
Kingella kingae RtxA Cytotoxin in the Context of Other RTX Toxins



Abstract
1. Introduction
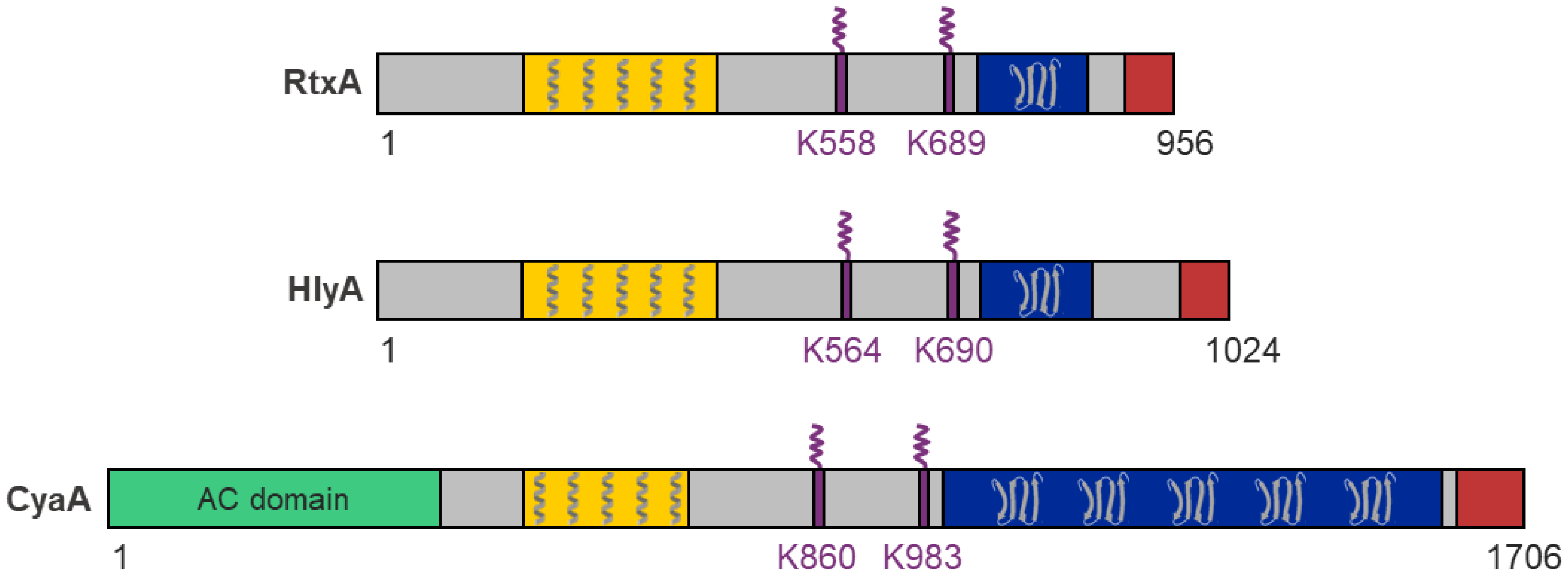
- A hydrophobic pore-forming domain in the N-terminal part of the molecule that harbors several putative transmembrane α-helices;
- An acylated segment where the RTX protoxin is activated and converted into the RTX toxin by a co-expressed toxin-activating acyltransferase that catalyzes the covalent posttranslational acylation of conserved lysine residues;
- A typical C-terminal calcium-binding RTX domain containing various numbers of the conserved glycine- and aspartate-rich nonapeptide repeats of a consensus sequence G-G-X-G-X-D-X-U-X (X represents any residue and U represents the hydrophobic residue leucine, valine or isoleucine), which form calcium-binding sites;
- A C-proximal unprocessed secretion signal for export of the RTX toxin from the bacterial cell by the type I secretion system (T1SS).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| RTX Toxin | Bacterium; Disease | Size (kDa) | Acylated Residues | Species and Cell Specificity 1 | Ref. |
|---|---|---|---|---|---|
| RtxA | Kingella kingae; Osteoarticular infections, endocarditis and others | 105 | K558 K689 | Broad: human epithelial and monocyte cell lines, mouse monocyte/macrophage cell line, rabbit fibroblast cell line, sheep erythrocytes | [1,23,36,37,39] |
| HlyA | Uropathogenic Escherichia coli; Urinary tract infections | 110 | K564 K690 | Broad: primary human epithelial cells and leukocytes, primary rat epithelial cells, primary porcine endothelial cells, human epithelial, promonocytic myeloid, T- and B-lymphocyte cell lines, porcine endothelial cell line, erythrocytes of various species | [40,41,42,43,44,45,46,47,48] |
| CyaA | Bordetella pertussis; Whooping cough | 177 | K860 K983 | Narrow: primary human myeloid cells, human monocyte and splenic myeloid dendritic cell lines, mouse macrophage cell line | [49,50,51,52,53,54,55,56,57] |
| LtxA | Aggregatibacter actinomycetemcomitans; Aggressive periodontitis | 116 | K562 K687 | Narrow: primary human monocytes, primary human and primate polymorphonuclear leukocytes, human T- and B-lymphocyte, monocyte, and promyeloblast cell lines | [58,59,60,61,62,63,64,65,66] |
| LktA | Mannheimia haemolytica; Pneumonic pasteurellosis | 102 | K554 K669 | Narrow: primary ruminant leukocytes and platelets, bovine B-lymphosarcoma cell line | [67,68,69,70,71] |
| ApxIA | Actinobacillus pleuropneumoniae; Porcine pleuropneumonia | 110 | K560 K686 | Broad: primary porcine alveolar macrophages and neutrophils, primary bovine and porcine endothelial cells, sheep, swine and horse erythrocytes | [72,73,74,75,76,77,78] |
| ApxIIIA | Actinobacillus pleuropneumoniae; Porcine pleuropneumonia | 120 | K571 K702 | Narrow: primary porcine and wild boar peripheral blood mononuclear cells, primary bovine and porcine endothelial cells | [73,77,79,80] |
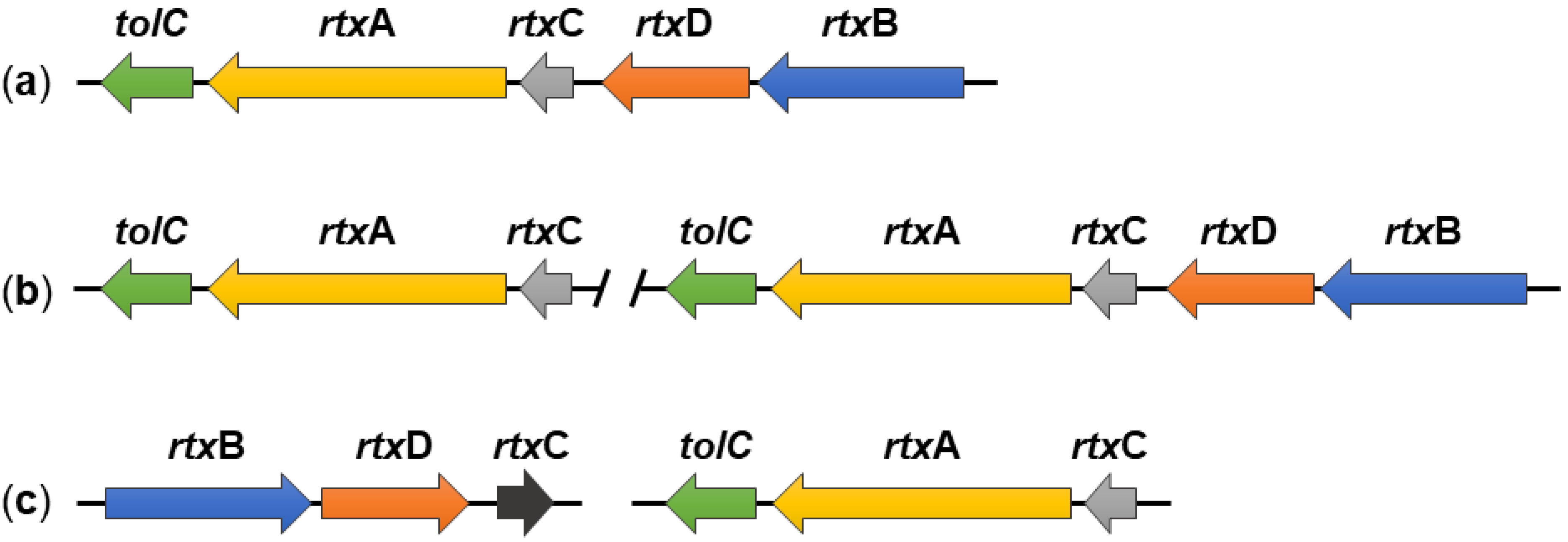
2. Genes Required for RtxA Production, Activation and Secretion
3. Polymorphism of the rtxA Gene
4. The rtxA Gene as a Diagnostic Marker of K. kingae
5. Regulation of rtxA Gene Expression by Phase Variation
6. General Structural Features of RtxA and Other RTX Toxins
6.1. N-Terminal Part with Pore-Forming Domain
6.2. Acylated Segment and Its Posttranslational Modification
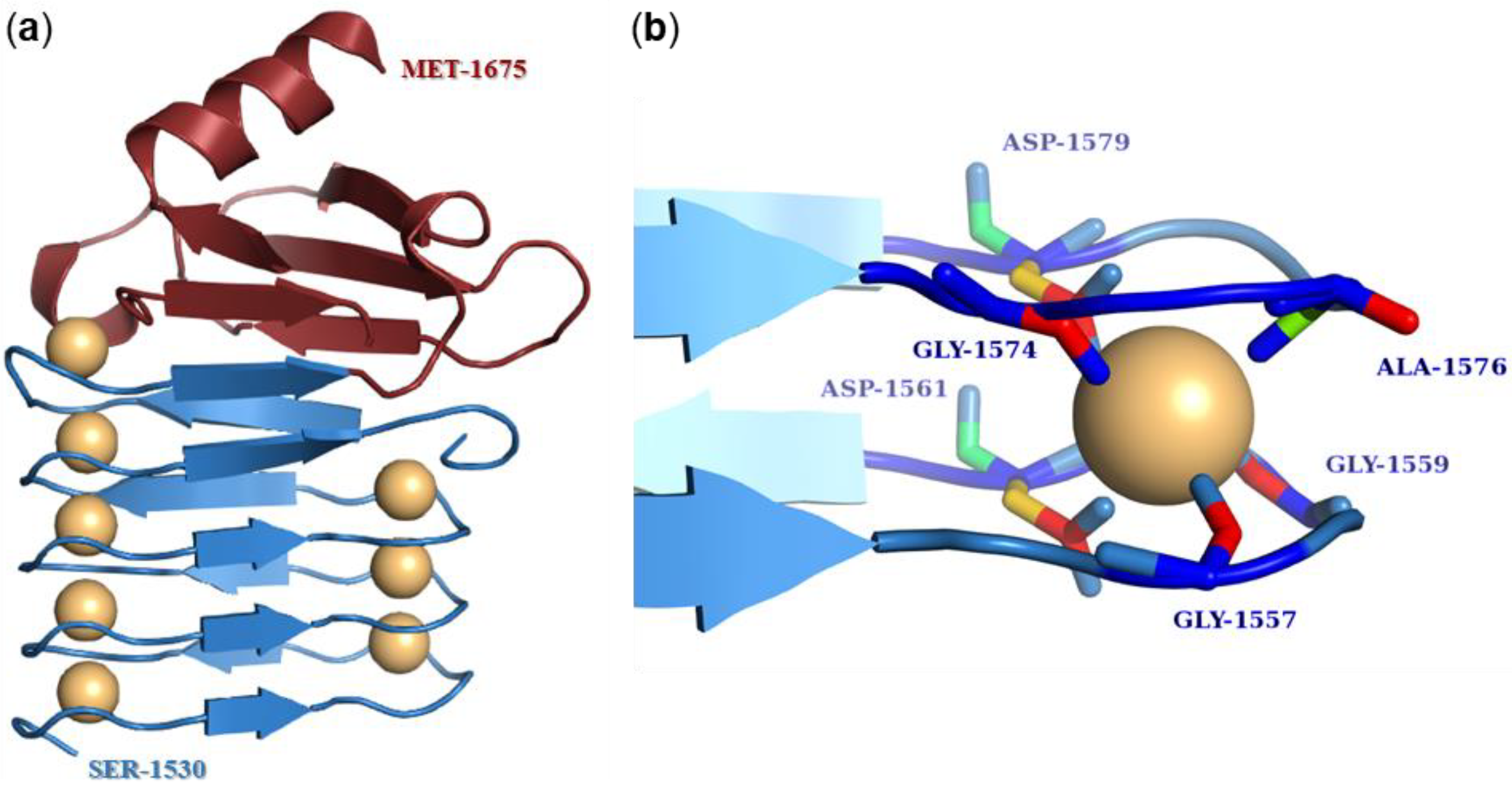
6.3. Calcium-Binding Repeat Domain
6.4. C-Terminal Secretion Signal
6.5. Adenylate Cyclase Domain and Linker Segment of CyaA
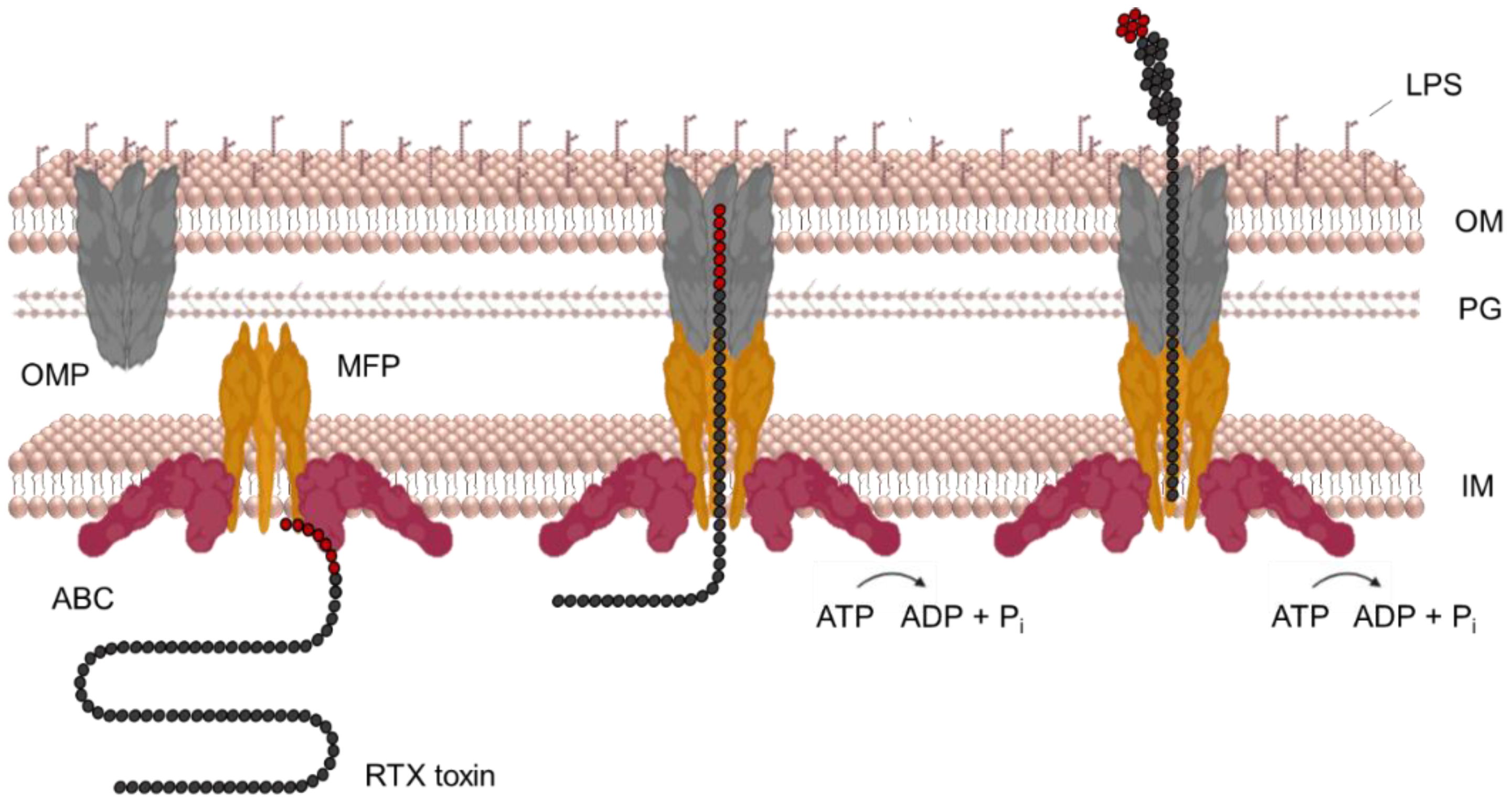
7. Secretion of RtxA and Other RTX Toxins
8. Interaction of RtxA and Other RTX Toxins with Target Cells
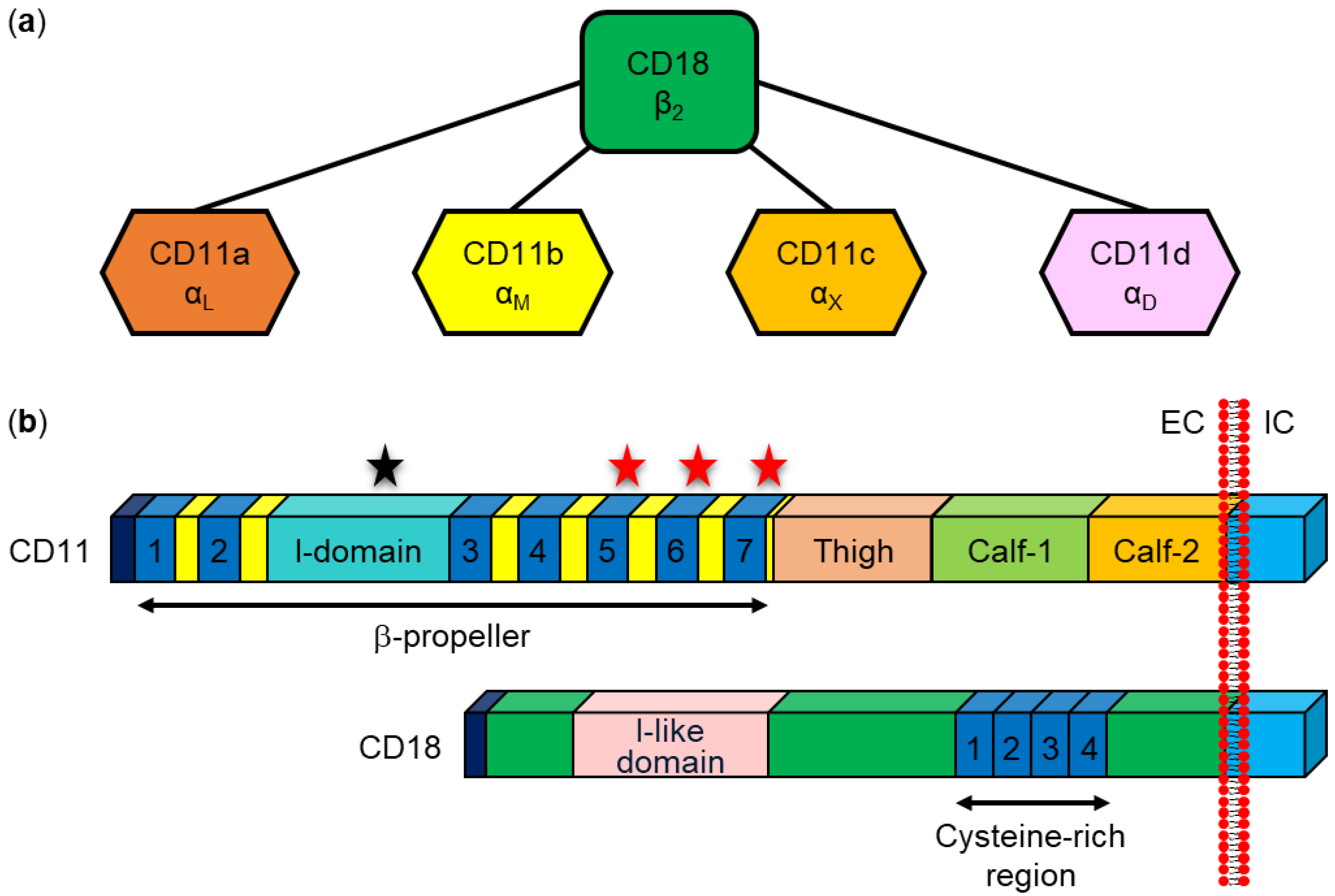
8.1. Interaction with Specific β2 Integrin Receptors
| RTX Toxin | β2 Integrin Subunit | Binding Site(s) on β2 Integrin Subunit | Ref. | Other Cell Surface Structures | Ref. |
|---|---|---|---|---|---|
| RtxA | None | [262] |
| [37,262] | |
| HlyA | CD18 | NA 1 | [48,263] |
| [264,265,266] |
| CyaA | CD11b |
| [53,57] |
| [55,267,268,269,270,271,272] |
| LtxA | CD18 |
| [48,64,263,273,274] |
| [63,65,275,276,277] |
| CD11a |
| [48,274,278,279] | |||
| LktA | CD18 |
| [70,280,281,282,283,284,285,286,287,288] | NA 1 | |
| ApxIIIA | CD18 | NA 1 | [257] | NA 1 |
8.2. β2 Integrin Receptor-Independent Interaction
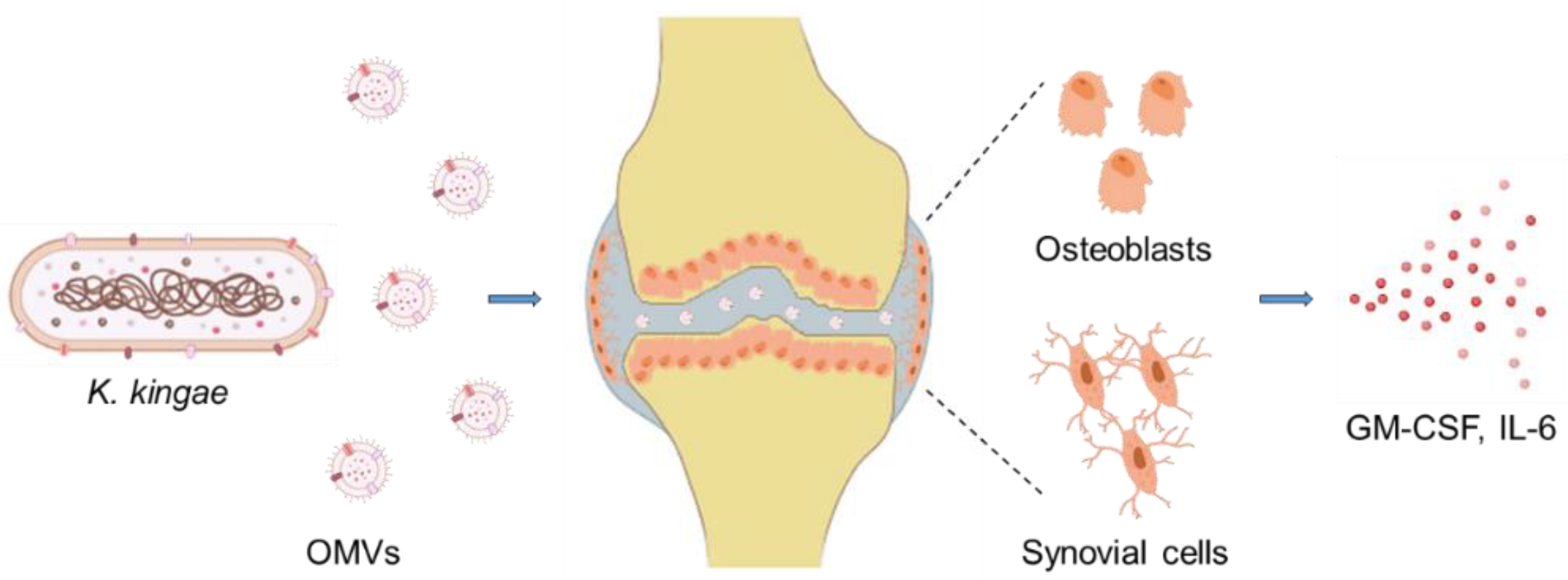
8.3. Interaction via Outer Membrane Vesicles
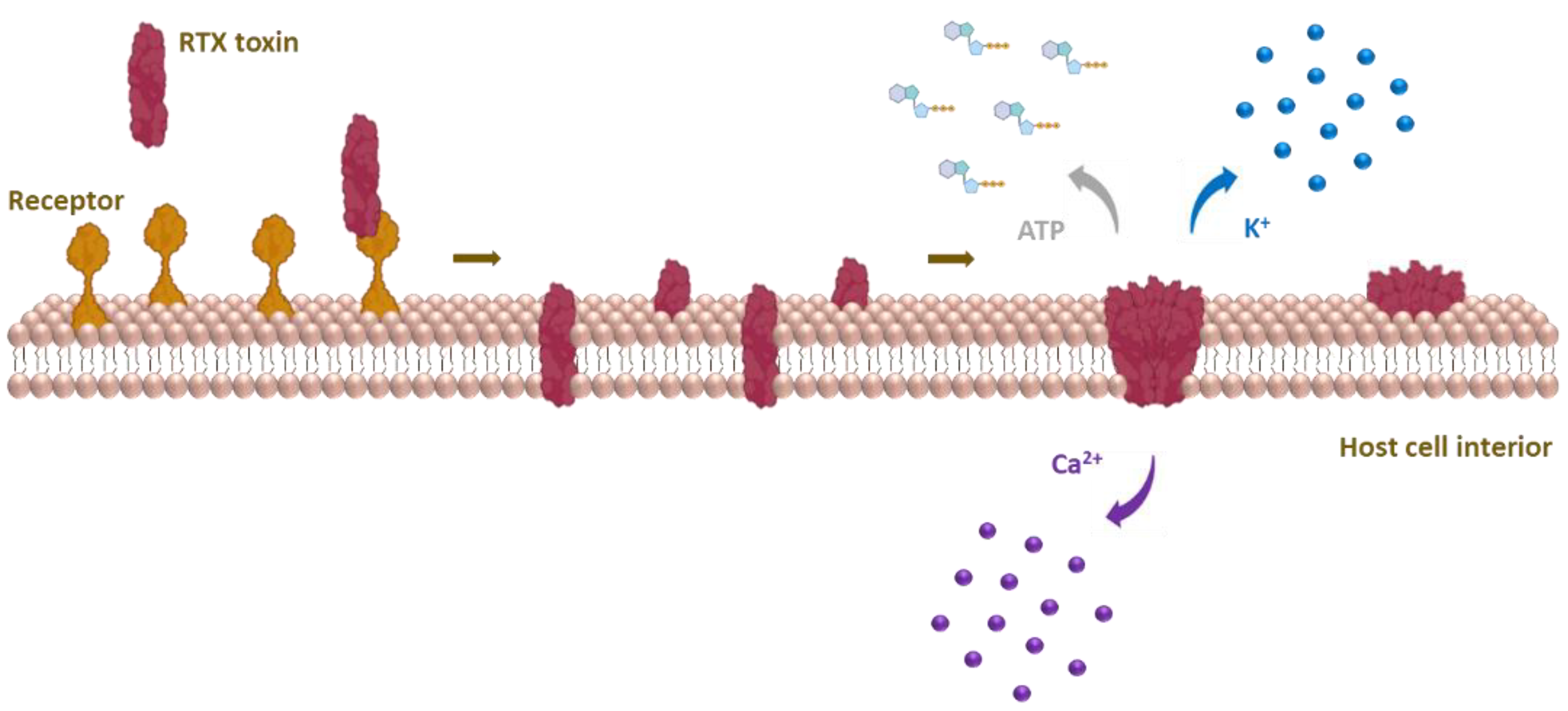
9. Formation of Membrane Pores by RtxA and Other RTX Toxins
- Complementation analysis of inactive mutants of CyaA [315] and HlyA [316] resulted in partial recovery of their hemolytic activities. The combination of truncated, non-overlapping CyaA and HlyA variants restored the ability of the toxins to permeabilize the cell membrane [315,316]. Experiments with truncated variants of CyaA suggested that functional complementation might occur via calcium-binding nonapeptide repeats in the C-terminal part of the toxin molecule [191].
- The acylation status of CyaA appears to modulate the propensity of the toxin to form oligomeric membrane pores, as hemolytic (pore-forming) activity was influenced by the attachment of various fatty acyl chains [133,148,149,150]. It was also suggested that the acyl chains in HlyA promote the protein–protein interactions necessary for oligomerization of the toxin [162].
- FRET analysis revealed selective self-association of CyaA molecules in solution leading to oligomeric complexes [314].
- The strongest evidence for oligomerization of RTX toxins was provided in 2009 when FRET analysis of HlyA revealed its oligomerization in membranes of sheep erythrocytes [162] and oligomeric complexes of CyaA formed in erythrocyte membranes were detected by immunogold labeling and blue native polyacrylamide gel electrophoresis [203].
| RTX Toxin | Pore Diameter (nm) | Single-Pore Conductance (pS) 1 | Single-Pore Lifetime (s) | Ref. |
|---|---|---|---|---|
| RtxA | ~1.9 | ~400, ~419 * | ~0.24 *, 2.12 * | [36,37] |
| HlyA | ~1–3 | ~500 | ~2 | [32,33,34] |
| CyaA | ~0.6–0.8 | ~9–11 | ~2 | [133,139,148,196,323,324] |
| LtxA | ~0.9 | ~406, 262, 118 | NA 2 | [141,319,320] |
10. Effects of RtxA and Other RTX Toxins on Host Cells
11. Conclusions
Author Contributions
Funding
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Yagupsky, P.; Porsch, E.; St Geme, J.W., 3rd. Kingella kingae: An emerging pathogen in young children. Pediatrics 2011, 127, 557–565. [Google Scholar] [CrossRef]
- Henriksen, S.D.; Bovre, K. Moraxella kingii sp.nov., a haemolytic, saccharolytic species of the genus Moraxella. J. Gen. Microbiol. 1968, 51, 377–385. [Google Scholar] [CrossRef] [PubMed]
- Yagupsky, P. Kingella kingae: Carriage, transmission, and disease. Clin. Microbiol. Rev. 2015, 28, 54–79. [Google Scholar] [CrossRef] [PubMed]
- Bøvre, K.; Henriksen, S.D.; Jonsson, V. Correction of the specific epithet kingii in the combinations Moraxella kingii Henriksen and Bøvre 1968 and Pseudomonas kingii Jonsson 1970 to kingae. Int. J. Syst. Evol. Microbiol. 1974, 24, 307. [Google Scholar] [CrossRef]
- Henriksen, S.D.; Bøvre, K. Transfer of Moraxella kingae Henriksen and Bøvre to the Genus Kingella gen. nov. in the Family Neisseriaceae. Int. J. Syst. Evol. Microbiol. 1976, 26, 447–450. [Google Scholar] [CrossRef]
- Ceroni, D.; Dubois-Ferrière, V.; Cherkaoui, A.; Lamah, L.; Renzi, G.; Lascombes, P.; Wilson, B.; Schrenzel, J. 30 years of study of Kingella kingae: Post tenebras, lux. Future Microbiol. 2013, 8, 233–245. [Google Scholar] [CrossRef]
- Principi, N.; Esposito, S. Kingella kingae infections in children. BMC Infect. Dis. 2015, 15, 260. [Google Scholar] [CrossRef]
- Gene, A.; Garcia-Garcia, J.J.; Sala, P.; Sierra, M.; Huguet, R. Enhanced culture detection of Kingella kingae, a pathogen of increasing clinical importance in pediatrics. Pediatr. Infect. Dis. J. 2004, 23, 886–888. [Google Scholar] [CrossRef]
- Moumile, K.; Merckx, J.; Glorion, C.; Berche, P.; Ferroni, A. Osteoarticular infections caused by Kingella kingae in children: Contribution of polymerase chain reaction to the microbiologic diagnosis. Pediatr. Infect. Dis. J. 2003, 22, 837–839. [Google Scholar] [CrossRef]
- Verdier, I.; Gayet-Ageron, A.; Ploton, C.; Taylor, P.; Benito, Y.; Freydiere, A.M.; Chotel, F.; Berard, J.; Vanhems, P.; Vandenesch, F. Contribution of a broad range polymerase chain reaction to the diagnosis of osteoarticular infections caused by Kingella kingae: Description of twenty-four recent pediatric diagnoses. Pediatr. Infect. Dis. J. 2005, 24, 692–696. [Google Scholar] [CrossRef]
- Dubnov-Raz, G.; Ephros, M.; Garty, B.Z.; Schlesinger, Y.; Maayan-Metzger, A.; Hasson, J.; Kassis, I.; Schwartz-Harari, O.; Yagupsky, P. Invasive pediatric Kingella kingae Infections: A nationwide collaborative study. Pediatr. Infect. Dis. J. 2010, 29, 639–643. [Google Scholar] [CrossRef] [PubMed]
- Yagupsky, P. Detection of Respiratory Colonization by Kingella kingae and the Novel Kingella negevensis Species in Children: Uses and Methodology. J. Clin. Microbiol. 2018, 56, e00633-18. [Google Scholar] [CrossRef]
- Gouveia, C.; Duarte, M.; Norte, S.; Arcangelo, J.; Pinto, M.; Correia, C.; Simoes, M.J.; Canhao, H.; Tavares, D. Kingella kingae Displaced S. aureus as the Most Common Cause of Acute Septic Arthritis in Children of All Ages. Pediatr. Infect. Dis. J. 2021, 40, 623–627. [Google Scholar] [CrossRef] [PubMed]
- Yagupsky, P.; Dagan, R.; Prajgrod, F.; Merires, M. Respiratory carriage of Kingella kingae among healthy children. Pediatr. Infect. Dis. J. 1995, 14, 673–678. [Google Scholar] [CrossRef]
- Yagupsky, P.; Weiss-Salz, I.; Fluss, R.; Freedman, L.; Peled, N.; Trefler, R.; Porat, N.; Dagan, R. Dissemination of Kingella kingae in the community and long-term persistence of invasive clones. Pediatr. Infect. Dis. J. 2009, 28, 707–710. [Google Scholar] [CrossRef]
- Yagupsky, P.; Peled, N.; Katz, O. Epidemiological features of invasive Kingella kingae infections and respiratory carriage of the organism. J. Clin. Microbiol. 2002, 40, 4180–4184. [Google Scholar] [CrossRef] [PubMed]
- Yagupsky, P.; Porat, N.; Pinco, E. Pharyngeal colonization by Kingella kingae in children with invasive disease. Pediatr. Infect. Dis. J. 2009, 28, 155–157. [Google Scholar] [CrossRef]
- Kehl-Fie, T.E.; Miller, S.E.; St Geme, J.W., 3rd. Kingella kingae expresses type IV pili that mediate adherence to respiratory epithelial and synovial cells. J. Bacteriol. 2008, 190, 7157–7163. [Google Scholar] [CrossRef]
- Porsch, E.A.; Kehl-Fie, T.E.; St Geme, J.W., 3rd. Modulation of Kingella kingae adherence to human epithelial cells by type IV Pili, capsule, and a novel trimeric autotransporter. mBio 2012, 3, e00372-12. [Google Scholar] [CrossRef]
- Basmaci, R.; Bonacorsi, S.; Ilharreborde, B.; Doit, C.; Lorrot, M.; Kahil, M.; Visseaux, B.; Houhou, N.; Bidet, P. High respiratory virus oropharyngeal carriage rate during Kingella kingae osteoarticular infections in children. Future Microbiol. 2015, 10, 9–14. [Google Scholar] [CrossRef]
- Yagupsky, P.; Dagan, R.; Howard, C.B.; Einhorn, M.; Kassis, I.; Simu, A. Clinical features and epidemiology of invasive Kingella kingae infections in southern Israel. Pediatrics 1993, 92, 800–804. [Google Scholar] [CrossRef] [PubMed]
- Sena, A.C.; Seed, P.; Nicholson, B.; Joyce, M.; Cunningham, C.K. Kingella kingae endocarditis and a cluster investigation among daycare attendees. Pediatr. Infect. Dis. J. 2010, 29, 86–88. [Google Scholar] [CrossRef] [PubMed]
- Kehl-Fie, T.E.; St Geme, J.W., 3rd. Identification and characterization of an RTX toxin in the emerging pathogen Kingella kingae. J. Bacteriol. 2007, 189, 430–436. [Google Scholar] [CrossRef] [PubMed]
- Ceroni, D.; Cherkaoui, A.; Ferey, S.; Kaelin, A.; Schrenzel, J. Kingella kingae osteoarticular infections in young children: Clinical features and contribution of a new specific real-time PCR assay to the diagnosis. J. Pediatr. Orthop. 2010, 30, 301–304. [Google Scholar] [CrossRef]
- Cherkaoui, A.; Ceroni, D.; Emonet, S.; Lefevre, Y.; Schrenzel, J. Molecular diagnosis of Kingella kingae osteoarticular infections by specific real-time PCR assay. J. Med. Microbiol. 2009, 58, 65–68. [Google Scholar] [CrossRef]
- Opota, O.; Laurent, S.; Pillonel, T.; Leger, M.; Trachsel, S.; Prod’hom, G.; Jaton, K.; Greub, G. Genomics of the new species Kingella negevensis: Diagnostic issues and identification of a locus encoding a RTX toxin. Microbes Infect. 2017, 19, 546–552. [Google Scholar] [CrossRef]
- El Houmami, N.; Bakour, S.; Bzdrenga, J.; Rathored, J.; Seligmann, H.; Robert, C.; Armstrong, N.; Schrenzel, J.; Raoult, D.; Yagupsky, P.; et al. Isolation and characterization of Kingella negevensis sp. nov., a novel Kingella species detected in a healthy paediatric population. Int. J. Syst. Evol. Microbiol. 2017, 67, 2370–2376. [Google Scholar] [CrossRef]
