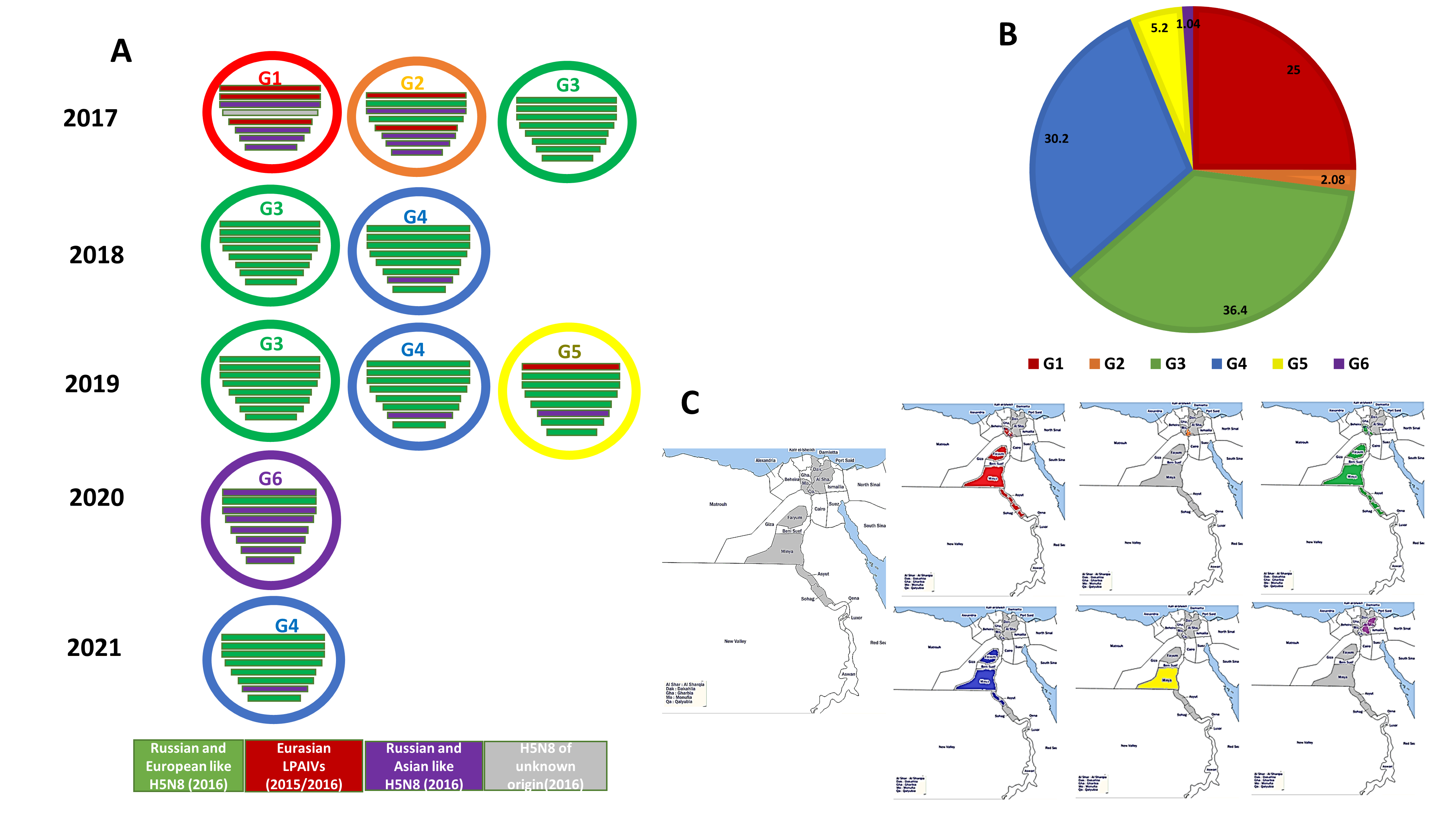
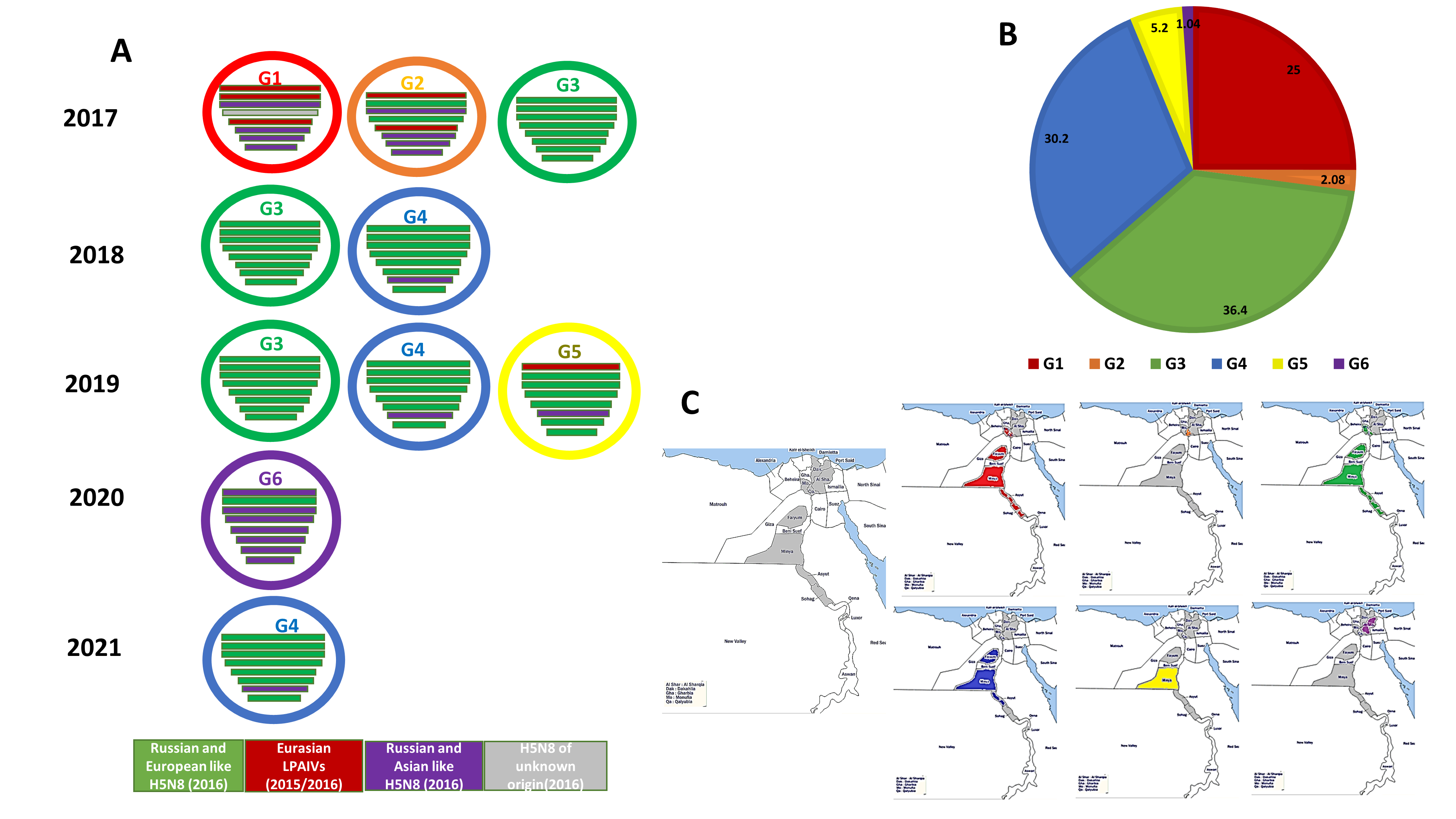
3.3. Genotyping of the Egyptian H5N8 Viruses
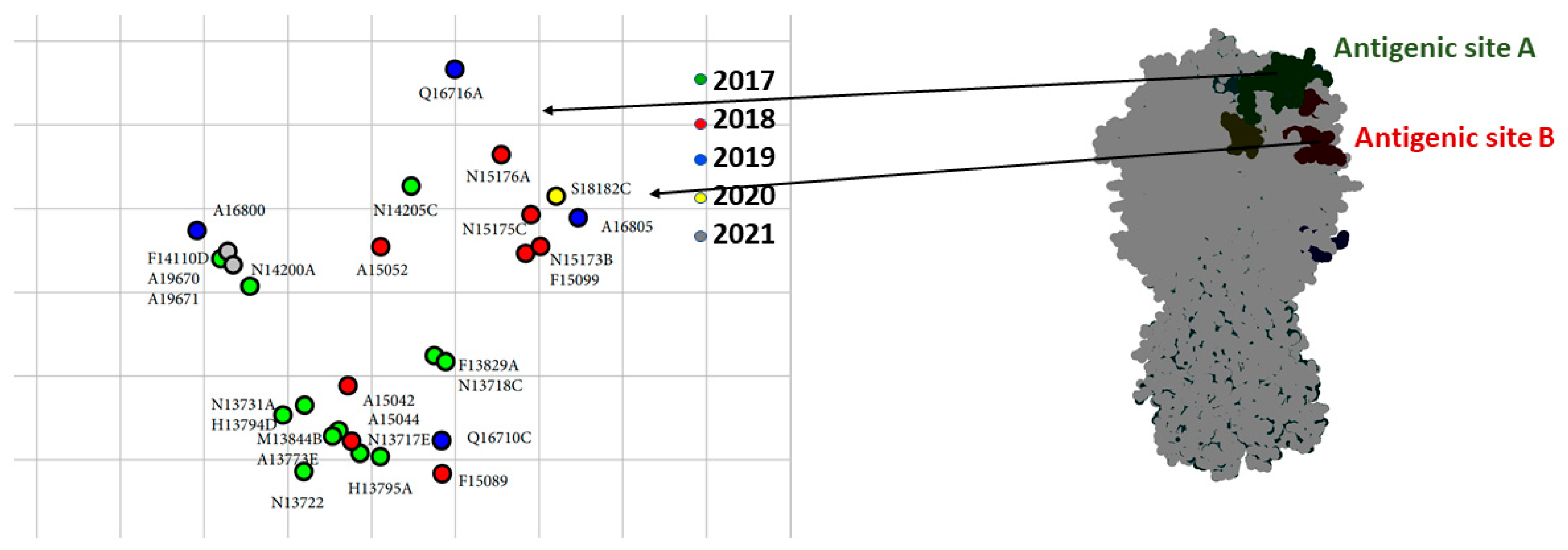
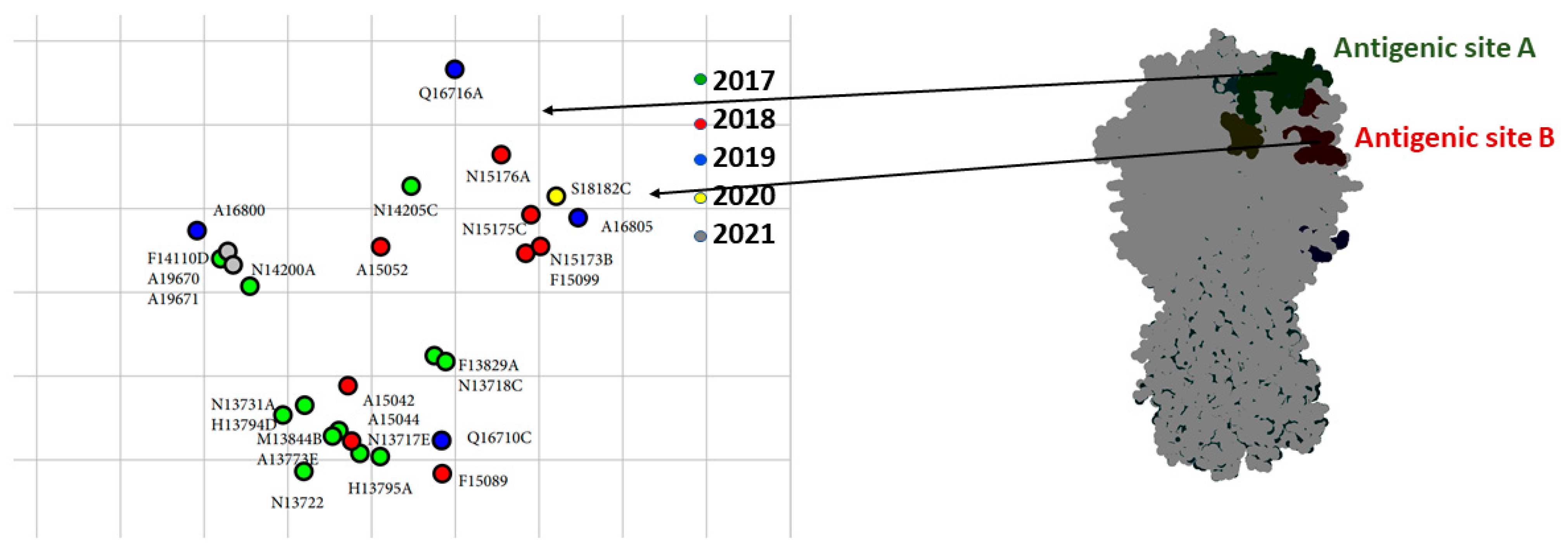
3.4. Antigenic Analysis
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pereira, H.G.; Tumova, B.; Law, V.G. Avian influenza a viruses. Bull. World Health Organ. 1965, 32, 855–860. [Google Scholar] [PubMed]
- Xu, X.; Subbarao, K.; Cox, N.J.; Guo, Y. Genetic characterization of the pathogenic influenza a/goose/guangdong/1/96 (h5n1) virus: Similarity of its hemagglutinin gene to those of h5n1 viruses from the 1997 outbreaks in hong kong. Virology 1999, 261, 15–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dhingra, M.S.; Artois, J.; Dellicour, S.; Lemey, P.; Dauphin, G.; Von Dobschuetz, S.; Van Boeckel, T.P.; Castellan, D.M.; Morzaria, S.; Gilbert, M. Geographical and historical patterns in the emergences of novel highly pathogenic avian influenza (hpai) h5 and h7 viruses in poultry. Front. Vet. Sci. 2018, 5, 84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El Shesheny, R.; Barman, S.; Feeroz, M.; Hasan, M.; Jones-Engel, L.; Franks, J.; Turner, J.; Seiler, P.; Walker, D.; Edwards, K.; et al. Genesis of influenza a(h5n8) viruses. Emerg. Infect. Dis. 2017, 23, 1368–1371. [Google Scholar] [CrossRef]
