
Individuals with Inflammatory Bowel Disease Have an Altered Gut Microbiome Composition of Fungi and Protozoa
 ,
,  ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Populations and Design
2.2. Processing Metagenomic Sequences
2.3. Identifying Eukaryotes in Gut Metagenomes
2.4. Statistical Analyses
2.4.1. Descriptive Taxonomic Analysis
2.4.2. Cohort Analysis
2.4.3. Regression Analysis
3. Results
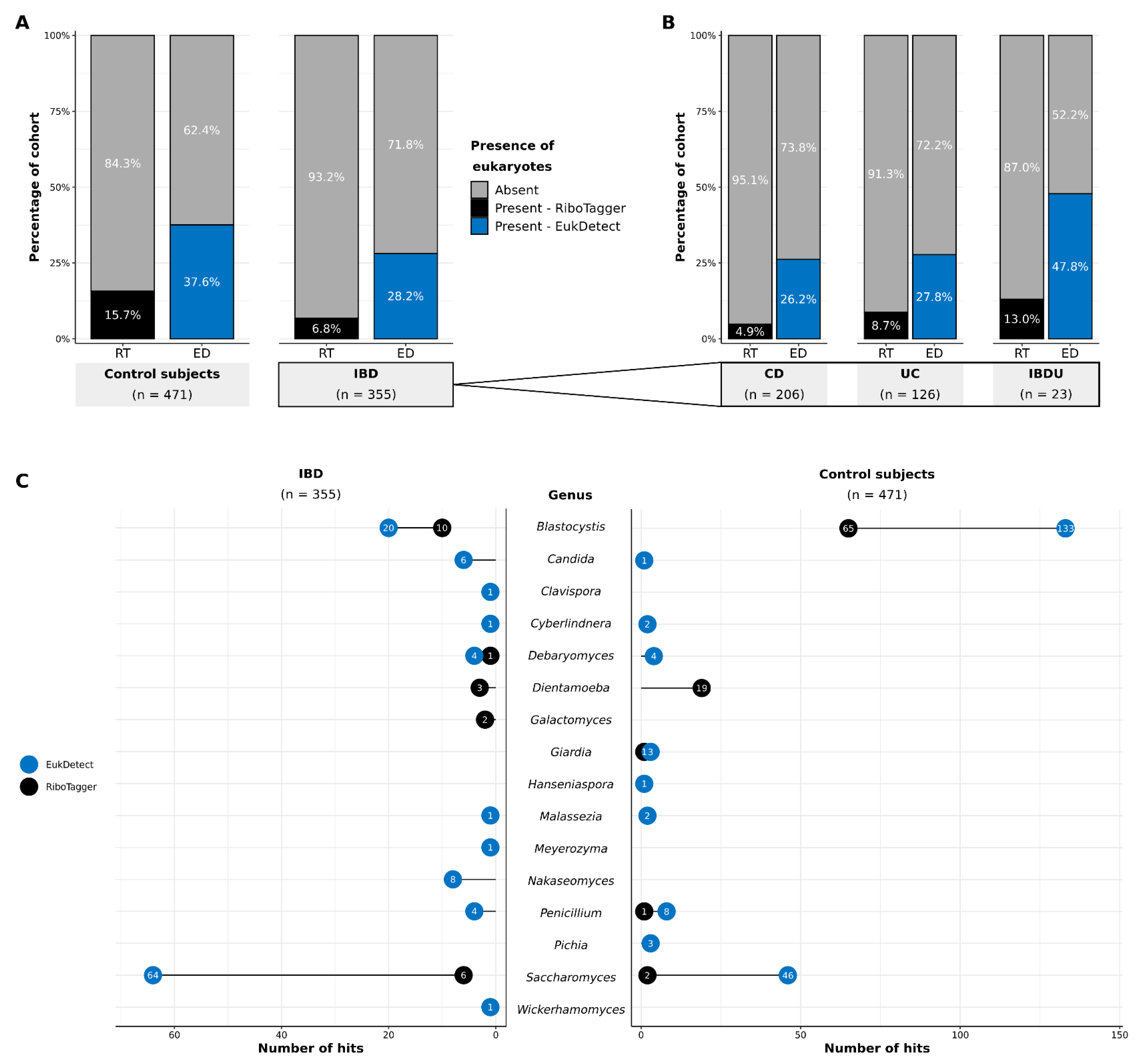
3.1. Detection of Eukaryotes in Gut Metagenomes
3.2. Distribution and Prevalence of Eukaryotes
3.2.1. Proportional Abundances
3.2.2. Species-Level Eukaryotic Prevalence
3.2.3. Shared and Unique Species
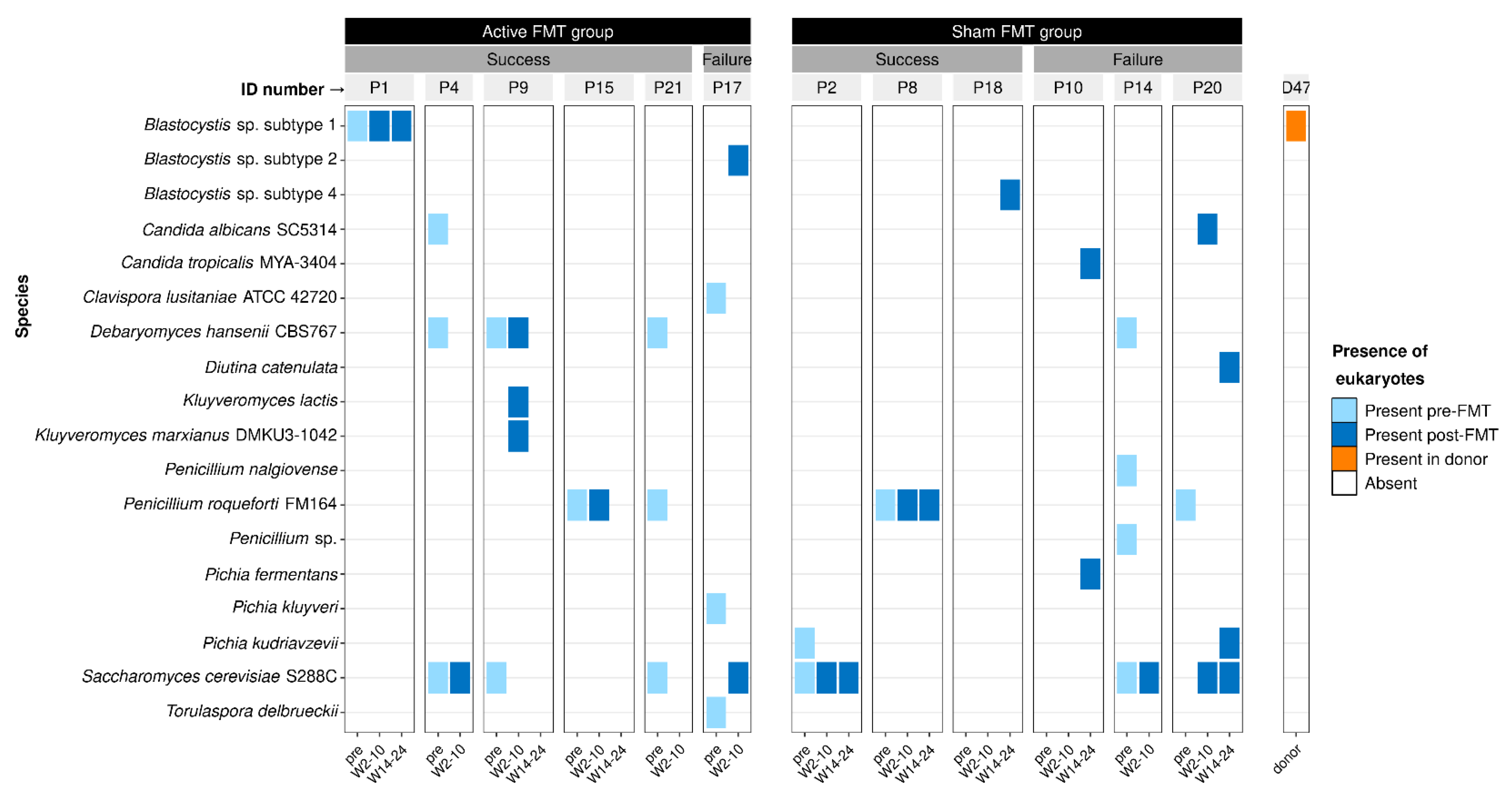
3.3. Effect of Faecal Microbiota Transplantation on Eukaryotes
3.4. Influence of Cohort Demographics on Gut Eukaryote Composition
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Podolsky, D.K. Inflammatory bowel disease. N. Engl. J. Med. 1991, 325, 928–937. [Google Scholar] [CrossRef] [PubMed]
- Khor, B.; Gardet, A.; Xavier, R.J. Genetics and pathogenesis of inflammatory bowel disease. Nature 2011, 474, 307–317. [Google Scholar] [CrossRef] [PubMed]