- Chang, D.W.; Nudell, Y.A.; Lau, J.; Zakharian, E.; Balashova, N.V. RTX toxin plays a key role in Kingella kingae virulence in an infant rat model. Infect. Immun. 2014, 82, 2318–2328. [Google Scholar] [CrossRef]
- Linhartova, I.; Bumba, L.; Masin, J.; Basler, M.; Osicka, R.; Kamanova, J.; Prochazkova, K.; Adkins, I.; Hejnova-Holubova, J.; Sadilkova, L.; et al. RTX proteins: A highly diverse family secreted by a common mechanism. FEMS Microbiol. Rev. 2010, 34, 1076–1112. [Google Scholar] [CrossRef]
- Linhartova, I.; Osicka, R.; Bumba, L.; Masin, J.; Sebo, P. RTX Toxins: A Review. In Microbial Toxins, 1st ed.; Toxinology; Gopalakrishnakone, P., Stiles, B., Alape-Girón, A., Dubreuil, J., Mandal, M., Eds.; Springer: Dordrecht, The Netherlands, 2015; pp. 1–29. [Google Scholar] [CrossRef]
- Jorgensen, S.E.; Mulcahy, P.F.; Wu, G.K.; Louis, C.F. Calcium accumulation in human and sheep erythrocytes that is induced by Escherichia coli hemolysin. Toxicon Off. J. Int. Soc. Toxinol. 1983, 21, 717–727. [Google Scholar] [CrossRef]
- Bhakdi, S.; Mackman, N.; Nicaud, J.M.; Holland, I.B. Escherichia coli hemolysin may damage target cell membranes by generating transmembrane pores. Infect. Immun. 1986, 52, 63–69. [Google Scholar] [CrossRef] [PubMed]
- Menestrina, G.; Mackman, N.; Holland, I.B.; Bhakdi, S. Escherichia coli haemolysin forms voltage-dependent ion channels in lipid membranes. Biochim. Biophys. Acta 1987, 905, 109–117. [Google Scholar] [CrossRef]
- Benz, R.; Schmid, A.; Wagner, W.; Goebel, W. Pore formation by the Escherichia coli hemolysin: Evidence for an association-dissociation equilibrium of the pore-forming aggregates. Infect. Immun. 1989, 57, 887–895. [Google Scholar] [CrossRef] [PubMed]
- Gray, M.; Szabo, G.; Otero, A.S.; Gray, L.; Hewlett, E. Distinct mechanisms for K+ efflux, intoxication, and hemolysis by Bordetella pertussis AC toxin. J. Biol. Chem. 1998, 273, 18260–18267. [Google Scholar] [CrossRef]
- Bárcena-Uribarri, I.; Benz, R.; Winterhalter, M.; Zakharian, E.; Balashova, N. Pore forming activity of the potent RTX-toxin produced by pediatric pathogen Kingella kingae: Characterization and comparison to other RTX-family members. Biochim. Biophys. Acta 2015, 1848, 1536–1544. [Google Scholar] [CrossRef]
- Osickova, A.; Balashova, N.; Masin, J.; Sulc, M.; Roderova, J.; Wald, T.; Brown, A.C.; Koufos, E.; Chang, E.H.; Giannakakis, A.; et al. Cytotoxic activity of Kingella kingae RtxA toxin depends on post-translational acylation of lysine residues and cholesterol binding. Emerg. Microb. Infect. 2018, 7, 178. [Google Scholar] [CrossRef]
- Mazzone, A.; Ricevuti, G. Leukocyte CD11/CD18 integrins: Biological and clinical relevance. Haematologica 1995, 80, 161–175. [Google Scholar]
- Maldonado, R.; Wei, R.; Kachlany, S.C.; Kazi, M.; Balashova, N.V. Cytotoxic effects of Kingella kingae outer membrane vesicles on human cells. Microb. Pathog. 2011, 51, 22–30. [Google Scholar] [CrossRef]
- Cavalieri, S.J.; Snyder, I.S. Effect of Escherichia coli alpha-hemolysin on human peripheral leukocyte function in vitro. Infect. Immun. 1982, 37, 966–974. [Google Scholar] [CrossRef]
- Felmlee, T.; Pellett, S.; Welch, R.A. Nucleotide sequence of an Escherichia coli chromosomal hemolysin. J. Bacteriol. 1985, 163, 94–105. [Google Scholar] [CrossRef]
- Keane, W.F.; Welch, R.; Gekker, G.; Peterson, P.K. Mechanism of Escherichia coli alpha-hemolysin-induced injury to isolated renal tubular cells. Am. J. Pathol. 1987, 126, 350–357. [Google Scholar] [PubMed]
- Bhakdi, S.; Greulich, S.; Muhly, M.; Eberspacher, B.; Becker, H.; Thiele, A.; Hugo, F. Potent leukocidal action of Escherichia coli hemolysin mediated by permeabilization of target cell membranes. J. Exp. Med. 1989, 169, 737–754. [Google Scholar] [CrossRef] [PubMed]
- Suttorp, N.; Floer, B.; Schnittler, H.; Seeger, W.; Bhakdi, S. Effects of Escherichia coli hemolysin on endothelial cell function. Infect. Immun. 1990, 58, 3796–3801. [Google Scholar] [CrossRef] [PubMed]
- Mobley, H.L.; Green, D.M.; Trifillis, A.L.; Johnson, D.E.; Chippendale, G.R.; Lockatell, C.V.; Jones, B.D.; Warren, J.W. Pyelonephritogenic Escherichia coli and killing of cultured human renal proximal tubular epithelial cells: Role of hemolysin in some strains. Infect. Immun. 1990, 58, 1281–1289. [Google Scholar] [CrossRef]
- O’Hanley, P.; Lalonde, G.; Ji, G. Alpha-hemolysin contributes to the pathogenicity of piliated digalactoside-binding Escherichia coli in the kidney: Efficacy of an alpha-hemolysin vaccine in preventing renal injury in the BALB/c mouse model of pyelonephritis. Infect. Immun. 1991, 59, 1153–1161. [Google Scholar] [CrossRef]
- Stanley, P.; Packman, L.C.; Koronakis, V.; Hughes, C. Fatty acylation of two internal lysine residues required for the toxic activity of Escherichia coli hemolysin. Science 1994, 266, 1992–1996. [Google Scholar] [CrossRef]
- Lally, E.T.; Kieba, I.R.; Sato, A.; Green, C.L.; Rosenbloom, J.; Korostoff, J.; Wang, J.F.; Shenker, B.J.; Ortlepp, S.; Robinson, M.K.; et al. RTX toxins recognize a beta2 integrin on the surface of human target cells. J. Biol. Chem. 1997, 272, 30463–30469. [Google Scholar] [CrossRef]
- Glaser, P.; Ladant, D.; Sezer, O.; Pichot, F.; Ullmann, A.; Danchin, A. The calmodulin-sensitive adenylate cyclase of Bordetella pertussis: Cloning and expression in Escherichia coli. Mol. Microbiol. 1988, 2, 19–30. [Google Scholar] [CrossRef]
- Bellalou, J.; Sakamoto, H.; Ladant, D.; Geoffroy, C.; Ullmann, A. Deletions affecting hemolytic and toxin activities of Bordetella pertussis adenylate cyclase. Infect. Immun. 1990, 58, 3242–3247. [Google Scholar] [CrossRef]
- Rogel, A.; Meller, R.; Hanski, E. Adenylate cyclase toxin from Bordetella pertussis. The relationship between induction of cAMP and hemolysis. J. Biol. Chem. 1991, 266, 3154–3161. [Google Scholar] [CrossRef]
- Hackett, M.; Guo, L.; Shabanowitz, J.; Hunt, D.F.; Hewlett, E.L. Internal lysine palmitoylation in adenylate cyclase toxin from Bordetella pertussis. Science 1994, 266, 433–435. [Google Scholar] [CrossRef] [PubMed]
- Guermonprez, P.; Khelef, N.; Blouin, E.; Rieu, P.; Ricciardi-Castagnoli, P.; Guiso, N.; Ladant, D.; Leclerc, C. The adenylate cyclase toxin of Bordetella pertussis binds to target cells via the alpha(M)beta(2) integrin (CD11b/CD18). J. Exp. Med. 2001, 193, 1035–1044. [Google Scholar] [CrossRef] [PubMed]
- Havlicek, V.; Higgins, L.; Chen, W.; Halada, P.; Sebo, P.; Sakamoto, H.; Hackett, M. Mass spectrometric analysis of recombinant adenylate cyclase toxin from Bordetella pertussis strain 18323/pHSP9. J. Mass Spectrom. 2001, 36, 384–391. [Google Scholar] [CrossRef] [PubMed]
- Morova, J.; Osicka, R.; Masin, J.; Sebo, P. RTX cytotoxins recognize beta2 integrin receptors through N-linked oligosaccharides. Proc. Natl. Acad. Sci. USA 2008, 105, 5355–5360. [Google Scholar] [CrossRef]
- Masin, J.; Fiser, R.; Linhartova, I.; Osicka, R.; Bumba, L.; Hewlett, E.L.; Benz, R.; Sebo, P. Differences in purinergic amplification of osmotic cell lysis by the pore-forming RTX toxins Bordetella pertussis CyaA and Actinobacillus pleuropneumoniae ApxIA: The role of pore size. Infect. Immun. 2013, 81, 4571–4582. [Google Scholar] [CrossRef]
- Osicka, R.; Osickova, A.; Hasan, S.; Bumba, L.; Cerny, J.; Sebo, P. Bordetella adenylate cyclase toxin is a unique ligand of the integrin complement receptor 3. eLife 2015, 4, e10766. [Google Scholar] [CrossRef]
- Taichman, N.S.; Dean, R.T.; Sanderson, C.J. Biochemical and morphological characterization of the killing of human monocytes by a leukotoxin derived from Actinobacillus actinomycetemcomitans. Infect. Immun. 1980, 28, 258–268. [Google Scholar] [CrossRef]
- Taichman, N.S.; Simpson, D.L.; Sakurada, S.; Cranfield, M.; DiRienzo, J.; Slots, J. Comparative studies on the biology of Actinobacillus actinomycetemcomitans leukotoxin in primates. Oral Microbiol. Immunol. 1987, 2, 97–104. [Google Scholar] [CrossRef]
- Simpson, D.L.; Berthold, P.; Taichman, N.S. Killing of human myelomonocytic leukemia and lymphocytic cell lines by Actinobacillus actinomycetemcomitans leukotoxin. Infect. Immun. 1988, 56, 1162–1166. [Google Scholar] [CrossRef]
- Balashova, N.V.; Crosby, J.A.; Al Ghofaily, L.; Kachlany, S.C. Leukotoxin confers beta-hemolytic activity to Actinobacillus actinomycetemcomitans. Infect. Immun. 2006, 74, 2015–2021. [Google Scholar] [CrossRef]
- Balashova, N.V.; Shah, C.; Patel, J.K.; Megalla, S.; Kachlany, S.C. Aggregatibacter actinomycetemcomitans LtxC is required for leukotoxin activity and initial interaction between toxin and host cells. Gene 2009, 443, 42–47. [Google Scholar] [CrossRef]
- Brown, A.C.; Balashova, N.V.; Epand, R.M.; Epand, R.F.; Bragin, A.; Kachlany, S.C.; Walters, M.J.; Du, Y.; Boesze-Battaglia, K.; Lally, E.T. Aggregatibacter actinomycetemcomitans leukotoxin utilizes a cholesterol recognition/amino acid consensus site for membrane association. J. Biol. Chem. 2013, 288, 23607–23621. [Google Scholar] [CrossRef] [PubMed]
- Reinholdt, J.; Poulsen, K.; Brinkmann, C.R.; Hoffmann, S.V.; Stapulionis, R.; Enghild, J.J.; Jensen, U.B.; Boesen, T.; Vorup-Jensen, T. Monodisperse and LPS-free Aggregatibacter actinomycetemcomitans leukotoxin: Interactions with human beta2 integrins and erythrocytes. Biochim. Biophys. Acta 2013, 1834, 546–558. [Google Scholar] [CrossRef] [PubMed]
- Munksgaard, P.S.; Skals, M.; Reinholdt, J.; Poulsen, K.; Jensen, M.R.; Yang, C.; Leipziger, J.; Vorup-Jensen, T.; Praetorius, H.A. Sialic acid residues are essential for cell lysis mediated by leukotoxin from Aggregatibacter actinomycetemcomitans. Infect. Immun. 2014, 82, 2219–2228. [Google Scholar] [CrossRef] [PubMed]
- Vega, B.A.; Schober, L.T.; Kim, T.; Belinka, B.A., Jr.; Kachlany, S.C. Aggregatibacter actinomycetemcomitans Leukotoxin (LtxA) Requires Death Receptor Fas, in Addition to LFA-1, To Trigger Cell Death in T Lymphocytes. Infect. Immun. 2019, 87, e00309-19. [Google Scholar] [CrossRef]
- Kaehler, K.L.; Markham, R.J.; Muscoplat, C.C.; Johnson, D.W. Evidence of species specificity in the cytocidal effects of Pasteurella haemolytica. Infect. Immun. 1980, 30, 615–616. [Google Scholar] [CrossRef]
- Clinkenbeard, K.D.; Upton, M.L. Lysis of bovine platelets by Pasteurella haemolytica leukotoxin. Am. J. Vet. Res. 1991, 52, 453–457. [Google Scholar]
- Murphy, G.L.; Whitworth, L.C.; Clinkenbeard, K.D.; Clinkenbeard, P.A. Hemolytic activity of the Pasteurella haemolytica leukotoxin. Infect. Immun. 1995, 63, 3209–3212. [Google Scholar] [CrossRef]
- Wang, J.F.; Kieba, I.R.; Korostoff, J.; Guo, T.L.; Yamaguchi, N.; Rozmiarek, H.; Billings, P.C.; Shenker, B.J.; Lally, E.T. Molecular and biochemical mechanisms of Pasteurella haemolytica leukotoxin-induced cell death. Microb. Pathog. 1998, 25, 317–331. [Google Scholar] [CrossRef]
- Batra, S.A.; Shanthalingam, S.; Munske, G.R.; Raghavan, B.; Kugadas, A.; Bavanthasivam, J.; Highlander, S.K.; Srikumaran, S. Acylation Enhances, but Is Not Required for, the Cytotoxic Activity of Mannheimia haemolytica Leukotoxin in Bighorn Sheep. Infect. Immun. 2015, 83, 3982–3988. [Google Scholar] [CrossRef]
- Frey, J.; Meier, R.; Gygi, D.; Nicolet, J. Nucleotide sequence of the hemolysin I gene from Actinobacillus pleuropneumoniae. Infect. Immun. 1991, 59, 3026–3032. [Google Scholar] [CrossRef] [PubMed]
- Serebrin, S.; Rosendal, S.; Valdivieso-Garcia, A.; Little, P.B. Endothelial cytotoxicity of Actinobacillus pleuropneumoniae. Res. Vet. Sci. 1991, 50, 18–22. [Google Scholar] [CrossRef]
- Kamp, E.M.; Popma, J.K.; Anakotta, J.; Smits, M.A. Identification of hemolytic and cytotoxic proteins of Actinobacillus pleuropneumoniae by use of monoclonal antibodies. Infect. Immun. 1991, 59, 3079–3085. [Google Scholar] [CrossRef]
- Van Leengoed, L.A.; Dickerson, H.W. Influence of calcium on secretion and activity of the cytolysins of Actinobacillus pleuropneumoniae. Infect. Immun. 1992, 60, 353–359. [Google Scholar] [CrossRef] [PubMed]
- Frey, J.; Bosse, J.T.; Chang, Y.F.; Cullen, J.M.; Fenwick, B.; Gerlach, G.F.; Gygi, D.; Haesebrouck, F.; Inzana, T.J.; Jansen, R.; et al. Actinobacillus pleuropneumoniae RTX-toxins: Uniform designation of haemolysins, cytolysins, pleurotoxin and their genes. J. Gen. Microbiol. 1993, 139, 1723–1728. [Google Scholar] [CrossRef]
- Stanley, P.; Koronakis, V.; Hughes, C. Acylation of Escherichia coli hemolysin: A unique protein lipidation mechanism underlying toxin function. Microbiol. Mol. Biol. Rev. MMBR 1998, 62, 309–333. [Google Scholar] [CrossRef]
- Ramjeet, M.; Cox, A.D.; Hancock, M.A.; Mourez, M.; Labrie, J.; Gottschalk, M.; Jacques, M. Mutation in the LPS outer core biosynthesis gene, galU, affects LPS interaction with the RTX toxins ApxI and ApxII and cytolytic activity of Actinobacillus pleuropneumoniae serotype 1. Mol. Microbiol. 2008, 70, 221–235. [Google Scholar] [CrossRef]