- Kwon, H.I.; Kim, E.H.; Kim, Y.I.; Park, S.J.; Si, Y.J.; Lee, I.W.; Nguyen, H.D.; Yu, K.M.; Yu, M.A.; Jung, J.H.; et al. Comparison of the pathogenic potential of highly pathogenic avian influenza (hpai) h5n6, and h5n8 viruses isolated in south korea during the 2016–2017 winter season. Emerg. Microbes Infect. 2018, 7, 29. [Google Scholar] [CrossRef]
- Kang, H.M.; Lee, E.K.; Song, B.M.; Heo, G.B.; Jung, J.; Jang, I.; Bae, Y.C.; Jung, S.C.; Lee, Y.J. Experimental infection of mandarin duck with highly pathogenic avian influenza a (h5n8 and h5n1) viruses. Vet. Microbiol. 2017, 198, 59–63. [Google Scholar] [CrossRef]
- Lee, D.-H.; Kwon, J.-H.; Noh, J.-Y.; Park, J.-K.; Yuk, S.-S.; Erdene-Ochir, T.-O.; Lee, J.-B.; Park, S.-Y.; Choi, I.-S.; Lee, S.-W.; et al. Pathogenicity of the korean h5n8 highly pathogenic avian influenza virus in commercial domestic poultry species. Avian Pathol. 2016, 45, 208–211. [Google Scholar] [CrossRef] [Green Version]
- FAO. Telemetry Studies, Egypt Ducks. Last Update: April 2010. Available online: http://www.fao.org/avianflu/en/wildlife/sat_telemetry_egy.htm (accessed on 10 November 2021).