- Jansen, R.; Briaire, J.; Kamp, E.M.; Gielkens, A.L.; Smits, M.A. Cloning and characterization of the Actinobacillus pleuropneumoniae-RTX-toxin III (ApxIII) gene. Infect. Immun. 1993, 61, 947–954. [Google Scholar] [CrossRef]
- Vanden Bergh, P.G.; Zecchinon, L.L.; Fett, T.; Desmecht, D. Probing of Actinobacillus pleuropneumoniae ApxIIIA toxin-dependent cytotoxicity towards mammalian peripheral blood mononucleated cells. BMC Res. Notes 2008, 1, 121. [Google Scholar] [CrossRef]
- Welch, R.A.; Pellett, S. Transcriptional organization of the Escherichia coli hemolysin genes. J. Bacteriol. 1988, 170, 1622–1630. [Google Scholar] [CrossRef]
- Wandersman, C.; Delepelaire, P. TolC, an Escherichia coli outer membrane protein required for hemolysin secretion. Proc. Natl. Acad. Sci. USA 1990, 87, 4776–4780. [Google Scholar] [CrossRef] [PubMed]
- Glaser, P.; Sakamoto, H.; Bellalou, J.; Ullmann, A.; Danchin, A. Secretion of cyclolysin, the calmodulin-sensitive adenylate cyclase-haemolysin bifunctional protein of Bordetella pertussis. EMBO J 1988, 7, 3997–4004. [Google Scholar] [CrossRef] [PubMed]
- Lehours, P.; Freydiere, A.M.; Richer, O.; Burucoa, C.; Boisset, S.; Lanotte, P.; Prere, M.F.; Ferroni, A.; Lafuente, C.; Vandenesch, F.; et al. The rtxA toxin gene of Kingella kingae: A pertinent target for molecular diagnosis of osteoarticular infections. J. Clin. Microbiol. 2011, 49, 1245–1250. [Google Scholar] [CrossRef] [PubMed]
- Read, A.F. The evolution of virulence. Trends. Microbiol. 1994, 2, 73–76. [Google Scholar] [CrossRef]
- Holden, M.T.; Feil, E.J.; Lindsay, J.A.; Peacock, S.J.; Day, N.P.; Enright, M.C.; Foster, T.J.; Moore, C.E.; Hurst, L.; Atkin, R.; et al. Complete genomes of two clinical Staphylococcus aureus strains: Evidence for the rapid evolution of virulence and drug resistance. Proc. Natl. Acad. Sci. USA 2004, 101, 9786–9791. [Google Scholar] [CrossRef]
- Amit, U.; Porat, N.; Basmaci, R.; Bidet, P.; Bonacorsi, S.; Dagan, R.; Yagupsky, P. Genotyping of invasive Kingella kingae isolates reveals predominant clones and association with specific clinical syndromes. Clin. Infect. Dis. 2012, 55, 1074–1079. [Google Scholar] [CrossRef]
- Basmaci, R.; Yagupsky, P.; Ilharreborde, B.; Guyot, K.; Porat, N.; Chomton, M.; Thiberge, J.M.; Mazda, K.; Bingen, E.; Bonacorsi, S.; et al. Multilocus sequence typing and rtxA toxin gene sequencing analysis of Kingella kingae isolates demonstrates genetic diversity and international clones. PLoS ONE 2012, 7, e38078. [Google Scholar] [CrossRef]
- Yagupsky, P. Kingella kingae: From medical rarity to an emerging paediatric pathogen. Lancet Infect. Dis. 2004, 4, 358–367. [Google Scholar] [CrossRef]
- Kiang, K.M.; Ogunmodede, F.; Juni, B.A.; Boxrud, D.J.; Glennen, A.; Bartkus, J.M.; Cebelinski, E.A.; Harriman, K.; Koop, S.; Faville, R.; et al. Outbreak of osteomyelitis/septic arthritis caused by Kingella kingae among child care center attendees. Pediatrics 2005, 116, e206–e213. [Google Scholar] [CrossRef]
- Rosey, A.L.; Abachin, E.; Quesnes, G.; Cadilhac, C.; Pejin, Z.; Glorion, C.; Berche, P.; Ferroni, A. Development of a broad-range 16S rDNA real-time PCR for the diagnosis of septic arthritis in children. J. Microbiol. Methods 2007, 68, 88–93. [Google Scholar] [CrossRef]
- Matta, M.; Wermert, D.; Podglajen, I.; Sanchez, O.; Buu-Hoi, A.; Gutmann, L.; Meyer, G.; Mainardi, J.L. Molecular diagnosis of Kingella kingae pericarditis by amplification and sequencing of the 16S rRNA gene. J. Clin. Microbiol. 2007, 45, 3133–3134. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Chometon, S.; Benito, Y.; Chaker, M.; Boisset, S.; Ploton, C.; Berard, J.; Vandenesch, F.; Freydiere, A.M. Specific real-time polymerase chain reaction places Kingella kingae as the most common cause of osteoarticular infections in young children. Pediatr. Infect. Dis. J. 2007, 26, 377–381. [Google Scholar] [CrossRef] [PubMed]
- Ilharreborde, B.; Bidet, P.; Lorrot, M.; Even, J.; Mariani-Kurkdjian, P.; Liguori, S.; Vitoux, C.; Lefevre, Y.; Doit, C.; Fitoussi, F.; et al. New real-time PCR-based method for Kingella kingae DNA detection: Application to samples collected from 89 children with acute arthritis. J. Clin. Microbiol. 2009, 47, 1837–1841. [Google Scholar] [CrossRef] [PubMed]
- Levy, P.Y.; Fournier, P.E.; Fenollar, F.; Raoult, D. Systematic PCR detection in culture-negative osteoarticular infections. Am. J. Med. 2013, 126, 1143.e25–1143.e33. [Google Scholar] [CrossRef]
- Ferroni, A.; Al Khoury, H.; Dana, C.; Quesne, G.; Berche, P.; Glorion, C.; Pejin, Z. Prospective survey of acute osteoarticular infections in a French paediatric orthopedic surgery unit. Clin. Microbiol. Infect. 2013, 19, 822–828. [Google Scholar] [CrossRef]
- Slinger, R.; Moldovan, I.; Bowes, J.; Chan, F. Polymerase chain reaction detection of Kingella kingae in children with culture-negative septic arthritis in eastern Ontario. Paediatr. Child Health 2016, 21, 79–82. [Google Scholar] [CrossRef]
- Haldar, M.; Butler, M.; Quinn, C.D.; Stratton, C.W.; Tang, Y.W.; Burnham, C.A. Evaluation of a real-time PCR assay for simultaneous detection of Kingella kingae and Staphylococcus aureus from synovial fluid in suspected septic arthritis. Ann. Lab. Med. 2014, 34, 313–316. [Google Scholar] [CrossRef][Green Version]
- Paakkonen, M. Septic arthritis in children: Diagnosis and treatment. Pediatr. Health Med. Ther. 2017, 8, 65–68. [Google Scholar] [CrossRef]
- Williams, N.; Cooper, C.; Cundy, P. Kingella kingae septic arthritis in children: Recognising an elusive pathogen. J. Child. Orthop. 2014, 8, 91–95. [Google Scholar] [CrossRef]
- El Houmami, N.; Bzdrenga, J.; Durand, G.A.; Minodier, P.; Seligmann, H.; Prudent, E.; Bakour, S.; Bonacorsi, S.; Raoult, D.; Yagupsky, P.; et al. Molecular Tests That Target the RTX Locus Do Not Distinguish between Kingella kingae and the Recently Described Kingella negevensis Species. J. Clin. Microbiol. 2017, 55, 3113–3122. [Google Scholar] [CrossRef]
- El Houmami, N.; Durand, G.A.; Bzdrenga, J.; Darmon, A.; Minodier, P.; Seligmann, H.; Raoult, D.; Fournier, P.E. A New Highly Sensitive and Specific Real-Time PCR Assay Targeting the Malate Dehydrogenase Gene of Kingella kingae and Application to 201 Pediatric Clinical Specimens. J. Clin. Microbiol. 2018, 56, e00505-18. [Google Scholar] [CrossRef] [PubMed]
- Klein, C.; Peltier, F.; Pluquet, E.; Haraux, E.; Gouron, R.; Joseph, C. Management of an outbreak of invasive Kingella kingae skeletal infections in a day care center. Arch. Pediatrie Organe Off. Soc. Fr. Pediatrie 2021, 28, 12–15. [Google Scholar] [CrossRef] [PubMed]
- Porsch, E.A.; Yagupsky, P.; St Geme, J.W., 3rd. Kingella negevensis shares multiple putative virulence factors with Kingella kingae. PLoS ONE 2020, 15, e0241511. [Google Scholar] [CrossRef]
- Henderson, I.R.; Owen, P.; Nataro, J.P. Molecular switches—The ON and OFF of bacterial phase variation. Mol. Microbiol. 1999, 33, 919–932. [Google Scholar] [CrossRef] [PubMed]
- Srikhanta, Y.N.; Maguire, T.L.; Stacey, K.J.; Grimmond, S.M.; Jennings, M.P. The phasevarion: A genetic system controlling coordinated, random switching of expression of multiple genes. Proc. Natl. Acad. Sci. USA 2005, 102, 5547–5551. [Google Scholar] [CrossRef] [PubMed]
- van der Woude, M.W. Phase variation: How to create and coordinate population diversity. Curr. Opin. Microbiol. 2011, 14, 205–211. [Google Scholar] [CrossRef]
- Snyder, L.; Champness, W.; Champness, W. Molecular Genetics of Bacteria; ASM Press: Washington, DC, USA, 1997; Volume 19. [Google Scholar]
- van der Woude, M.W.; Baumler, A.J. Phase and antigenic variation in bacteria. Clin. Microbiol. Rev. 2004, 17, 581–611. [Google Scholar] [CrossRef]
- Hood, D.W.; Deadman, M.E.; Jennings, M.P.; Bisercic, M.; Fleischmann, R.D.; Venter, J.C.; Moxon, E.R. DNA repeats identify novel virulence genes in Haemophilus influenzae. Proc. Natl. Acad. Sci. USA 1996, 93, 11121–11125. [Google Scholar] [CrossRef]
- Seib, K.L.; Peak, I.R.; Jennings, M.P. Phase variable restriction-modification systems in Moraxella catarrhalis. FEMS Immunol. Med Microbiol. 2002, 32, 159–165. [Google Scholar] [CrossRef]
- Srikhanta, Y.N.; Fox, K.L.; Jennings, M.P. The phasevarion: Phase variation of type III DNA methyltransferases controls coordinated switching in multiple genes. Nat. Rev. Microbiol. 2010, 8, 196–206. [Google Scholar] [CrossRef]
- Srikhanta, Y.N.; Dowideit, S.J.; Edwards, J.L.; Falsetta, M.L.; Wu, H.J.; Harrison, O.B.; Fox, K.L.; Seib, K.L.; Maguire, T.L.; Wang, A.H.; et al. Phasevarions mediate random switching of gene expression in pathogenic Neisseria. PLoS Pathog. 2009, 5, e1000400. [Google Scholar] [CrossRef]
- Srikhanta, Y.N.; Gorrell, R.J.; Steen, J.A.; Gawthorne, J.A.; Kwok, T.; Grimmond, S.M.; Robins-Browne, R.M.; Jennings, M.P. Phasevarion mediated epigenetic gene regulation in Helicobacter pylori. PLoS ONE 2011, 6, e27569. [Google Scholar] [CrossRef] [PubMed]
- Gauntlett, J.C.; Nilsson, H.O.; Fulurija, A.; Marshall, B.J.; Benghezal, M. Phase-variable restriction/modification systems are required for Helicobacter pylori colonization. Gut Pathog. 2014, 6, 35. [Google Scholar] [CrossRef] [PubMed]
- Srikhanta, Y.N.; Fung, K.Y.; Pollock, G.L.; Bennett-Wood, V.; Howden, B.P.; Hartland, E.L. Phasevarion-Regulated Virulence in the Emerging Pediatric Pathogen Kingella kingae. Infect. Immun. 2017, 85, e00319-17. [Google Scholar] [CrossRef]
- Wallin, R.P.; Lundqvist, A.; More, S.H.; von Bonin, A.; Kiessling, R.; Ljunggren, H.G. Heat-shock proteins as activators of the innate immune system. Trends Immunol. 2002, 23, 130–135. [Google Scholar] [CrossRef]
- Welch, R.A. Pore-forming cytolysins of gram-negative bacteria. Mol. Microbiol. 1991, 5, 521–528. [Google Scholar] [CrossRef] [PubMed]
- Welch, R.A. RTX toxin structure and function: A story of numerous anomalies and few analogies in toxin biology. Curr. Top Microbiol. Immunol. 2001, 257, 85–111. [Google Scholar] [CrossRef]
- Ludwig, A.; Goebel, W. Structure and mode of action of RTX toxins. In The Comprehensive Sourcebook of Bacterial Protein Toxins, 3rd ed.; Popoff, M.R., Alouf, J.E., Eds.; Elsevier Academic Press: London, UK, 2006; pp. 547–569. [Google Scholar] [CrossRef]
- Benz, R. Channel formation by RTX-toxins of pathogenic bacteria: Basis of their biological activity. Biochim. Biophys. Acta 2016, 1858, 526–537. [Google Scholar] [CrossRef]
- Masin, J.; Osicka, R.; Bumba, L.; Sebo, P. Bordetella adenylate cyclase toxin: A unique combination of a pore-forming moiety with a cell-invading adenylate cyclase enzyme. Pathog. Dis. 2015, 73, ftv075. [Google Scholar] [CrossRef]
- Novak, J.; Cerny, O.; Osickova, A.; Linhartova, I.; Masin, J.; Bumba, L.; Sebo, P.; Osicka, R. Structure-Function Relationships Underlying the Capacity of Bordetella Adenylate Cyclase Toxin to Disarm Host Phagocytes. Toxins 2017, 9, 300. [Google Scholar] [CrossRef]
- Ludwig, A.; Jarchau, T.; Benz, R.; Goebel, W. The repeat domain of Escherichia coli haemolysin (HlyA) is responsible for its Ca2+-dependent binding to erythrocytes. Mol. Gen. Genet. 1988, 214, 553–561. [Google Scholar] [CrossRef] [PubMed]
- Coote, J.G. Structural and functional relationships among the RTX toxin determinants of gram-negative bacteria. FEMS Microbiol. Rev. 1992, 8, 137–161. [Google Scholar] [CrossRef] [PubMed]
- Valeva, A.; Siegel, I.; Wylenzek, M.; Wassenaar, T.M.; Weis, S.; Heinz, N.; Schmitt, R.; Fischer, C.; Reinartz, R.; Bhakdi, S.; et al. Putative identification of an amphipathic alpha-helical sequence in hemolysin of Escherichia coli (HlyA) involved in transmembrane pore formation. Biol. Chem. 2008, 389, 1201–1207. [Google Scholar] [CrossRef]
- Ludwig, A.; Schmid, A.; Benz, R.; Goebel, W. Mutations affecting pore formation by haemolysin from Escherichia coli. Mol. Gen. Genet. 1991, 226, 198–208. [Google Scholar] [CrossRef]
- Erb, K.; Vogel, M.; Wagner, W.; Goebel, W. Alkaline phosphatase which lacks its own signal sequence becomes enzymatically active when fused to N-terminal sequences of Escherichia coli haemolysin (HlyA). Mol. Gen. Genet. 1987, 208, 88–93. [Google Scholar] [CrossRef]
- Wiles, T.J.; Mulvey, M.A. The RTX pore-forming toxin α-hemolysin of uropathogenic Escherichia coli: Progress and perspectives. Future Microbiol. 2013, 8, 73–84. [Google Scholar] [CrossRef]
- Ladant, D.; Ullmann, A. Bordetella pertussis adenylate cyclase: A toxin with multiple talents. Trends Microbiol. 1999, 7, 172–176. [Google Scholar] [CrossRef]
- Hyland, C.; Vuillard, L.; Hughes, C.; Koronakis, V. Membrane interaction of Escherichia coli hemolysin: Flotation and insertion-dependent labeling by phospholipid vesicles. J. Bacteriol. 2001, 183, 5364–5370. [Google Scholar] [CrossRef]
- Eisenberg, D.; Schwarz, E.; Komaromy, M.; Wall, R. Analysis of membrane and surface protein sequences with the hydrophobic moment plot. J. Mol. Biol. 1984, 179, 125–142. [Google Scholar] [CrossRef]
- Benz, R.; Maier, E.; Ladant, D.; Ullmann, A.; Sebo, P. Adenylate cyclase toxin (CyaA) of Bordetella pertussis. Evidence for the formation of small ion-permeable channels and comparison with HlyA of Escherichia coli. J. Biol. Chem. 1994, 269, 27231–27239. [Google Scholar] [CrossRef]
- Osickova, A.; Osicka, R.; Maier, E.; Benz, R.; Sebo, P. An amphipathic alpha-helix including glutamates 509 and 516 is crucial for membrane translocation of adenylate cyclase toxin and modulates formation and cation selectivity of its membrane channels. J. Biol. Chem. 1999, 274, 37644–37650. [Google Scholar] [CrossRef]
- Basler, M.; Knapp, O.; Masin, J.; Fiser, R.; Maier, E.; Benz, R.; Sebo, P.; Osicka, R. Segments crucial for membrane translocation and pore-forming activity of Bordetella adenylate cyclase toxin. J. Biol. Chem. 2007, 282, 12419–12429. [Google Scholar] [CrossRef]
- Roderova, J.; Osickova, A.; Sukova, A.; Mikusova, G.; Fiser, R.; Sebo, P.; Osicka, R.; Masin, J. Residues 529 to 549 participate in membrane penetration and pore-forming activity of the Bordetella adenylate cyclase toxin. Sci. Rep. 2019, 9, 5758. [Google Scholar] [CrossRef] [PubMed]
- Powthongchin, B.; Angsuthanasombat, C. Effects on haemolytic activity of single proline substitutions in the Bordetella pertussis CyaA pore-forming fragment. Arch. Microbiol. 2009, 191, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Juntapremjit, S.; Thamwiriyasati, N.; Kurehong, C.; Prangkio, P.; Shank, L.; Powthongchin, B.; Angsuthanasombat, C. Functional importance of the Gly cluster in transmembrane helix 2 of the Bordetella pertussis CyaA-hemolysin: Implications for toxin oligomerization and pore formation. Toxicon Off. J. Int. Soc. Toxinol. 2015, 106, 14–19. [Google Scholar] [CrossRef]
- Masin, J.; Roderova, J.; Osickova, A.; Novak, P.; Bumba, L.; Fiser, R.; Sebo, P.; Osicka, R. The conserved tyrosine residue 940 plays a key structural role in membrane interaction of Bordetella adenylate cyclase toxin. Sci. Rep. 2017, 7, 9330. [Google Scholar] [CrossRef]
- Wald, T.; Petry-Podgorska, I.; Fiser, R.; Matousek, T.; Dedina, J.; Osicka, R.; Sebo, P.; Masin, J. Quantification of potassium levels in cells treated with Bordetella adenylate cyclase toxin. Anal. Biochem. 2014, 450, 57–62. [Google Scholar] [CrossRef]
- Skals, M.; Bjaelde, R.G.; Reinholdt, J.; Poulsen, K.; Vad, B.S.; Otzen, D.E.; Leipziger, J.; Praetorius, H.A. Bacterial RTX toxins allow acute ATP release from human erythrocytes directly through the toxin pore. J. Biol. Chem. 2014, 289, 19098–19109. [Google Scholar] [CrossRef]
- Goebel, W.; Hedgpeth, J. Cloning and functional characterization of the plasmid-encoded hemolysin determinant of Escherichia coli. J. Bacteriol. 1982, 151, 1290–1298. [Google Scholar] [CrossRef]
- Mackman, N.; Nicaud, J.M.; Gray, L.; Holland, I.B. Genetical and functional organisation of the Escherichia coli haemolysin determinant 2001. Mol. Gen. Genet. 1985, 201, 282–288. [Google Scholar] [CrossRef]
- Barry, E.M.; Weiss, A.A.; Ehrmann, I.E.; Gray, M.C.; Hewlett, E.L.; Goodwin, M.S. Bordetella pertussis adenylate cyclase toxin and hemolytic activities require a second gene, cyaC, for activation. J. Bacteriol. 1991, 173, 720–726. [Google Scholar] [CrossRef] [PubMed]
- Sebo, P.; Glaser, P.; Sakamoto, H.; Ullmann, A. High-level synthesis of active adenylate cyclase toxin of Bordetella pertussis in a reconstructed Escherichia coli system. Gene 1991, 104, 19–24. [Google Scholar] [CrossRef]
- Issartel, J.P.; Koronakis, V.; Hughes, C. Activation of Escherichia coli prohaemolysin to the mature toxin by acyl carrier protein-dependent fatty acylation. Nature 1991, 351, 759–761. [Google Scholar] [CrossRef]
- Lim, K.B.; Walker, C.R.; Guo, L.; Pellett, S.; Shabanowitz, J.; Hunt, D.F.; Hewlett, E.L.; Ludwig, A.; Goebel, W.; Welch, R.A.; et al. Escherichia coli alpha-hemolysin (HlyA) is heterogeneously acylated in vivo with 14-, 15-, and 17-carbon fatty acids. J. Biol. Chem. 2000, 275, 36698–36702. [Google Scholar] [CrossRef]
- Osickova, A.; Khaliq, H.; Masin, J.; Jurnecka, D.; Sukova, A.; Fiser, R.; Holubova, J.; Stanek, O.; Sebo, P.; Osicka, R. Acyltransferase-mediated selection of the length of the fatty acyl chain and of the acylation site governs activation of bacterial RTX toxins. J. Biol. Chem. 2020, 295, 9268–9280. [Google Scholar] [CrossRef] [PubMed]
- Hackett, M.; Walker, C.B.; Guo, L.; Gray, M.C.; Van Cuyk, S.; Ullmann, A.; Shabanowitz, J.; Hunt, D.F.; Hewlett, E.L.; Sebo, P. Hemolytic, but not cell-invasive activity, of adenylate cyclase toxin is selectively affected by differential fatty-acylation in Escherichia coli. J. Biol. Chem. 1995, 270, 20250–20253. [Google Scholar] [CrossRef]
- Basar, T.; Havlicek, V.; Bezouskova, S.; Halada, P.; Hackett, M.; Sebo, P. The conserved lysine 860 in the additional fatty-acylation site of Bordetella pertussis adenylate cyclase is crucial for toxin function independently of its acylation status. J. Biol. Chem. 1999, 274, 10777–10783. [Google Scholar] [CrossRef]
- Basar, T.; Havlicek, V.; Bezouskova, S.; Hackett, M.; Sebo, P. Acylation of lysine 983 is sufficient for toxin activity of Bordetella pertussis adenylate cyclase. Substitutions of alanine 140 modulate acylation site selectivity of the toxin acyltransferase CyaC. J. Biol. Chem. 2001, 276, 348–354. [Google Scholar] [CrossRef]
- Gygi, D.; Nicolet, J.; Frey, J.; Cross, M.; Koronakis, V.; Hughes, C. Isolation of the Actinobacillus pleuropneumoniae haemolysin gene and the activation and secretion of the prohaemolysin by the HlyC, HlyB and HlyD proteins of Escherichia coli. Mol. Microbiol. 1990, 4, 123–128. [Google Scholar] [CrossRef]
- Forestier, C.; Welch, R.A. Nonreciprocal complementation of the hlyC and lktC genes of the Escherichia coli hemolysin and Pasteurella haemolytica leukotoxin determinants. Infect. Immun. 1990, 58, 828–832. [Google Scholar] [CrossRef]
- Westrop, G.; Hormozi, K.; da Costa, N.; Parton, R.; Coote, J. Structure-function studies of the adenylate cyclase toxin of Bordetella pertussis and the leukotoxin of Pasteurella haemolytica by heterologous C protein activation and construction of hybrid proteins. J. Bacteriol. 1997, 179, 871–879. [Google Scholar] [CrossRef][Green Version]
- Greene, N.P.; Crow, A.; Hughes, C.; Koronakis, V. Structure of a bacterial toxin-activating acyltransferase. Proc. Natl. Acad. Sci. USA 2015, 112, E3058–E3066. [Google Scholar] [CrossRef]
- Ludwig, A.; Garcia, F.; Bauer, S.; Jarchau, T.; Benz, R.; Hoppe, J.; Goebel, W. Analysis of the in vivo activation of hemolysin (HlyA) from Escherichia coli. J. Bacteriol. 1996, 178, 5422–5430. [Google Scholar] [CrossRef]
- Masin, J.; Basler, M.; Knapp, O.; El-Azami-El-Idrissi, M.; Maier, E.; Konopasek, I.; Benz, R.; Leclerc, C.; Sebo, P. Acylation of lysine 860 allows tight binding and cytotoxicity of Bordetella adenylate cyclase on CD11b-expressing cells. Biochemistry 2005, 44, 12759–12766. [Google Scholar] [CrossRef]
- Karst, J.C.; Ntsogo Enguene, V.Y.; Cannella, S.E.; Subrini, O.; Hessel, A.; Debard, S.; Ladant, D.; Chenal, A. Calcium, acylation, and molecular confinement favor folding of Bordetella pertussis adenylate cyclase CyaA toxin into a monomeric and cytotoxic form. J. Biol. Chem. 2014, 289, 30702–30716. [Google Scholar] [CrossRef]
- O’Brien, D.P.; Cannella, S.E.; Voegele, A.; Raoux-Barbot, D.; Davi, M.; Douche, T.; Matondo, M.; Brier, S.; Ladant, D.; Chenal, A. Post-translational acylation controls the folding and functions of the CyaA RTX toxin. FASEB J. 2019, 33, 10065–10076. [Google Scholar] [CrossRef]
- El-Azami-El-Idrissi, M.; Bauche, C.; Loucka, J.; Osicka, R.; Sebo, P.; Ladant, D.; Leclerc, C. Interaction of Bordetella pertussis adenylate cyclase with CD11b/CD18: Role of toxin acylation and identification of the main integrin interaction domain. J. Biol. Chem. 2003, 278, 38514–38521. [Google Scholar] [CrossRef]
- Herlax, V.; Bakas, L. Acyl chains are responsible for the irreversibility in the Escherichia coli alpha-hemolysin binding to membranes. Chem. Phys. Lipids 2003, 122, 185–190. [Google Scholar] [CrossRef]
- Herlax, V.; Mate, S.; Rimoldi, O.; Bakas, L. Relevance of fatty acid covalently bound to Escherichia coli alpha-hemolysin and membrane microdomains in the oligomerization process. J. Biol. Chem. 2009, 284, 25199–25210. [Google Scholar] [CrossRef]
- Baumann, U.; Wu, S.; Flaherty, K.M.; McKay, D.B. Three-dimensional structure of the alkaline protease of Pseudomonas aeruginosa: A two-domain protein with a calcium binding parallel beta roll motif. EMBO J. 1993, 12, 3357–3364. [Google Scholar] [CrossRef]
- Chenal, A.; Sotomayor Pérez, A.C.; Ladant, D. Structure and function of RTX toxins. In The Comprehensive Sourcebook of Bacterial Protein Toxins, 4th ed.; Alouf, J., Ladant, D., Popoff, M.R., Eds.; Elsevier: Amsterdam, The Netherlands, 2015; pp. 677–718. [Google Scholar] [CrossRef]
- Osicka, R.; Osickova, A.; Basar, T.; Guermonprez, P.; Rojas, M.; Leclerc, C.; Sebo, P. Delivery of CD8+ T-cell epitopes into major histocompatibility complex class I antigen presentation pathway by Bordetella pertussis adenylate cyclase: Delineation of cell invasive structures and permissive insertion sites. Infect. Immun. 2000, 68, 247–256. [Google Scholar] [CrossRef]
- Boehm, D.F.; Welch, R.A.; Snyder, I.S. Domains of Escherichia coli hemolysin (HlyA) involved in binding of calcium and erythrocyte membranes. Infect. Immun. 1990, 58, 1959–1964. [Google Scholar] [CrossRef]
- Bumba, L.; Masin, J.; Macek, P.; Wald, T.; Motlova, L.; Bibova, I.; Klimova, N.; Bednarova, L.; Veverka, V.; Kachala, M.; et al. Calcium-Driven Folding of RTX Domain beta-Rolls Ratchets Translocation of RTX Proteins through Type I Secretion Ducts. Mol. Cell 2016, 62, 47–62. [Google Scholar] [CrossRef]
- Motlova, L.; Klimova, N.; Fiser, R.; Sebo, P.; Bumba, L. Continuous Assembly of beta-Roll Structures Is Implicated in the Type I-Dependent Secretion of Large Repeat-in-Toxins (RTX) Proteins. J. Mol. Biol. 2020, 432, 5696–5710. [Google Scholar] [CrossRef]
- Blenner, M.A.; Shur, O.; Szilvay, G.R.; Cropek, D.M.; Banta, S. Calcium-induced folding of a beta roll motif requires C-terminal entropic stabilization. J. Mol. Biol. 2010, 400, 244–256. [Google Scholar] [CrossRef]
- Bauche, C.; Chenal, A.; Knapp, O.; Bodenreider, C.; Benz, R.; Chaffotte, A.; Ladant, D. Structural and functional characterization of an essential RTX subdomain of Bordetella pertussis adenylate cyclase toxin. J. Biol. Chem. 2006, 281, 16914–16926. [Google Scholar] [CrossRef]
- Chenal, A.; Guijarro, J.I.; Raynal, B.; Delepierre, M.; Ladant, D. RTX calcium binding motifs are intrinsically disordered in the absence of calcium: Implication for protein secretion. J. Biol. Chem. 2009, 284, 1781–1789. [Google Scholar] [CrossRef]
- Chenal, A.; Karst, J.C.; Sotomayor Perez, A.C.; Wozniak, A.K.; Baron, B.; England, P.; Ladant, D. Calcium-induced folding and stabilization of the intrinsically disordered RTX domain of the CyaA toxin. Biophys. J. 2010, 99, 3744–3753. [Google Scholar] [CrossRef]
- Hewlett, E.L.; Gray, L.; Allietta, M.; Ehrmann, I.; Gordon, V.M.; Gray, M.C. Adenylate cyclase toxin from Bordetella pertussis. Conformational change associated with toxin activity. J. Biol. Chem. 1991, 266, 17503–17508. [Google Scholar] [CrossRef]
- Schindel, C.; Zitzer, A.; Schulte, B.; Gerhards, A.; Stanley, P.; Hughes, C.; Koronakis, V.; Bhakdi, S.; Palmer, M. Interaction of Escherichia coli hemolysin with biological membranes. A study using cysteine scanning mutagenesis. Eur. J. Biochem. 2001, 268, 800–808. [Google Scholar] [CrossRef]
- Sanchez-Magraner, L.; Cortajarena, A.L.; Garcia-Pacios, M.; Arrondo, J.L.; Agirre, J.; Guerin, D.M.; Goni, F.M.; Ostolaza, H. Interdomain Ca2+ effects in Escherichia coli alpha-haemolysin: Ca2+ binding to the C-terminal domain stabilizes both C- and N-terminal domains. Biochim. Biophys. Acta 2010, 1798, 1225–1233. [Google Scholar] [CrossRef]
- Snyder, I.S.; Zwadyk, P. Some factors affecting production and assay of Escherichia coli haemolysins. J. Gen. Microbiol. 1969, 55, 139–143. [Google Scholar] [CrossRef][Green Version]
- Short, E.C.; Kurtz, H.J. Properties of the Hemolytic Activities of Escherichia coli. Infect. Immun. 1971, 3, 678–687. [Google Scholar] [CrossRef]
- Dobereiner, A.; Schmid, A.; Ludwig, A.; Goebel, W.; Benz, R. The effects of calcium and other polyvalent cations on channel formation by Escherichia coli alpha-hemolysin in red blood cells and lipid bilayer membranes. Eur. J. Biochem. 1996, 240, 454–460. [Google Scholar] [CrossRef]