- Selim, A.A.; Erfan, A.M.; Hagag, N.; Zanaty, A.; Samir, A.H.; Samy, M.; Abdelhalim, A.; Arafa, A.A.; Soliman, M.A.; Shaheen, M.; et al. Highly pathogenic avian influenza virus (h5n8) clade 2.3.4.4 infection in migratory birds, Egypt. Emerg. Infect. Dis. 2017, 23, 1048–1051. [Google Scholar] [CrossRef] [Green Version]
- Kandeil, A.; Kayed, A.; Moatasim, Y.; Webby, R.J.; McKenzie, P.P.; Kayali, G.; Ali, M.A. Genetic characterization of highly pathogenic avian influenza a h5n8 viruses isolated from wild birds in Egypt. J. Gen. Virol. 2017, 98, 1573–1586. [Google Scholar] [CrossRef]
- Yehia, N.; Naguib, M.M.; Li, R.; Hagag, N.; El-Husseiny, M.; Mosaad, Z.; Nour, A.; Rabea, N.; Hasan, W.M.; Hassan, M.K.; et al. Multiple introductions of reassorted highly pathogenic avian influenza viruses (h5n8) clade 2.3.4.4b causing outbreaks in wild birds and poultry in Egypt. Infect. Genet. Evol. 2018, 58, 56–65. [Google Scholar] [CrossRef]
- Moatasim, Y.; Kandeil, A.; Aboulhoda, B.E.; El-Shesheny, R.; Alkhazindar, M.; AbdElSalam, E.T.; Kutkat, O.; Kamel, M.N.; El Taweel, A.N.; Mostafa, A.; et al. Comparative virological and pathogenic characteristics of avian influenza h5n8 viruses