- Rhodes, C.R.; Gray, M.C.; Watson, J.M.; Muratore, T.L.; Kim, S.B.; Hewlett, E.L.; Grisham, C.M. Structural consequences of divalent metal binding by the adenylyl cyclase toxin of Bordetella pertussis. Arch. Biochem. Biophys. 2001, 395, 169–176. [Google Scholar] [CrossRef]
- Rose, T.; Sebo, P.; Bellalou, J.; Ladant, D. Interaction of calcium with Bordetella pertussis adenylate cyclase toxin. Characterization of multiple calcium-binding sites and calcium-induced conformational changes. J. Biol. Chem. 1995, 270, 26370–26376. [Google Scholar] [CrossRef]
- Soloaga, A.; Ramirez, J.M.; Goni, F.M. Reversible denaturation, self-aggregation, and membrane activity of Escherichia coli alpha-hemolysin, a protein stable in 6 M urea. Biochemistry 1998, 37, 6387–6393. [Google Scholar] [CrossRef]
- Thomas, S.; Bakkes, P.J.; Smits, S.H.; Schmitt, L. Equilibrium folding of pro-HlyA from Escherichia coli reveals a stable calcium ion dependent folding intermediate. Biochim. Biophys. Acta 2014, 1844, 1500–1510. [Google Scholar] [CrossRef]
- Goldsmith, J.A.; DiVenere, A.M.; Maynard, J.A.; McLellan, J.S. Structural basis for antibody binding to adenylate cyclase toxin reveals RTX linkers as neutralization-sensitive epitopes. PLoS Pathog. 2021, 17, e1009920. [Google Scholar] [CrossRef]
- Sotomayor Perez, A.C.; Karst, J.C.; Davi, M.; Guijarro, J.I.; Ladant, D.; Chenal, A. Characterization of the regions involved in the calcium-induced folding of the intrinsically disordered RTX motifs from the Bordetella pertussis adenylate cyclase toxin. J. Mol. Biol. 2010, 397, 534–549. [Google Scholar] [CrossRef]
- Wang, X.; Stapleton, J.A.; Klesmith, J.R.; Hewlett, E.L.; Whitehead, T.A.; Maynard, J.A. Fine Epitope Mapping of Two Antibodies Neutralizing the Bordetella Adenylate Cyclase Toxin. Biochemistry 2017, 56, 1324–1336. [Google Scholar] [CrossRef] [PubMed]
- Nicaud, J.M.; Mackman, N.; Gray, L.; Holland, I.B. The C-terminal, 23 kDa peptide of E. coli haemolysin 2001 contains all the information necessary for its secretion by the haemolysin (Hly) export machinery. FEBS Lett. 1986, 204, 331–335. [Google Scholar] [CrossRef]
- Masure, H.R.; Au, D.C.; Gross, M.K.; Donovan, M.G.; Storm, D.R. Secretion of the Bordetella pertussis adenylate cyclase from Escherichia coli containing the hemolysin operon. Biochemistry 1990, 29, 140–145. [Google Scholar] [CrossRef] [PubMed]
- Sebo, P.; Ladant, D. Repeat sequences in the Bordetella pertussis adenylate cyclase toxin can be recognized as alternative carboxy-proximal secretion signals by the Escherichia coli alpha-haemolysin translocator. Mol. Microbiol. 1993, 9, 999–1009. [Google Scholar] [CrossRef] [PubMed]
- Mackman, N.; Baker, K.; Gray, L.; Haigh, R.; Nicaud, J.M.; Holland, I.B. Release of a chimeric protein into the medium from Escherichia coli using the C-terminal secretion signal of haemolysin. EMBO J. 1987, 6, 2835–2841. [Google Scholar] [CrossRef]
- Koronakis, V.; Koronakis, E.; Hughes, C. Isolation and analysis of the C-terminal signal directing export of Escherichia coli hemolysin protein across both bacterial membranes. EMBO J. 1989, 8, 595–605. [Google Scholar] [CrossRef]
- Bejerano, M.; Nisan, I.; Ludwig, A.; Goebel, W.; Hanski, E. Characterization of the C-terminal domain essential for toxic activity of adenylate cyclase toxin. Mol. Microbiol. 1999, 31, 381–392. [Google Scholar] [CrossRef]
- Stanley, P.; Koronakis, V.; Hughes, C. Mutational analysis supports a role for multiple structural features in the C-terminal secretion signal of Escherichia coli haemolysin. Mol. Microbiol. 1991, 5, 2391–2403. [Google Scholar] [CrossRef]
- Blight, M.A.; Chervaux, C.; Holland, I.B. Protein secretion pathway in Escherichia coli. Curr. Opin. Biotechnol. 1994, 5, 468–474. [Google Scholar] [CrossRef]
- Hui, D.; Morden, C.; Zhang, F.; Ling, V. Combinatorial analysis of the structural requirements of the Escherichia coli hemolysin signal sequence. J. Biol. Chem. 2000, 275, 2713–2720. [Google Scholar] [CrossRef]
- Jumpertz, T.; Chervaux, C.; Racher, K.; Zouhair, M.; Blight, M.A.; Holland, I.B.; Schmitt, L. Mutations affecting the extreme C terminus of Escherichia coli haemolysin A reduce haemolytic activity by altering the folding of the toxin. Microbiology 2010, 156, 2495–2505. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Masin, J.; Osickova, A.; Sukova, A.; Fiser, R.; Halada, P.; Bumba, L.; Linhartova, I.; Osicka, R.; Sebo, P. Negatively charged residues of the segment linking the enzyme and cytolysin moieties restrict the membrane-permeabilizing capacity of adenylate cyclase toxin. Sci. Rep. 2016, 6, 29137. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, H.; Bellalou, J.; Sebo, P.; Ladant, D. Bordetella pertussis adenylate cyclase toxin. Structural and functional independence of the catalytic and hemolytic activities. J. Biol. Chem. 1992, 267, 13598–13602. [Google Scholar] [CrossRef]
- Masin, J.; Osickova, A.; Jurnecka, D.; Klimova, N.; Khaliq, H.; Sebo, P.; Osicka, R. Retargeting from the CR3 to the LFA-1 receptor uncovers the adenylyl cyclase enzyme-translocating segment of Bordetella adenylate cyclase toxin. J. Biol. Chem. 2020, 295, 9349–9365. [Google Scholar] [CrossRef]
- Wolff, J.; Cook, G.H.; Goldhammer, A.R.; Berkowitz, S.A. Calmodulin activates prokaryotic adenylate cyclase. Proc. Natl. Acad. Sci. USA 1980, 77, 3841–3844. [Google Scholar] [CrossRef]
- Gentile, F.; Raptis, A.; Knipling, L.G.; Wolff, J. Bordetella pertussis adenylate cyclase. Penetration into host cells. Eur. J. Biochem. 1988, 175, 447–453. [Google Scholar] [CrossRef]
- Rogel, A.; Hanski, E. Distinct steps in the penetration of adenylate cyclase toxin of Bordetella pertussis into sheep erythrocytes. Translocation of the toxin across the membrane. J. Biol. Chem. 1992, 267, 22599–22605. [Google Scholar] [CrossRef]
- Osickova, A.; Masin, J.; Fayolle, C.; Krusek, J.; Basler, M.; Pospisilova, E.; Leclerc, C.; Osicka, R.; Sebo, P. Adenylate cyclase toxin translocates across target cell membrane without forming a pore. Mol. Microbiol. 2010, 75, 1550–1562. [Google Scholar] [CrossRef]
- Vojtova-Vodolanova, J.; Basler, M.; Osicka, R.; Knapp, O.; Maier, E.; Cerny, J.; Benada, O.; Benz, R.; Sebo, P. Oligomerization is involved in pore formation by Bordetella adenylate cyclase toxin. FASEB J. 2009, 23, 2831–2843. [Google Scholar] [CrossRef]
- Ladant, D. Interaction of Bordetella pertussis adenylate cyclase with calmodulin. Identification of two separated calmodulin-binding domains. J. Biol. Chem. 1988, 263, 2612–2618. [Google Scholar] [CrossRef]
- Guo, Q.; Shen, Y.; Lee, Y.S.; Gibbs, C.S.; Mrksich, M.; Tang, W.J. Structural basis for the interaction of Bordetella pertussis adenylyl cyclase toxin with calmodulin. EMBO J. 2005, 24, 3190–3201. [Google Scholar] [CrossRef] [PubMed]
- Subrini, O.; Sotomayor-Perez, A.C.; Hessel, A.; Spiaczka-Karst, J.; Selwa, E.; Sapay, N.; Veneziano, R.; Pansieri, J.; Chopineau, J.; Ladant, D.; et al. Characterization of a membrane-active peptide from the Bordetella pertussis CyaA toxin. J. Biol. Chem. 2013, 288, 32585–32598. [Google Scholar] [CrossRef] [PubMed]
- Sukova, A.; Bumba, L.; Srb, P.; Veverka, V.; Stanek, O.; Holubova, J.; Chmelik, J.; Fiser, R.; Sebo, P.; Masin, J. Negative charge of the AC-to-Hly linking segment modulates calcium-dependent membrane activities of Bordetella adenylate cyclase toxin. Biochim. Biophys. Acta Biomembr. 2020, 1862, 183310. [Google Scholar] [CrossRef] [PubMed]
- Voegele, A.; Sadi, M.; O’Brien, D.P.; Gehan, P.; Raoux-Barbot, D.; Davi, M.; Hoos, S.; Brule, S.; Raynal, B.; Weber, P.; et al. A High-Affinity Calmodulin-Binding Site in the CyaA Toxin Translocation Domain is Essential for Invasion of Eukaryotic Cells. Adv. Sci. 2021, 8, 2003630. [Google Scholar] [CrossRef] [PubMed]
- Delepelaire, P. Type I secretion in gram-negative bacteria. Biochim. Biophys. Acta 2004, 1694, 149–161. [Google Scholar] [CrossRef]
- Gray, L.; Mackman, N.; Nicaud, J.M.; Holland, I.B. The carboxy-terminal region of haemolysin 2001 is required for secretion of the toxin from Escherichia coli. Mol. Gen. Genet. 1986, 205, 127–133. [Google Scholar] [CrossRef] [PubMed]
- Felmlee, T.; Welch, R.A. Alterations of amino acid repeats in the Escherichia coli hemolysin affect cytolytic activity and secretion. Proc. Natl. Acad. Sci. USA 1988, 85, 5269–5273. [Google Scholar] [CrossRef]
- Juranka, P.; Zhang, F.; Kulpa, J.; Endicott, J.; Blight, M.; Holland, I.B.; Ling, V. Characterization of the hemolysin transporter, HlyB, using an epitope insertion. J. Biol. Chem. 1992, 267, 3764–3770. [Google Scholar] [CrossRef]
- Thanabalu, T.; Koronakis, E.; Hughes, C.; Koronakis, V. Substrate-induced assembly of a contiguous channel for protein export from E. coli: Reversible bridging of an inner-membrane translocase to an outer membrane exit pore. EMBO J. 1998, 17, 6487–6496. [Google Scholar] [CrossRef]
- Balakrishnan, L.; Hughes, C.; Koronakis, V. Substrate-triggered recruitment of the TolC channel-tunnel during type I export of hemolysin by Escherichia coli. J. Mol. Biol. 2001, 313, 501–510. [Google Scholar] [CrossRef]
- Higgins, C.F.; Hiles, I.D.; Salmond, G.P.; Gill, D.R.; Downie, J.A.; Evans, I.J.; Holland, I.B.; Gray, L.; Buckel, S.D.; Bell, A.W.; et al. A family of related ATP-binding subunits coupled to many distinct biological processes in bacteria. Nature 1986, 323, 448–450. [Google Scholar] [CrossRef] [PubMed]
- Holland, I.B.; Peherstorfer, S.; Kanonenberg, K.; Lenders, M.; Reimann, S.; Schmitt, L. Type I Protein Secretion-Deceptively Simple yet with a Wide Range of Mechanistic Variability across the Family. EcoSal Plus 2016, 7, 1–46. [Google Scholar] [CrossRef] [PubMed]
- Wagner, W.; Vogel, M.; Goebel, W. Transport of hemolysin across the outer membrane of Escherichia coli requires two functions. J. Bacteriol. 1983, 154, 200–210. [Google Scholar] [CrossRef] [PubMed]
- Hartlein, M.; Schiessl, S.; Wagner, W.; Rdest, U.; Kreft, J.; Goebel, W. Transport of hemolysin by Escherichia coli. J. Cell. Biochem. 1983, 22, 87–97. [Google Scholar] [CrossRef] [PubMed]
- Pimenta, A.L.; Young, J.; Holland, I.B.; Blight, M.A. Antibody analysis of the localisation, expression and stability of HlyD, the MFP component of the E. coli haemolysin translocator. Mol. Gen. Genet. 1999, 261, 122–132. [Google Scholar] [CrossRef]
- Mackman, N.; Nicaud, J.M.; Gray, L.; Holland, I.B. Identification of polypeptides required for the export of haemolysin 2001 from E. coli. Mol. Gen. Genet. 1985, 201, 529–536. [Google Scholar] [CrossRef]
- Schmitt, L.; Benabdelhak, H.; Blight, M.A.; Holland, I.B.; Stubbs, M.T. Crystal structure of the nucleotide-binding domain of the ABC-transporter haemolysin B: Identification of a variable region within ABC helical domains. J. Mol. Biol. 2003, 330, 333–342. [Google Scholar] [CrossRef]
- Lecher, J.; Schwarz, C.K.; Stoldt, M.; Smits, S.H.; Willbold, D.; Schmitt, L. An RTX transporter tethers its unfolded substrate during secretion via a unique N-terminal domain. Structure 2012, 20, 1778–1787. [Google Scholar] [CrossRef]
- Holland, I.B.; Schmitt, L.; Young, J. Type 1 protein secretion in bacteria, the ABC-transporter dependent pathway (review). Mol. Membr. Biol. 2005, 22, 29–39. [Google Scholar] [CrossRef]
- Wang, R.C.; Seror, S.J.; Blight, M.; Pratt, J.M.; Broome-Smith, J.K.; Holland, I.B. Analysis of the membrane organization of an Escherichia coli protein translocator, HlyB, a member of a large family of prokaryote and eukaryote surface transport proteins. J. Mol. Biol. 1991, 217, 441–454. [Google Scholar] [CrossRef]
- Gentschev, I.; Goebel, W. Topological and functional studies on HlyB of Escherichia coli. Mol. Gen. Genet. 1992, 232, 40–48. [Google Scholar] [CrossRef] [PubMed]
- Gerlach, J.H.; Endicott, J.A.; Juranka, P.F.; Henderson, G.; Sarangi, F.; Deuchars, K.L.; Ling, V. Homology between P-glycoprotein and a bacterial haemolysin transport protein suggests a model for multidrug resistance. Nature 1986, 324, 485–489. [Google Scholar] [CrossRef] [PubMed]
- Higgins, C.F. ABC transporters: From microorganisms to man. Annu. Rev. Cell Biol. 1992, 8, 67–113. [Google Scholar] [CrossRef] [PubMed]
- Benabdelhak, H.; Kiontke, S.; Horn, C.; Ernst, R.; Blight, M.A.; Holland, I.B.; Schmitt, L. A specific interaction between the NBD of the ABC-transporter HlyB and a C-terminal fragment of its transport substrate haemolysin A. J. Mol. Biol. 2003, 327, 1169–1179. [Google Scholar] [CrossRef]
- Springer, W.; Goebel, W. Synthesis and secretion of hemolysin by Escherichia coli. J. Bacteriol. 1980, 144, 53–59. [Google Scholar] [CrossRef] [PubMed]
- Koronakis, V.; Koronakis, E.; Hughes, C. Comparison of the haemolysin secretion protein HlyB from Proteus vulgaris and Escherichia coli; site-directed mutagenesis causing impairment of export function. Mol. Gen. Genet. 1988, 213, 551–555. [Google Scholar] [CrossRef]