detected in wild birds and domestic poultry in Egypt during the winter of 2016/2017. Viruses 2019, 11, 990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kandeil, A.; Hicks, J.T.; Young, S.G.; El Taweel, A.N.; Kayed, A.S.; Moatasim, Y.; Kutkat, O.; Bagato, O.; McKenzie, P.P.; Cai, Z.; et al. Active surveillance and genetic evolution of avian influenza viruses in Egypt, 2016–2018. Emerg. Microbes Infect. 2019, 8, 1370–1382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kayali, G.; Kandeil, A.; El-Shesheny, R.; Kayed, A.S.; Gomaa, M.M.; Maatouq, A.M.; Shehata, M.M.; Moatasim, Y.; Bagato, O.; Cai, Z.; et al. Active surveillance for avian influenza virus, Egypt, 2010–2012. Emerg. Infect. Dis. 2014, 20, 542–551. [Google Scholar] [CrossRef] [PubMed]
- Centers for Disease Control and Prevention (CDC). CDC Realtime RT-PCR (rRTPCR) Protocol for Detection and Characterization of Influenza (Version 2007); CDC REF. No. I-007-05; Centers for Disease Control and Prevention (CDC): Atlanta, GA, USA, 2007.
- WHO. Manual for the Laboratory Diagnosis and Virological Surveillance of Influenza. 2011. Available online: http://whqlibdoc.who.int/publications/2011/9789241548090_eng.pdf (accessed on 10 November 2021).