- Koronakis, E.; Hughes, C.; Milisav, I.; Koronakis, V. Protein exporter function and in vitro ATPase activity are correlated in ABC-domain mutants of HlyB. Mol. Microbiol. 1995, 16, 87–96. [Google Scholar] [CrossRef]
- Koronakis, V.; Hughes, C.; Koronakis, E. Energetically distinct early and late stages of HlyB/HlyD-dependent secretion across both Escherichia coli membranes. EMBO J. 1991, 10, 3263–3272. [Google Scholar] [CrossRef]
- Rees, D.C.; Johnson, E.; Lewinson, O. ABC transporters: The power to change. Nat. Rev. Mol. Cell Biol. 2009, 10, 218–227. [Google Scholar] [CrossRef]
- Schulein, R.; Gentschev, I.; Schlor, S.; Gross, R.; Goebel, W. Identification and characterization of two functional domains of the hemolysin translocator protein HlyD. Mol. Gen. Genet. 1994, 245, 203–211. [Google Scholar] [CrossRef]
- Lee, M.; Jun, S.Y.; Yoon, B.Y.; Song, S.; Lee, K.; Ha, N.C. Membrane fusion proteins of type I secretion system and tripartite efflux pumps share a binding motif for TolC in gram-negative bacteria. PLoS ONE 2012, 7, e40460. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.S.; Song, S.; Lee, M.; Lee, S.; Lee, K.; Ha, N.C. Crystal Structure of a Soluble Fragment of the Membrane Fusion Protein HlyD in a Type I Secretion System of Gram-Negative Bacteria. Structure 2016, 24, 477–485. [Google Scholar] [CrossRef] [PubMed]
- Schulein, R.; Gentschev, I.; Mollenkopf, H.J.; Goebel, W. A topological model for the haemolysin translocator protein HlyD. Mol. Gen. Genet. 1992, 234, 155–163. [Google Scholar] [CrossRef]
- Pimenta, A.L.; Racher, K.; Jamieson, L.; Blight, M.A.; Holland, I.B. Mutations in HlyD, part of the type 1 translocator for hemolysin secretion, affect the folding of the secreted toxin. J. Bacteriol. 2005, 187, 7471–7480. [Google Scholar] [CrossRef] [PubMed]
- Koronakis, V.; Sharff, A.; Koronakis, E.; Luisi, B.; Hughes, C. Crystal structure of the bacterial membrane protein TolC central to multidrug efflux and protein export. Nature 2000, 405, 914–919. [Google Scholar] [CrossRef] [PubMed]
- Koronakis, V.; Li, J.; Koronakis, E.; Stauffer, K. Structure of TolC, the outer membrane component of the bacterial type I efflux system, derived from two-dimensional crystals. Mol. Microbiol. 1997, 23, 617–626. [Google Scholar] [CrossRef]
- Andersen, C.; Hughes, C.; Koronakis, V. Electrophysiological behavior of the TolC channel-tunnel in planar lipid bilayers. J. Membr. Biol. 2002, 185, 83–92. [Google Scholar] [CrossRef]
- Eswaran, J.; Hughes, C.; Koronakis, V. Locking TolC entrance helices to prevent protein translocation by the bacterial type I export apparatus. J. Mol. Biol. 2003, 327, 309–315. [Google Scholar] [CrossRef]
- Pei, X.Y.; Hinchliffe, P.; Symmons, M.F.; Koronakis, E.; Benz, R.; Hughes, C.; Koronakis, V. Structures of sequential open states in a symmetrical opening transition of the TolC exit duct. Proc. Natl. Acad. Sci. USA 2011, 108, 2112–2117. [Google Scholar] [CrossRef]
- Andersen, C.; Koronakis, E.; Hughes, C.; Koronakis, V. An aspartate ring at the TolC tunnel entrance determines ion selectivity and presents a target for blocking by large cations. Mol. Microbiol. 2002, 44, 1131–1139. [Google Scholar] [CrossRef]
- Young, R.; Bremer, H. Polypeptide-chain-elongation rate in Escherichia coli B/r as a function of growth rate. Biochem. J. 1976, 160, 185–194. [Google Scholar] [CrossRef] [PubMed]
- Bakkes, P.J.; Jenewein, S.; Smits, S.H.; Holland, I.B.; Schmitt, L. The rate of folding dictates substrate secretion by the Escherichia coli hemolysin type 1 secretion system. J. Biol. Chem. 2010, 285, 40573–40580. [Google Scholar] [CrossRef]
- Lenders, M.H.; Weidtkamp-Peters, S.; Kleinschrodt, D.; Jaeger, K.E.; Smits, S.H.; Schmitt, L. Directionality of substrate translocation of the hemolysin A Type I secretion system. Sci. Rep. 2015, 5, 12470. [Google Scholar] [CrossRef]
- O’Brien, D.P.; Perez, A.C.S.; Karst, J.; Cannella, S.E.; Enguene, V.Y.N.; Hessel, A.; Raoux-Barbot, D.; Voegele, A.; Subrini, O.; Davi, M.; et al. Calcium-dependent disorder-to-order transitions are central to the secretion and folding of the CyaA toxin of Bordetella pertussis, the causative agent of whooping cough. Toxicon Off. J. Int. Soc. Toxinol. 2018, 149, 37–44. [Google Scholar] [CrossRef] [PubMed]
- Jones, H.E.; Holland, I.B.; Baker, H.L.; Campbell, A.K. Slow changes in cytosolic free Ca2+ in Escherichia coli highlight two putative influx mechanisms in response to changes in extracellular calcium. Cell Calcium 1999, 25, 265–274. [Google Scholar] [CrossRef]
- Brown, E.M.; MacLeod, R.J. Extracellular calcium sensing and extracellular calcium signaling. Physiol. Rev. 2001, 81, 239–297. [Google Scholar] [CrossRef] [PubMed]
- Lenders, M.H.; Beer, T.; Smits, S.H.; Schmitt, L. In vivo quantification of the secretion rates of the hemolysin A Type I secretion system. Sci. Rep. 2016, 6, 33275. [Google Scholar] [CrossRef] [PubMed]
- Lepesheva, A.; Osickova, A.; Holubova, J.; Jurnecka, D.; Knoblochova, S.; Espinosa-Vinals, C.; Bumba, L.; Skopova, K.; Fiser, R.; Osicka, R.; et al. Different roles of conserved tyrosine residues of the acylated domains in folding and activity of RTX toxins. Sci. Rep. 2021, 11, 19814. [Google Scholar] [CrossRef] [PubMed]
- Nicaud, J.M.; Mackman, N.; Gray, L.; Holland, I.B. Characterisation of HlyC and mechanism of activation and secretion of haemolysin from E. coli 2001. FEBS Lett. 1985, 187, 339–344. [Google Scholar] [CrossRef]
- Vakharia, H.; German, G.J.; Misra, R. Isolation and characterization of Escherichia coli tolC mutants defective in secreting enzymatically active alpha-hemolysin. J. Bacteriol. 2001, 183, 6908–6916. [Google Scholar] [CrossRef]
- Kanonenberg, K.; Spitz, O.; Erenburg, I.N.; Beer, T.; Schmitt, L. Type I secretion system-it takes three and a substrate. FEMS Microbiol. Lett. 2018, 365, fny094. [Google Scholar] [CrossRef] [PubMed]
- Valeva, A.; Walev, I.; Kemmer, H.; Weis, S.; Siegel, I.; Boukhallouk, F.; Wassenaar, T.M.; Chavakis, T.; Bhakdi, S. Binding of Escherichia coli hemolysin and activation of the target cells is not receptor-dependent. J. Biol. Chem. 2005, 280, 36657–36663. [Google Scholar] [CrossRef] [PubMed]
- Vanden Bergh, P.G.; Zecchinon, L.L.; Fett, T.; Desmecht, D. Porcine CD18 mediates Actinobacillus pleuropneumoniae ApxIII species-specific toxicity. Vet. Res. 2009, 40, 33. [Google Scholar] [CrossRef]
- Ristow, L.C.; Welch, R.A. RTX Toxins Ambush Immunity’s First Cellular Responders. Toxins 2019, 11, 720. [Google Scholar] [CrossRef]
- Arnaout, M.A. Structure and function of the leukocyte adhesion molecules CD11/CD18. Blood 1990, 75, 1037–1050. [Google Scholar] [CrossRef] [PubMed]
- Fagerholm, S.C.; Guenther, C.; Llort Asens, M.; Savinko, T.; Uotila, L.M. Beta2-Integrins and Interacting Proteins in Leukocyte Trafficking, Immune Suppression, and Immunodeficiency Disease. Front. Immunol. 2019, 10, 254. [Google Scholar] [CrossRef]
- Kishimoto, T.K.; Hollander, N.; Roberts, T.M.; Anderson, D.C.; Springer, T.A. Heterogeneous mutations in the beta subunit common to the LFA-1, Mac-1, and p150,95 glycoproteins cause leukocyte adhesion deficiency. Cell 1987, 50, 193–202. [Google Scholar] [CrossRef]
- Rahman, W.U.; Osickova, A.; Klimova, N.; Lora, J.; Balashova, N.; Osicka, R. Binding of Kingella kingae RtxA Toxin Depends on Cell Surface Oligosaccharides, but Not on beta2 Integrins. Int. J. Mol. Sci. 2020, 21, 9092. [Google Scholar] [CrossRef]
- Ristow, L.C.; Tran, V.; Schwartz, K.J.; Pankratz, L.; Mehle, A.; Sauer, J.D.; Welch, R.A. The Extracellular Domain of the beta2 Integrin beta Subunit (CD18) Is Sufficient for Escherichia coli Hemolysin and Aggregatibacter actinomycetemcomitans Leukotoxin Cytotoxic Activity. mBio 2019, 10, e01459-19. [Google Scholar] [CrossRef]
- Cortajarena, A.L.; Goni, F.M.; Ostolaza, H. Glycophorin as a receptor for Escherichia coli alpha-hemolysin in erythrocytes. J. Biol. Chem. 2001, 276, 12513–12519. [Google Scholar] [CrossRef]
- Cortajarena, A.L.; Goni, F.M.; Ostolaza, H. A receptor-binding region in Escherichia coli alpha-haemolysin. J. Biol. Chem. 2003, 278, 19159–19163. [Google Scholar] [CrossRef] [PubMed]
- Vazquez, R.F.; Maté, S.M.; Bakás, L.S.; Fernández, M.M.; Malchiodi, E.L.; Herlax, V.S. Novel evidence for the specific interaction between cholesterol and α-haemolysin of Escherichia coli. Biochem. J. 2014, 458, 481–489. [Google Scholar] [CrossRef] [PubMed]
- Martin, C.; Requero, M.A.; Masin, J.; Konopasek, I.; Goni, F.M.; Sebo, P.; Ostolaza, H. Membrane restructuring by Bordetella pertussis adenylate cyclase toxin, a member of the RTX toxin family. J. Bacteriol. 2004, 186, 3760–3765. [Google Scholar] [CrossRef]
- Vojtova, J.; Kofronova, O.; Sebo, P.; Benada, O. Bordetella adenylate cyclase toxin induces a cascade of morphological changes of sheep erythrocytes and localizes into clusters in erythrocyte membranes. Microsc. Res. Tech. 2006, 69, 119–129. [Google Scholar] [CrossRef]
- Gonzalez Bullon, D.; Uribe, K.B.; Amuategi, J.; Martin, C.; Ostolaza, H. Cholesterol stimulates the lytic activity of Adenylate Cyclase Toxin on lipid membranes by promoting toxin oligomerization and formation of pores with a greater effective size. FEBS J. 2021, 288, 6795–6814. [Google Scholar] [CrossRef]
- Gable, P.; Eaton, J.; Confer, D. Intoxication of human phagocytes by Bordetella adenylate cyclase toxin: Implication of a ganglioside receptor. In Clinical Research; Slack Inc.: Thorofare, NJ, USA, 1985; p. A844. [Google Scholar]
- Gordon, V.M.; Young, W.W., Jr.; Lechler, S.M.; Gray, M.C.; Leppla, S.H.; Hewlett, E.L. Adenylate cyclase toxins from Bacillus anthracis and Bordetella pertussis. Different processes for interaction with and entry into target cells. J. Biol. Chem. 1989, 264, 14792–14796. [Google Scholar] [CrossRef]
- Mrówczyńska, L.; Bobrowska-Hägerstrand, M.; Lindqvist, C.; Hägerstrand, H. Bordetella Adenylate Cyclase Toxin Can Bind Ganglioside GM1. BIO 2011, 1, 67–71. [Google Scholar] [CrossRef]
- Dileepan, T.; Kachlany, S.C.; Balashova, N.V.; Patel, J.; Maheswaran, S.K. Human CD18 is the functional receptor for Aggregatibacter actinomycetemcomitans leukotoxin. Infect. Immun. 2007, 75, 4851–4856. [Google Scholar] [CrossRef]
- Nygren, P.; Balashova, N.; Brown, A.C.; Kieba, I.; Dhingra, A.; Boesze-Battaglia, K.; Lally, E.T. Aggregatibacter actinomycetemcomitans leukotoxin causes activation of lymphocyte function-associated antigen 1. Cell Microbiol. 2019, 21, e12967. [Google Scholar] [CrossRef]
- Forman, M.S.; Nishikubo, J.B.; Han, R.K.; Le, A.; Balashova, N.V.; Kachlany, S.C. Gangliosides block Aggregatibacter Actinomycetemcomitans leukotoxin (LtxA)-mediated hemolysis. Toxins 2010, 2, 2824–2836. [Google Scholar] [CrossRef]
- Li, H.; Papadopoulos, V. Peripheral-type benzodiazepine receptor function in cholesterol transport. Identification of a putative cholesterol recognition/interaction amino acid sequence and consensus pattern. Endocrinology 1998, 139, 4991–4997. [Google Scholar] [CrossRef]
- Brown, A.C.; Koufos, E.; Balashova, N.V.; Boesze-Battaglia, K.; Lally, E.T. Inhibition of LtxA toxicity by blocking cholesterol binding with peptides. Mol. Oral Microbiol. 2016, 31, 94–105. [Google Scholar] [CrossRef]
- Kieba, I.R.; Fong, K.P.; Tang, H.Y.; Hoffman, K.E.; Speicher, D.W.; Klickstein, L.B.; Lally, E.T. Aggregatibacter actinomycetemcomitans leukotoxin requires beta-sheets 1 and 2 of the human CD11a beta-propeller for cytotoxicity. Cell Microbiol. 2007, 9, 2689–2699. [Google Scholar] [CrossRef] [PubMed]
- Krueger, E.; Hayes, S.; Chang, E.H.; Yutuc, S.; Brown, A.C. Receptor-Based Peptides for Inhibition of Leukotoxin Activity. ACS Infect. Dis. 2018, 4, 1073–1081. [Google Scholar] [CrossRef]
- Li, J.; Clinkenbeard, K.D.; Ritchey, J.W. Bovine CD18 identified as a species specific receptor for Pasteurella haemolytica leukotoxin. Vet. Microbiol. 1999, 67, 91–97. [Google Scholar] [CrossRef]
- Jeyaseelan, S.; Hsuan, S.L.; Kannan, M.S.; Walcheck, B.; Wang, J.F.; Kehrli, M.E.; Lally, E.T.; Sieck, G.C.; Maheswaran, S.K. Lymphocyte function-associated antigen 1 is a receptor for Pasteurella haemolytica leukotoxin in bovine leukocytes. Infect. Immun. 2000, 68, 72–79. [Google Scholar] [CrossRef] [PubMed]
- Deshpande, M.S.; Ambagala, T.C.; Ambagala, A.P.; Kehrli, M.E., Jr.; Srikumaran, S. Bovine CD18 is necessary and sufficient to mediate Mannheimia (Pasteurella) haemolytica leukotoxin-induced cytolysis. Infect. Immun. 2002, 70, 5058–5064. [Google Scholar] [CrossRef]
- Dileepan, T.; Thumbikat, P.; Walcheck, B.; Kannan, M.S.; Maheswaran, S.K. Recombinant expression of bovine LFA-1 and characterization of its role as a receptor for Mannheimia haemolytica leukotoxin. Microb. Pathog. 2005, 38, 249–257. [Google Scholar] [CrossRef]