- Chen, G.W.; Chang, S.C.; Mok, C.K.; Lo, Y.L.; Kung, Y.N.; Huang, J.H.; Shih, Y.H.; Wang, J.Y.; Chiang, C.; Chen, C.J.; et al. Genomic signatures of human versus avian influenza a viruses. Emerg. Infect. Dis. 2006, 12, 1353–1360. [Google Scholar] [CrossRef] [PubMed]
- Rolling, T.; Koerner, I.; Zimmermann, P.; Holz, K.; Haller, O.; Staeheli, P.; Kochs, G. Adaptive mutations resulting in enhanced polymerase activity contribute to high virulence of influenza a virus in mice. J. Virol. 2009, 83, 6673–6680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Z.; Chen, H.; Jiao, P.; Deng, G.; Tian, G.; Li, Y.; Hoffmann, E.; Webster, R.G.; Matsuoka, Y.; Yu, K. Molecular basis of replication of duck h5n1 influenza viruses in a mammalian mouse model. J. Virol. 2005, 79, 12058–12064. [Google Scholar] [CrossRef] [Green Version]
- Jiao, P.; Tian, G.; Li, Y.; Deng, G.; Jiang, Y.; Liu, C.; Liu, W.; Bu, Z.; Kawaoka, Y.; Chen, H. A single-amino-acid substitution in the ns1 protein changes the pathogenicity of h5n1 avian influenza viruses in mice. J. Virol. 2008, 82, 1146–1154. [Google Scholar] [CrossRef] [Green Version]
- Lycett, S.J.; Ward, M.J.; Lewis, F.I.; Poon, A.F.; Kosakovsky Pond, S.L.; Brown, A.J. Detection of mammalian virulence determinants in highly pathogenic avian influenza h5n1 viruses: Multivariate analysis of published data. J. Virol. 2009, 83, 9901–9910. [Google Scholar] [CrossRef] [Green Version]
- Durrant, M.G.; Eggett, D.L.; Busath, D.D. Investigation of a recent rise of dual amantadine-resistance mutations in the influenza a m2 sequence. BMC Genet. 2015, 16, S3. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Sun, Y.; Xu, Q.; Tan, Y.; Pu, J.; Yang, H.; Brown, E.G.; Liu, J. Mouse-adapted h9n2 influenza a virus pb2 protein m147l and e627k mutations are critical for high virulence. PLoS ONE 2012, 7, e40752. [Google Scholar] [CrossRef]
- Teng, Q.; Zhang, X.; Xu, D.; Zhou, J.; Dai, X.; Chen, Z.; Li, Z. Characterization of an h3n2 canine influenza virus isolated from tibetan mastiffs in china. Vet. Microbiol. 2013, 162, 345–352. [Google Scholar] [CrossRef] [PubMed]
- Mok, C.K.P.; Lee, H.H.Y.; Lestra, M.; Nicholls, J.M.; Chan, M.C.W.; Sia, S.F.; Zhu, H.; Poon, L.L.M.; Guan, Y.; Peiris, J.S.M. Amino acid substitutions in polymerase basic protein 2 gene contribute to the pathogenicity of the novel a/h7n9 influenza virus in mammalian hosts. J. Virol. 2014, 88, 3568–3576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, M.S.; Deng, M.C.; Lin, Y.J.; Chang, C.Y.; Shieh, H.K.; Shiau, J.Z.; Huang, C.C. Characterization of an h5n1 avian influenza virus from taiwan. Vet. Microbiol. 2007, 124, 193–201. [Google Scholar] [CrossRef] [PubMed]
- Otte, A.; Sauter, M.; Daxer, M.A.; McHardy, A.C.; Klingel, K.; Gabriel, G. Adaptive mutations that occurred during circulation in humans of h1n1 influenza virus in the 2009 pandemic enhance virulence in mice. J. Virol. 2015, 89, 7329–7337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, L.; Wang, C.; Luo, J.; Li, M.; Liu, H.; Zhao, N.; Huang, J.; Zhu, X.; Ma, G.; Yuan, G.; et al. Amino acid substitution k470r in the nucleoprotein increases the virulence of h5n1 influenza a virus in mammals. Front. Microbiol. 2017, 8, 1308. [Google Scholar] [CrossRef] [PubMed]
- Dankar, S.K.; Wang, S.; Ping, J.; Forbes, N.E.; Keleta, L.; Li, Y.; Brown, E.G. Influenza a virus ns1 gene mutations f103l and m106i increase replication and virulence. Virol. J. 2011, 8, 13. [Google Scholar] [CrossRef] [Green Version]
- Subbarao, K.; Shaw, M.W. Molecular aspects of avian influenza (h5n1) viruses isolated from humans. Rev. Med. Virol. 2000, 10, 337–348. [Google Scholar] [CrossRef]
- Shaw, M.; Cooper, L.; Xu, X.; Thompson, W.; Krauss, S.; Guan, Y.; Zhou, N.; Klimov, A.; Cox, N.; Webster, R.; et al. Molecular changes associated with the transmission of avian influenza a h5n1 and h9n2 viruses to humans. J. Med. Virol. 2002, 66, 107–114. [Google Scholar] [CrossRef]
- Guilligay, D.; Tarendeau, F.; Resa-Infante, P.; Coloma, R.; Crepin, T.; Sehr, P.; Lewis, J.; Ruigrok, R.W.; Ortin, J.; Hart, D.J.; et al. The structural basis for cap binding by influenza virus polymerase subunit pb2. Nat. Struct. Mol. Biol. 2008, 15, 500–506. [Google Scholar] [CrossRef]
- Kuzuhara, T.; Kise, D.; Yoshida, H.; Horita, T.; Murazaki, Y.; Nishimura, A.; Echigo, N.; Utsunomiya, H.; Tsuge, H. Structural basis of the influenza a virus rna polymerase pb2 rna-binding domain containing the pathogenicity-determinant lysine 627 residue. J. Biol. Chem. 2009, 284, 6855–6860. [Google Scholar] [CrossRef] [Green Version]
- Naffakh, N.; Tomoiu, A.; Rameix-Welti, M.A.; van der Werf, S. Host restriction of avian influenza viruses at the level of the ribonucleoproteins. Annu. Rev. Microbiol. 2008, 62, 403–424. [Google Scholar] [CrossRef] [PubMed]
- Taubenberger, J.K.; Reid, A.H.; Lourens, R.M.; Wang, R.; Jin, G.; Fanning, T.G. Characterization of the 1918 influenza virus polymerase genes. Nature 2005, 437, 889–893. [Google Scholar] [CrossRef] [PubMed]
- Wanitchang, A.; Jengarn, J.; Jongkaewwattana, A. The n terminus of pa polymerase of swine-origin influenza virus h1n1 determines its compatibility with pb2 and pb1 subunits through a strain-specific amino acid serine 186. Virus Res. 2011, 155, 325–333. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Tang, G.; Huang, Y.; Yu, C.; Li, S.; Zhuang, L.; Fu, L.; Wang, S.; Li, N.; Li, X.; et al. A returning migrant worker with avian influenza a (h7n9) virus infection in Guizhou, China: A case report. J. Med. Case Rep. 2015, 9, 109. [Google Scholar] [CrossRef] [Green Version]
- Brown, E.G.; Liu, H.; Kit, L.C.; Baird, S.; Nesrallah, M. Pattern of mutation in the genome of influenza a virus on adaptation to increased virulence in the mouse lung: Identification of functional themes. Proc. Natl. Acad. Sci. USA 2001, 98, 6883–6888. [Google Scholar] [CrossRef] [Green Version]
- Finkelstein, D.B.; Mukatira, S.; Mehta, P.K.; Obenauer, J.C.; Su, X.; Webster, R.G.; Naeve, C.W. Persistent host markers in pandemic and h5n1 influenza viruses. J. Virol. 2007, 81, 10292–10299. [Google Scholar] [CrossRef] [Green Version]
- Yamaji, R.; Yamada, S.; Le, M.Q.; Ito, M.; Sakai-Tagawa, Y.; Kawaoka, Y. Mammalian adaptive mutations of the pa protein of highly pathogenic avian h5n1 influenza virus. J. Virol. 2015, 89, 4117–4125. [Google Scholar] [CrossRef] [Green Version]
- Gabriel, G.; Dauber, B.; Wolff, T.; Planz, O.; Klenk, H.-D.; Stech, J. The viral polymerase mediates adaptation of an avian influenza virus to a mammalian host. Proc. Natl. Acad. Sci. USA 2005, 102, 18590–18595. [Google Scholar] [CrossRef] [Green Version]
- Van Poucke, S.; Doedt, J.; Baumann, J.; Qiu, Y.; Matrosovich, T.; Klenk, H.D.; Van Reeth, K.; Matrosovich, M. Role of substitutions in the hemagglutinin in the emergence of the 1968 pandemic influenza virus. J. Virol. 2015, 89, 12211–12216. [Google Scholar] [CrossRef] [Green Version]
- Katz, J.M.; Lu, X.; Tumpey, T.M.; Smith, C.B.; Shaw, M.W.; Subbarao, K. Molecular correlates of influenza a h5n1 virus pathogenesis in mice. J. Virol. 2000, 74, 10807–10810. [Google Scholar] [CrossRef] [Green Version]
- Pan, C.; Jiang, S. E14-f55 combination in m2 protein: A putative molecular determinant responsible for swine-origin influenza a virus transmission in humans. PLoS Curr. 2009, 1, RRN1044. [Google Scholar] [CrossRef] [PubMed]