- Gopinath, R.S.; Ambagala, T.C.; Deshpande, M.S.; Donis, R.O.; Srikumaran, S. Mannheimia (Pasteurella) haemolytica leukotoxin binding domain lies within amino acids 1 to 291 of bovine CD18. Infect. Immun. 2005, 73, 6179–6182. [Google Scholar] [CrossRef]
- Thumbikat, P.; Dileepan, T.; Kannan, M.S.; Maheswaran, S.K. Characterization of Mannheimia (Pasteurella) haemolytica leukotoxin interaction with bovine alveolar macrophage beta2 integrins. Vet. Res. 2005, 36, 771–786. [Google Scholar] [CrossRef]
- Dassanayake, R.P.; Maheswaran, S.K.; Srikumaran, S. Monomeric expression of bovine beta2-integrin subunits reveals their role in Mannheimia haemolytica leukotoxin-induced biological effects. Infect. Immun. 2007, 75, 5004–5010. [Google Scholar] [CrossRef] [PubMed]
- Dileepan, T.; Kannan, M.S.; Walcheck, B.; Maheswaran, S.K. Integrin-EGF-3 domain of bovine CD18 is critical for Mannheimia haemolytica leukotoxin species-specific susceptibility. FEMS Microbiol. Lett. 2007, 274, 67–72. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Shanthalingam, S.; Srikumaran, S. Intact signal peptide of CD18, the beta-subunit of beta2-integrins, renders ruminants susceptible to Mannheimia haemolytica leukotoxin. Proc. Natl. Acad. Sci. USA 2009, 106, 15448–15453. [Google Scholar] [CrossRef] [PubMed]
- Dileepan, T.; Kannan, M.S.; Walcheck, B.; Thumbikat, P.; Maheswaran, S.K. Mapping of the binding site for Mannheimia haemolytica leukotoxin within bovine CD18. Infect. Immun. 2005, 73, 5233–5237. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Shanthalingam, S.; Tibary, A.; Beever, J.E.; Kasinathan, P.; Brown, W.C.; Srikumaran, S. Precise gene editing paves the way for derivation of Mannheimia haemolytica leukotoxin-resistant cattle. Proc. Natl. Acad. Sci. USA 2016, 113, 13186–13190. [Google Scholar] [CrossRef]
- Hasan, S.; Osickova, A.; Bumba, L.; Novak, P.; Sebo, P.; Osicka, R. Interaction of Bordetella adenylate cyclase toxin with complement receptor 3 involves multivalent glycan binding. FEBS Lett. 2015, 589, 374–379. [Google Scholar] [CrossRef]
- Wald, T.; Osickova, A.; Masin, J.; Liskova, P.M.; Petry-Podgorska, I.; Matousek, T.; Sebo, P.; Osicka, R. Transmembrane segments of complement receptor 3 do not participate in cytotoxic activities but determine receptor structure required for action of Bordetella adenylate cyclase toxin. Pathog. Dis. 2016, 74, ftw008. [Google Scholar] [CrossRef]
- Bumba, L.; Masin, J.; Fiser, R.; Sebo, P. Bordetella adenylate cyclase toxin mobilizes its beta2 integrin receptor into lipid rafts to accomplish translocation across target cell membrane in two steps. PLoS Pathog. 2010, 6, e1000901. [Google Scholar] [CrossRef]
- Paccani, S.R.; Finetti, F.; Davi, M.; Patrussi, L.; D’Elios, M.M.; Ladant, D.; Baldari, C.T. The Bordetella pertussis adenylate cyclase toxin binds to T cells via LFA-1 and induces its disengagement from the immune synapse. J. Exp. Med. 2011, 208, 1317–1330. [Google Scholar] [CrossRef]
- Avila-Campos, M.J. Haemolytic activity of Actinobacillus actinomycetemcomitans strains on different blood types. Rev. Inst. Med. Trop. Sao Paulo 1995, 37, 215–217. [Google Scholar] [CrossRef]
- Kimizuka, R.; Miura, T.; Okuda, K. Characterization of Actinobacillus actinomycetemcomitans hemolysin. Microbiol. Immunol. 1996, 40, 717–723. [Google Scholar] [CrossRef]
- Haubek, D.; Dirienzo, J.M.; Tinoco, E.M.; Westergaard, J.; Lopez, N.J.; Chung, C.P.; Poulsen, K.; Kilian, M. Racial tropism of a highly toxic clone of Actinobacillus actinomycetemcomitans associated with juvenile periodontitis. J. Clin. Microbiol. 1997, 35, 3037–3042. [Google Scholar] [CrossRef] [PubMed]
- Kiguchi, K.; Henning-Chubb, C.B.; Huberman, E. Glycosphingolipid patterns of peripheral blood lymphocytes, monocytes, and granulocytes are cell specific. J. Biochem. 1990, 107, 8–14. [Google Scholar] [CrossRef]
- Kachlany, S.C.; Schwartz, A.B.; Balashova, N.V.; Hioe, C.E.; Tuen, M.; Le, A.; Kaur, M.; Mei, Y.; Rao, J. Anti-leukemia activity of a bacterial toxin with natural specificity for LFA-1 on white blood cells. Leuk. Res. 2010, 34, 777–785. [Google Scholar] [CrossRef] [PubMed]
- Fong, K.P.; Pacheco, C.M.; Otis, L.L.; Baranwal, S.; Kieba, I.R.; Harrison, G.; Hersh, E.V.; Boesze-Battaglia, K.; Lally, E.T. Actinobacillus actinomycetemcomitans leukotoxin requires lipid microdomains for target cell cytotoxicity. Cell Microbiol. 2006, 8, 1753–1767. [Google Scholar] [CrossRef] [PubMed]
- Baier, C.J.; Fantini, J.; Barrantes, F.J. Disclosure of cholesterol recognition motifs in transmembrane domains of the human nicotinic acetylcholine receptor. Sci. Rep. 2011, 1, 69. [Google Scholar] [CrossRef]
- Kuehn, M.J.; Kesty, N.C. Bacterial outer membrane vesicles and the host-pathogen interaction. Genes Dev. 2005, 19, 2645–2655. [Google Scholar] [CrossRef]
- Soderblom, T.; Oxhamre, C.; Wai, S.N.; Uhlen, P.; Aperia, A.; Uhlin, B.E.; Richter-Dahlfors, A. Effects of the Escherichia coli toxin cytolysin A on mucosal immunostimulation via epithelial Ca2+ signalling and Toll-like receptor 4. Cell Microbiol. 2005, 7, 779–788. [Google Scholar] [CrossRef]
- Kulp, A.; Kuehn, M.J. Biological functions and biogenesis of secreted bacterial outer membrane vesicles. Annu. Rev. Microbiol. 2010, 64, 163–184. [Google Scholar] [CrossRef]
- Kaparakis-Liaskos, M.; Ferrero, R.L. Immune modulation by bacterial outer membrane vesicles. Nat. Rev. Immunol. 2015, 15, 375–387. [Google Scholar] [CrossRef]
- Roier, S.; Zingl, F.G.; Cakar, F.; Durakovic, S.; Kohl, P.; Eichmann, T.O.; Klug, L.; Gadermaier, B.; Weinzerl, K.; Prassl, R.; et al. A novel mechanism for the biogenesis of outer membrane vesicles in Gram-negative bacteria. Nat. Commun. 2016, 7, 10515. [Google Scholar] [CrossRef]
- Toyofuku, M.; Nomura, N.; Eberl, L. Types and origins of bacterial membrane vesicles. Nat. Rev. Microbiol. 2019, 17, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Hozbor, D.; Rodriguez, M.E.; Fernandez, J.; Lagares, A.; Guiso, N.; Yantorno, O. Release of outer membrane vesicles from Bordetella pertussis. Curr. Microbiol. 1999, 38, 273–278. [Google Scholar] [CrossRef] [PubMed]
- Kato, S.; Kowashi, Y.; Demuth, D.R. Outer membrane-like vesicles secreted by Actinobacillus actinomycetemcomitans are enriched in leukotoxin. Microb. Pathog. 2002, 32, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Balsalobre, C.; Silvan, J.M.; Berglund, S.; Mizunoe, Y.; Uhlin, B.E.; Wai, S.N. Release of the type I secreted alpha-haemolysin via outer membrane vesicles from Escherichia coli. Mol. Microbiol. 2006, 59, 99–112. [Google Scholar] [CrossRef] [PubMed]
- Donato, G.M.; Goldsmith, C.S.; Paddock, C.D.; Eby, J.C.; Gray, M.C.; Hewlett, E.L. Delivery of Bordetella pertussis adenylate cyclase toxin to target cells via outer membrane vesicles. FEBS Lett. 2012, 586, 459–465. [Google Scholar] [CrossRef] [PubMed]
- Nice, J.B.; Balashova, N.V.; Kachlany, S.C.; Koufos, E.; Krueger, E.; Lally, E.T.; Brown, A.C. Aggregatibacter actinomycetemcomitans Leukotoxin Is Delivered to Host Cells in an LFA-1-Indepdendent Manner When Associated with Outer Membrane Vesicles. Toxins 2018, 10, 414. [Google Scholar] [CrossRef]
- Szabo, G.; Gray, M.C.; Hewlett, E.L. Adenylate cyclase toxin from Bordetella pertussis produces ion conductance across artificial lipid bilayers in a calcium- and polarity-dependent manner. J. Biol. Chem. 1994, 269, 22496–22499. [Google Scholar] [CrossRef]
- Lee, S.J.; Gray, M.C.; Zu, K.; Hewlett, E.L. Oligomeric behavior of Bordetella pertussis adenylate cyclase toxin in solution. Arch. Biochem. Biophys. 2005, 438, 80–87. [Google Scholar] [CrossRef]
- Iwaki, M.; Ullmann, A.; Sebo, P. Identification by in vitro complementation of regions required for cell-invasive activity of Bordetella pertussis adenylate cyclase toxin. Mol. Microbiol. 1995, 17, 1015–1024. [Google Scholar] [CrossRef]
- Ludwig, A.; Benz, R.; Goebel, W. Oligomerization of Escherichia coli haemolysin (HlyA) is involved in pore formation. Mol. Gen. Genet. 1993, 241, 89–96. [Google Scholar] [CrossRef] [PubMed]
- Moayeri, M.; Welch, R.A. Effects of temperature, time, and toxin concentration on lesion formation by the Escherichia coli hemolysin. Infect. Immun. 1994, 62, 4124–4134. [Google Scholar] [CrossRef] [PubMed]
- Moayeri, M.; Welch, R.A. Prelytic and lytic conformations of erythrocyte-associated Escherichia coli hemolysin. Infect. Immun. 1997, 65, 2233–2239. [Google Scholar] [CrossRef] [PubMed]
- Lear, J.D.; Furblur, U.G.; Lally, E.T.; Tanaka, J.C. Actinobacillus actinomycetemcomitans leukotoxin forms large conductance, voltage-gated ion channels when incorporated into planar lipid bilayers. Biochim. Biophys. Acta 1995, 1238, 34–41. [Google Scholar] [CrossRef]
- Iwase, M.; Lally, E.T.; Berthold, P.; Korchak, H.M.; Taichman, N.S. Effects of cations and osmotic protectants on cytolytic activity of Actinobacillus actinomycetemcomitans leukotoxin. Infect. Immun. 1990, 58, 1782–1788. [Google Scholar] [CrossRef]
- Karakelian, D.; Lear, J.D.; Lally, E.T.; Tanaka, J.C. Characterization of Actinobacillus actinomycetemcomitans leukotoxin pore formation in HL60 cells. Biochim. Biophys. Acta 1998, 1406, 175–187. [Google Scholar] [CrossRef]
- Brown, A.C.; Boesze-Battaglia, K.; Du, Y.; Stefano, F.P.; Kieba, I.R.; Epand, R.F.; Kakalis, L.; Yeagle, P.L.; Epand, R.M.; Lally, E.T. Aggregatibacter actinomycetemcomitans leukotoxin cytotoxicity occurs through bilayer destabilization. Cell Microbiol. 2012, 14, 869–881. [Google Scholar] [CrossRef]
- Ehrmann, I.E.; Gray, M.C.; Gordon, V.M.; Gray, L.S.; Hewlett, E.L. Hemolytic activity of adenylate cyclase toxin from Bordetella pertussis. FEBS Lett. 1991, 278, 79–83. [Google Scholar] [CrossRef]
- Knapp, O.; Maier, E.; Masin, J.; Sebo, P.; Benz, R. Pore formation by the Bordetella adenylate cyclase toxin in lipid bilayer membranes: Role of voltage and pH. Biochim. Biophys. Acta 2008, 1778, 260–269. [Google Scholar] [CrossRef]
- Menestrina, G. Escherichia coli hemolysin permeabilizes small unilamellar vesicles loaded with calcein by a single-hit mechanism. FEBS Lett. 1988, 232, 217–220. [Google Scholar] [CrossRef]
- Ostolaza, H.; Bartolome, B.; Ortiz de Zarate, I.; de la Cruz, F.; Goni, F.M. Release of lipid vesicle contents by the bacterial protein toxin alpha-haemolysin. Biochim. Biophys. Acta 1993, 1147, 81–88. [Google Scholar] [CrossRef]
- Menestrina, G.; Dalla Serra, M.; Pederzolli, C.; Bregante, M.; Gambale, F. Bacterial hemolysins and leukotoxins affect target cells by forming large exogenous pores into their plasma membrane: Escherichia coli hemolysin A as a case example. Biosci. Rep. 1995, 15, 543–551. [Google Scholar] [CrossRef]
- Skopova, K.; Tomalova, B.; Kanchev, I.; Rossmann, P.; Svedova, M.; Adkins, I.; Bibova, I.; Tomala, J.; Masin, J.; Guiso, N.; et al. Cyclic AMP-Elevating Capacity of Adenylate Cyclase Toxin-Hemolysin Is Sufficient for Lung Infection but Not for Full Virulence of Bordetella pertussis. Infect. Immun. 2017, 85, e00937-16. [Google Scholar] [CrossRef] [PubMed]
- Holubova, J.; Juhasz, A.; Masin, J.; Stanek, O.; Jurnecka, D.; Osickova, A.; Sebo, P.; Osicka, R. Selective Enhancement of the Cell-Permeabilizing Activity of Adenylate Cyclase Toxin Does Not Increase Virulence of Bordetella pertussis. Int. J. Mol. Sci. 2021, 22, 11655. [Google Scholar] [CrossRef] [PubMed]
- Betsou, F.; Sebo, P.; Guiso, N. CyaC-mediated activation is important not only for toxic but also for protective activities of Bordetella pertussis adenylate cyclase-hemolysin. Infect. Immun. 1993, 61, 3583–3589. [Google Scholar] [CrossRef]
- Cannella, S.E.; Ntsogo Enguene, V.Y.; Davi, M.; Malosse, C.; Sotomayor Perez, A.C.; Chamot-Rooke, J.; Vachette, P.; Durand, D.; Ladant, D.; Chenal, A. Stability, structural and functional properties of a monomeric, calcium-loaded adenylate cyclase toxin, CyaA, from Bordetella pertussis. Sci. Rep. 2017, 7, 42065. [Google Scholar] [CrossRef]
- Fiser, R.; Masin, J.; Basler, M.; Krusek, J.; Spulakova, V.; Konopasek, I.; Sebo, P. Third activity of Bordetella adenylate cyclase (AC) toxin-hemolysin. Membrane translocation of AC domain polypeptide promotes calcium influx into CD11b+ monocytes independently of the catalytic and hemolytic activities. J. Biol. Chem. 2007, 282, 2808–2820. [Google Scholar] [CrossRef] [PubMed]
- Seeger, W.; Walter, H.; Suttorp, N.; Muhly, M.; Bhakdi, S. Thromboxane-mediated hypertension and vascular leakage evoked by low doses of Escherichia coli hemolysin in rabbit lungs. J. Clin. Investig. 1989, 84, 220–227. [Google Scholar] [CrossRef]
- Bhakdi, S.; Muhly, M.; Korom, S.; Schmidt, G. Effects of Escherichia coli hemolysin on human monocytes. Cytocidal action and stimulation of interleukin 1 release. J. Clin. Investig. 1990, 85, 1746–1753. [Google Scholar] [CrossRef]
- Grimminger, F.; Sibelius, U.; Bhakdi, S.; Suttorp, N.; Seeger, W. Escherichia coli hemolysin is a potent inductor of phosphoinositide hydrolysis and related metabolic responses in human neutrophils. J. Clin. Investig. 1991, 88, 1531–1539. [Google Scholar] [CrossRef]