- Soubies, S.M.; Volmer, C.; Croville, G.; Loupias, J.; Peralta, B.; Costes, P.; Lacroux, C.; Guerin, J.L.; Volmer, R. Species-specific contribution of the four c-terminal amino acids of influenza a virus ns1 protein to virulence. J. Virol. 2010, 84, 6733–6747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orozovic, G.; Orozovic, K.; Lennerstrand, J.; Olsen, B. Detection of resistance mutations to antivirals oseltamivir and zanamivir in avian influenza a viruses isolated from wild birds. PLoS ONE 2011, 6, e16028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salaheldin, A.H.; El-Hamid, H.S.; Elbestawy, A.R.; Veits, J.; Hafez, H.M.; Mettenleiter, T.C.; Abdelwhab, E.M. Multiple introductions of influenza a(h5n8) virus into poultry, Egypt, 2017. Emerg. Infect. Dis. 2018, 24, 943–946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yehia, N.; Hassan, W.M.M.; Sedeek, A.; Elhusseiny, M.H. Genetic variability of avian influenza virus subtype h5n8 in Egypt in 2017 and 2018. Arch. Virol. 2020, 165, 1357–1366. [Google Scholar] [CrossRef]
- Hagag, N.M.; Erfan, A.M.; El-Husseiny, M.; Shalaby, A.G.; Saif, M.A.; Tawakol, M.M.; Nour, A.A.; Selim, A.A.; Arafa, A.S.; Hassan, M.K.; et al. Isolation of a novel reassortant highly pathogenic avian influenza (h5n2) virus in Egypt. Viruses 2019, 11, 565. [Google Scholar] [CrossRef] [Green Version]
- Hassan, K.E.; King, J.; El-Kady, M.; Afifi, M.; Abozeid, H.H.; Pohlmann, A.; Beer, M.; Harder, T. Novel reassortant highly pathogenic avian influenza a(h5n2) virus in broiler chickens, Egypt. Emerg. Infect. Dis. 2020, 26, 129–133. [Google Scholar] [CrossRef]
- Yehia, N.; Erfan, A.M.; Adel, A.; El-Tayeb, A.; Hassan, W.M.M.; Samy, A.; Abd El-Hack, M.E.; El-Saadony, M.T.; El-Tarabily, K.A.; Ahmed, K.A. Pathogenicity of three genetically distinct and highly pathogenic Egyptian H5N8 avian influenza viruses in chickens. Poult. Sci. 2022, 101, 101662. [Google Scholar] [CrossRef]
- Gomaa, M.R.; El Rifay, A.S.; Abu Zeid, D.; Elabd, M.A.; Elabd, E.; Kandeil, A.; Shama, N.M.A.; Kamel, M.N.; Marouf, M.A.; Barakat, A.; et al. Incidence and seroprevalence of avian influenza in a cohort of backyard poultry growers, Egypt, August 2015–March 2019. Emerg. Infect. Dis. 2020, 26, 2129–2136. [Google Scholar] [CrossRef]
- Kandeil, A.; Sabir, J.S.M.; Abdelaal, A.; Mattar, E.H.; El-Taweel, A.N.; Sabir, M.J.; Khalil, A.A.; Webby, R.; Kayali, G.; Ali, M.A. Efficacy of commercial vaccines against newly emerging avian influenza h5n8 virus in Egypt. Sci. Rep. 2018, 8, 9697. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein | aa Site | Virulent | Avirulent | Percentage | References |
|---|---|---|---|---|---|
| PB2 | 627 | K | E | E (100%) | [23] |
| 147 | L | M | I (100%) | [23] | |
| 250 | G | V | V (100%) | [23] | |
| 504 | V | I | V (100%) | [18] | |
| 701 | N | D | D (100%) | [24] | |
| 591 | K | Q | Q (100%) | [25] | |
| PB1 | 317 | I | M/V | M (100%) | [17,26] |
| PA | 127 | V | I | V (96.88%), I (3.12%) | [21] |
| 672 | L | F | L (100%) | [19] | |
| 100 | R | V | V (100%) | [27] | |
| 550 | L | I | L (100%) | [18] | |
| NP | 470 | R | K | K (100%) | [28] |
| M2 | 64 | S/A/F | P | S (100%) | [21] |
| 69 | P | L | P (100%) | [21] | |
| NS1 | 42 | S | A/P | S (100%) | [20] |
| 92 | E | D | D (100%) | [26] | |
| 103 | L | F | F (97.9%), L (2.1%) | [29] | |
| 106 | I | M | M (100%) | [29] | |
| 189 | N | D/G | D (95.84%), N (4.16%) | [30] | |
| PDZ motif (227–230) | Presence | Deletion | Presence (22.9%), Deletion (77.1%) | ||
| NS2 | 31 | I | M | M (98%), (2%) I | [30] |
| 56 | Y | H/L | H (100%) | [30] |