- Konig, B.; Konig, W. Induction and suppression of cytokine release (tumour necrosis factor-alpha; interleukin-6, interleukin-1 beta) by Escherichia coli pathogenicity factors (adhesions, alpha-haemolysin). Immunology 1993, 78, 526–533. [Google Scholar] [PubMed]
- Konig, B.; Ludwig, A.; Goebel, W.; Konig, W. Pore formation by the Escherichia coli alpha-hemolysin: Role for mediator release from human inflammatory cells. Infect. Immun. 1994, 62, 4611–4617. [Google Scholar] [CrossRef] [PubMed]
- Ohguchi, M.; Ishisaki, A.; Okahashi, N.; Koide, M.; Koseki, T.; Yamato, K.; Noguchi, T.; Nishihara, T. Actinobacillus actinomycetemcomitans toxin induces both cell cycle arrest in the G2/M phase and apoptosis. Infect. Immun. 1998, 66, 5980–5987. [Google Scholar] [CrossRef] [PubMed]
- Claesson, R.; Johansson, A.; Belibasakis, G.; Hanstrom, L.; Kalfas, S. Release and activation of matrix metalloproteinase 8 from human neutrophils triggered by the leukotoxin of Actinobacillus actinomycetemcomitans. J. Periodontal Res. 2002, 37, 353–359. [Google Scholar] [CrossRef]
- Wiles, T.J.; Dhakal, B.K.; Eto, D.S.; Mulvey, M.A. Inactivation of host Akt/protein kinase B signaling by bacterial pore-forming toxins. Mol. Biol. Cell 2008, 19, 1427–1438. [Google Scholar] [CrossRef]
- Kloft, N.; Busch, T.; Neukirch, C.; Weis, S.; Boukhallouk, F.; Bobkiewicz, W.; Cibis, I.; Bhakdi, S.; Husmann, M. Pore-forming toxins activate MAPK p38 by causing loss of cellular potassium. Biochem. Biophys. Res. Commun. 2009, 385, 503–506. [Google Scholar] [CrossRef]
- Dunne, A.; Ross, P.J.; Pospisilova, E.; Masin, J.; Meaney, A.; Sutton, C.E.; Iwakura, Y.; Tschopp, J.; Sebo, P.; Mills, K.H. Inflammasome activation by adenylate cyclase toxin directs Th17 responses and protection against Bordetella pertussis. J. Immunol. 2010, 185, 1711–1719. [Google Scholar] [CrossRef]
- Hilbert, D.W.; Paulish-Miller, T.E.; Tan, C.K.; Carey, A.J.; Ulett, G.C.; Mordechai, E.; Adelson, M.E.; Gygax, S.E.; Trama, J.P. Clinical Escherichia coli isolates utilize alpha-hemolysin to inhibit in vitro epithelial cytokine production. Microb. Infect. 2012, 14, 628–638. [Google Scholar] [CrossRef]
- Dhakal, B.K.; Mulvey, M.A. The UPEC pore-forming toxin alpha-hemolysin triggers proteolysis of host proteins to disrupt cell adhesion, inflammatory, and survival pathways. Cell Host Microb. 2012, 11, 58–69. [Google Scholar] [CrossRef]
- Dietmann, A.; Millonig, A.; Combes, V.; Couraud, P.O.; Kachlany, S.C.; Grau, G.E. Effects of Aggregatibacter actinomycetemcomitans leukotoxin on endothelial cells. Microb. Pathog. 2013, 61–62, 43–50. [Google Scholar] [CrossRef]
- Svedova, M.; Masin, J.; Fiser, R.; Cerny, O.; Tomala, J.; Freudenberg, M.; Tuckova, L.; Kovar, M.; Dadaglio, G.; Adkins, I.; et al. Pore-formation by adenylate cyclase toxoid activates dendritic cells to prime CD8+ and CD4+ T cells. Immunol. Cell Biol. 2016, 94, 322–333. [Google Scholar] [CrossRef] [PubMed]
- Jeyaseelan, S.; Kannan, M.S.; Briggs, R.E.; Thumbikat, P.; Maheswaran, S.K. Mannheimia haemolytica leukotoxin activates a nonreceptor tyrosine kinase signaling cascade in bovine leukocytes, which induces biological effects. Infect. Immun. 2001, 69, 6131–6139. [Google Scholar] [CrossRef] [PubMed]
- Troeger, H.; Richter, J.F.; Beutin, L.; Gunzel, D.; Dobrindt, U.; Epple, H.J.; Gitter, A.H.; Zeitz, M.; Fromm, M.; Schulzke, J.D. Escherichia coli alpha-haemolysin induces focal leaks in colonic epithelium: A novel mechanism of bacterial translocation. Cell Microbiol. 2007, 9, 2530–2540. [Google Scholar] [CrossRef]
- Bucker, R.; Schulz, E.; Gunzel, D.; Bojarski, C.; Lee, I.F.; John, L.J.; Wiegand, S.; Janssen, T.; Wieler, L.H.; Dobrindt, U.; et al. alpha-Haemolysin of Escherichia coli in IBD: A potentiator of inflammatory activity in the colon. Gut 2014, 63, 1893–1901. [Google Scholar] [CrossRef] [PubMed]
- Johansson, A.; Claesson, R.; Hanstrom, L.; Sandstrom, G.; Kalfas, S. Polymorphonuclear leukocyte degranulation induced by leukotoxin from Actinobacillus actinomycetemcomitans. J. Periodontal Res. 2000, 35, 85–92. [Google Scholar] [CrossRef] [PubMed]
- Kelk, P.; Claesson, R.; Hanstrom, L.; Lerner, U.H.; Kalfas, S.; Johansson, A. Abundant secretion of bioactive interleukin-1beta by human macrophages induced by Actinobacillus actinomycetemcomitans leukotoxin. Infect. Immun. 2005, 73, 453–458. [Google Scholar] [CrossRef] [PubMed]
- Kelk, P.; Claesson, R.; Chen, C.; Sjostedt, A.; Johansson, A. IL-1beta secretion induced by Aggregatibacter (Actinobacillus) actinomycetemcomitans is mainly caused by the leukotoxin. Int. J. Med Microbiol. IJMM 2008, 298, 529–541. [Google Scholar] [CrossRef]
- Hiyoshi, T.; Domon, H.; Maekawa, T.; Nagai, K.; Tamura, H.; Takahashi, N.; Yonezawa, D.; Miyoshi, T.; Yoshida, A.; Tabeta, K.; et al. Aggregatibacter actinomycetemcomitans induces detachment and death of human gingival epithelial cells and fibroblasts via elastase release following leukotoxin-dependent neutrophil lysis. Microbiol. Immunol. 2019, 63, 100–110. [Google Scholar] [CrossRef]
- May, A.K.; Sawyer, R.G.; Gleason, T.; Whitworth, A.; Pruett, T.L. In vivo cytokine response to Escherichia coli alpha-hemolysin determined with genetically engineered hemolytic and nonhemolytic E. coli variants. Infect. Immun. 1996, 64, 2167–2171. [Google Scholar] [CrossRef]
- Gleason, T.G.; Houlgrave, C.W.; May, A.K.; Crabtree, T.D.; Sawyer, R.G.; Denham, W.; Norman, J.G.; Pruett, T.L. Hemolytically active (acylated) alpha-hemolysin elicits interleukin-1beta (IL-1beta) but augments the lethality of Escherichia coli by an IL-1- and tumor necrosis factor-independent mechanism. Infect. Immun. 1998, 66, 4215–4221. [Google Scholar] [CrossRef]
- Murthy, A.M.V.; Phan, M.D.; Peters, K.M.; Nhu, N.T.K.; Welch, R.A.; Ulett, G.C.; Schembri, M.A.; Sweet, M.J. Regulation of hemolysin in uropathogenic Escherichia coli fine-tunes killing of human macrophages. Virulence 2018, 9, 967–980. [Google Scholar] [CrossRef] [PubMed]
- Verma, V.; Kumar, P.; Gupta, S.; Yadav, S.; Dhanda, R.S.; Thorlacius, H.; Yadav, M. alpha-Hemolysin of uropathogenic E. coli regulates NLRP3 inflammasome activation and mitochondrial dysfunction in THP-1 macrophages. Sci. Rep. 2020, 10, 12653. [Google Scholar] [CrossRef] [PubMed]
- Uhlén, P.; Laestadius, A.; Jahnukainen, T.; Söderblom, T.; Bäckhed, F.; Celsi, G.; Brismar, H.; Normark, S.; Aperia, A.; Richter-Dahlfors, A. Alpha-haemolysin of uropathogenic E. coli induces Ca2+ oscillations in renal epithelial cells. Nature 2000, 405, 694–697. [Google Scholar] [CrossRef] [PubMed]
- Koschinski, A.; Repp, H.; Unver, B.; Dreyer, F.; Brockmeier, D.; Valeva, A.; Bhakdi, S.; Walev, I. Why Escherichia coli alpha-hemolysin induces calcium oscillations in mammalian cells-the pore is on its own. FASEB J. 2006, 20, 973–975. [Google Scholar] [CrossRef] [PubMed]
- Huffman, D.L.; Abrami, L.; Sasik, R.; Corbeil, J.; van der Goot, F.G.; Aroian, R.V. Mitogen-activated protein kinase pathways defend against bacterial pore-forming toxins. Proc. Natl. Acad. Sci. USA 2004, 101, 10995–11000. [Google Scholar] [CrossRef]
- Skals, M.; Jorgensen, N.R.; Leipziger, J.; Praetorius, H.A. Alpha-hemolysin from Escherichia coli uses endogenous amplification through P2X receptor activation to induce hemolysis. Proc. Natl. Acad. Sci. USA 2009, 106, 4030–4035. [Google Scholar] [CrossRef]
- Munksgaard, P.S.; Vorup-Jensen, T.; Reinholdt, J.; Soderstrom, C.M.; Poulsen, K.; Leipziger, J.; Praetorius, H.A.; Skals, M. Leukotoxin from Aggregatibacter actinomycetemcomitans causes shrinkage and P2X receptor-dependent lysis of human erythrocytes. Cell Microbiol. 2012, 14, 1904–1920. [Google Scholar] [CrossRef]
- Fagerberg, S.K.; Jakobsen, M.R.; Skals, M.; Praetorius, H.A. Inhibition of P2X Receptors Protects Human Monocytes against Damage by Leukotoxin from Aggregatibacter actinomycetemcomitans and alpha-Hemolysin from Escherichia coli. Infect. Immun. 2016, 84, 3114–3130. [Google Scholar] [CrossRef]
- Prince, D.J.; Patel, D.; Kachlany, S.C. Leukotoxin (LtxA/Leukothera) induces ATP expulsion via pannexin-1 channels and subsequent cell death in malignant lymphocytes. Sci. Rep. 2021, 11, 18086. [Google Scholar] [CrossRef]
- Therkildsen, J.R.; Christensen, M.G.; Tingskov, S.J.; Wehmoller, J.; Norregaard, R.; Praetorius, H.A. Lack of P2X7 Receptors Protects against Renal Fibrosis after Pyelonephritis with alpha-Hemolysin-Producing Escherichia coli. Am. J. Pathol. 2019, 189, 1201–1211. [Google Scholar] [CrossRef]
- Schulz, E.; Schumann, M.; Schneemann, M.; Dony, V.; Fromm, A.; Nagel, O.; Schulzke, J.D.; Bucker, R. Escherichia coli Alpha-Hemolysin HlyA Induces Host Cell Polarity Changes, Epithelial Barrier Dysfunction and Cell Detachment in Human Colon Carcinoma Caco-2 Cell Model via PTEN-Dependent Dysregulation of Cell Junctions. Toxins 2021, 13, 520. [Google Scholar] [CrossRef] [PubMed]
- Confer, D.L.; Eaton, J.W. Phagocyte impotence caused by an invasive bacterial adenylate cyclase. Science 1982, 217, 948–950. [Google Scholar] [CrossRef] [PubMed]
- Friedman, R.L.; Fiederlein, R.L.; Glasser, L.; Galgiani, J.N. Bordetella pertussis adenylate cyclase: Effects of affinity-purified adenylate cyclase on human polymorphonuclear leukocyte functions. Infect. Immun. 1987, 55, 135–140. [Google Scholar] [CrossRef] [PubMed]
- Eby, J.C.; Gray, M.C.; Hewlett, E.L. Cyclic AMP-mediated suppression of neutrophil extracellular trap formation and apoptosis by the Bordetella pertussis adenylate cyclase toxin. Infect. Immun. 2014, 82, 5256–5269. [Google Scholar] [CrossRef]
- Ahmad, J.N.; Cerny, O.; Linhartova, I.; Masin, J.; Osicka, R.; Sebo, P. cAMP signalling of Bordetella adenylate cyclase toxin through the SHP-1 phosphatase activates the BimEL-Bax pro-apoptotic cascade in phagocytes. Cell Microbiol. 2016, 18, 384–398. [Google Scholar] [CrossRef] [PubMed]
- Cerny, O.; Anderson, K.E.; Stephens, L.R.; Hawkins, P.T.; Sebo, P. cAMP Signaling of Adenylate Cyclase Toxin Blocks the Oxidative Burst of Neutrophils through Epac-Mediated Inhibition of Phospholipase C Activity. J. Immunol. 2017, 198, 1285–1296. [Google Scholar] [CrossRef] [PubMed]
- Hasan, S.; Rahman, W.U.; Sebo, P.; Osicka, R. Distinct Spatiotemporal Distribution of Bacterial Toxin-Produced Cellular cAMP Differentially Inhibits Opsonophagocytic Signaling. Toxins 2019, 11, 362. [Google Scholar] [CrossRef]
- Ahmad, J.N.; Holubova, J.; Benada, O.; Kofronova, O.; Stehlik, L.; Vasakova, M.; Sebo, P. Bordetella Adenylate Cyclase Toxin Inhibits Monocyte-to-Macrophage Transition and Dedifferentiates Human Alveolar Macrophages into Monocyte-like Cells. mBio 2019, 10, e01743-19. [Google Scholar] [CrossRef]
- Ahmad, J.N.; Sebo, P. Bacterial RTX toxins and host immunity. Curr. Opin. Infect. Dis. 2021, 34, 187–196. [Google Scholar] [CrossRef]
- Fiser, R.; Masin, J.; Bumba, L.; Pospisilova, E.; Fayolle, C.; Basler, M.; Sadilkova, L.; Adkins, I.; Kamanova, J.; Cerny, J.; et al. Calcium influx rescues adenylate cyclase-hemolysin from rapid cell membrane removal and enables phagocyte permeabilization by toxin pores. PLoS Pathog. 2012, 8, e1002580. [Google Scholar] [CrossRef]
- Basler, M.; Masin, J.; Osicka, R.; Sebo, P. Pore-forming and enzymatic activities of Bordetella pertussis adenylate cyclase toxin synergize in promoting lysis of monocytes. Infect. Immun. 2006, 74, 2207–2214. [Google Scholar] [CrossRef] [PubMed]
- Hewlett, E.L.; Donato, G.M.; Gray, M.C. Macrophage cytotoxicity produced by adenylate cyclase toxin from Bordetella pertussis: More than just making cyclic AMP! Mol. Microbiol. 2006, 59, 447–459. [Google Scholar] [CrossRef] [PubMed]









| K. kingae Genes | modK1 ON 1 | modK1 OFF 1 | Host Genes | modK1 ON 1 | modK1 OFF 1 |
|---|---|---|---|---|---|
| rtxA |  |  | IL-1β | | |
| groEL | | | TNF | | |
| dnaK | | | IL-8 | | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Filipi, K.; Rahman, W.U.; Osickova, A.; Osicka, R. Kingella kingae RtxA Cytotoxin in the Context of Other RTX Toxins. Microorganisms 2022, 10, 518. https://doi.org/10.3390/microorganisms10030518
Filipi K, Rahman WU, Osickova A, Osicka R. Kingella kingae RtxA Cytotoxin in the Context of Other RTX Toxins. Microorganisms. 2022; 10(3):518. https://doi.org/10.3390/microorganisms10030518
Chicago/Turabian StyleFilipi, Katerina, Waheed Ur Rahman, Adriana Osickova, and Radim Osicka. 2022. "Kingella kingae RtxA Cytotoxin in the Context of Other RTX Toxins" Microorganisms 10, no. 3: 518. https://doi.org/10.3390/microorganisms10030518
APA StyleFilipi, K., Rahman, W. U., Osickova, A., & Osicka, R. (2022). Kingella kingae RtxA Cytotoxin in the Context of Other RTX Toxins. Microorganisms, 10(3), 518. https://doi.org/10.3390/microorganisms10030518