| Viral Protein | aa Site | Avian Preference | Mammalian Preference | Egyptian Strains | References |
|---|---|---|---|---|---|
| PB2 | 44 | A | S | A (100%) | [17,31] |
| 64 | M | T | M (100%) | [32] | |
| 81 | T | M | T (100%) | [31] | |
| 199 | A | S | A (100%) | [17,31] | |
| 591 | Q | K | Q (100%) | [25] | |
| 627 | E | K | E (100%) | [23] | |
| 661 | A | T | A (92.7%), V (7.3%) | [33] | |
| 701 | D | N | D (100%) | [24] | |
| 702 | K | R | K (100%) | [33] | |
| PB1 | 13 | L | P | P (100%) | [34] |
| 336 | V | I | V (100%) | [17] | |
| 375 | N | S | N (100%) | [35] | |
| PA | 28 | P | L | P (100%) | [36] |
| 55 | D | N | D (100%) | [17,31] | |
| 57 | R | Q | R (100%) | [17] | |
| 100 | V | A | V (100%) | [37] | |
| 133 | E | G | E (100%) | [38] | |
| 225 | S | C | S (100%) | [39] | |
| 241 | C | Y | C (100%) | [40] | |
| 268 | L | I | L (100%) | [39] | |
| 356 | K | R | K (100%) | [17] | |
| 382 | E | D | E (100%) | [31,35] | |
| 404 | A | S | A (100%) | [17] | |
| 409 | S | N | S (100%) | [17,31] | |
| 552 | T | S | T (100%) | [39] | |
| 615 | K | L | K (29.2%), R (70.8%) | [41] | |
| HA | 222 | Q | L | Q (100%) | [42] |
| 224 | G | S | G (100%) | [42] | |
| NP | 33 | V | I | V (94%), I (5%), D (1%) | [17,31] |
| 61 | I | L | I (100%) | [31,39] | |
| 109 | I | V | I (100%) | [17] | |
| 136 | L | M | L (100%) | [31] | |
| 214 | R | K | R (100%) | [17,31] | |
| 313 | F | Y | F (100%) | [17,31] | |
| 357 | Q | K | Q (100%) | [17] | |
| 372 | E | D | E (100%) | [17] | |
| 398 | K | Q | Q (100%) | [17] | |
| 455 | D | E | D (94.8%), N (3.1%), E (2.1%) | [17] | |
| M1 | 15 | V | I | V (100%) | [43] |
| 115 | V | I | V (89.6%), I (9.4%), T (1%) | [39] | |
| 121 | T | A | T (100%) | [39] | |
| 137 | T | A | T (100%) | [31,39] | |
| M2 | 11 | T | I | T (100%) | [17] |
| 16 | E | G/D | E (94.8%), G (5.2%) | [31] | |
| 20 | S | N | S (100%) | [17,31] | |
| 28 | I | I/V | I (100%) | [31] | |
| 57 | Y | H | Y (100%) | [17] | |
| 55 | L | F | L (100%) | [44] | |
| 86 | V | A | V (100%) | [17] | |
| NS1 | 227 | E | K/R | G (22.9%), deletion (77.1%) | [45] |
| full length | 217 | 230 | 230 (22.9%) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kandeil, A.; Moatasim, Y.; El Taweel, A.; El Sayes, M.; Rubrum, A.; Jeevan, T.; McKenzie, P.P.; Webby, R.J.; Ali, M.A.; Kayali, G.; et al. Genetic and Antigenic Characteristics of Highly Pathogenic Avian Influenza A(H5N8) Viruses Circulating in Domestic Poultry in Egypt, 2017–2021. Microorganisms 2022, 10, 595. https://doi.org/10.3390/microorganisms10030595
Kandeil A, Moatasim Y, El Taweel A, El Sayes M, Rubrum A, Jeevan T, McKenzie PP, Webby RJ, Ali MA, Kayali G, et al. Genetic and Antigenic Characteristics of Highly Pathogenic Avian Influenza A(H5N8) Viruses Circulating in Domestic Poultry in Egypt, 2017–2021. Microorganisms. 2022; 10(3):595. https://doi.org/10.3390/microorganisms10030595
Chicago/Turabian StyleKandeil, Ahmed, Yassmin Moatasim, Ahmed El Taweel, Mohamed El Sayes, Adam Rubrum, Trushar Jeevan, Pamela P. McKenzie, Richard J. Webby, Mohamed A. Ali, Ghazi Kayali, and et al. 2022. "Genetic and Antigenic Characteristics of Highly Pathogenic Avian Influenza A(H5N8) Viruses Circulating in Domestic Poultry in Egypt, 2017–2021" Microorganisms 10, no. 3: 595. https://doi.org/10.3390/microorganisms10030595
APA StyleKandeil, A., Moatasim, Y., El Taweel, A., El Sayes, M., Rubrum, A., Jeevan, T., McKenzie, P. P., Webby, R. J., Ali, M. A., Kayali, G., & El-Shesheny, R. (2022). Genetic and Antigenic Characteristics of Highly Pathogenic Avian Influenza A(H5N8) Viruses Circulating in Domestic Poultry in Egypt, 2017–2021. Microorganisms, 10(3), 595. https://doi.org/10.3390/microorganisms10030595