- Ho, S.-M.; Lewis, J.D.; Mayer, A.E.; Bernstein, C.N.; Plevy, E.S.; Chuang, E.; Rappaport, S.M.; Croitoru, K.; Korzenik, J.R.; Krischer, J.; et al. Challenges in IBD Research: Environmental Triggers. Inflamm. Bowel Dis. 2019, 25, S13–S23. [Google Scholar] [CrossRef] [PubMed]
- Loddo, I.; Romano, C. Inflammatory Bowel Disease: Genetics, Epigenetics, and Pathogenesis. Front. Immunol. 2015, 6, 551. [Google Scholar] [CrossRef]
- Marchesi, J.R.; Ravel, J. The vocabulary of microbiome research: A proposal. Microbiome 2015, 3, 31. [Google Scholar] [CrossRef]
- Kostic, A.D.; Xavier, R.J.; Gevers, D. The Microbiome in Inflammatory Bowel Disease: Current Status and the Future Ahead. Gastroenterology 2014, 146, 1489–1499. [Google Scholar] [CrossRef]
- Ni, J.; Wu, G.D.; Albenberg, L.; Tomov, V.T. Gut microbiota and IBD: Causation or correlation? Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 573–584. [Google Scholar] [CrossRef]
- Khan, I.; Ullah, N.; Zha, L.; Bai, Y.; Khan, A.; Zhao, T.; Che, T.; Zhang, C. Alteration of Gut Microbiota in Inflammatory Bowel Disease (IBD): Cause or Consequence? IBD Treatment Targeting the Gut Microbiome. Pathogens 2019, 8, 126. [Google Scholar] [CrossRef]
- Lopez, J.; Grinspan, A. Fecal Microbiota Transplantation for Inflammatory Bowel Disease. Gastroenterol. Hepatol. 2016, 12, 374–379. [Google Scholar]
- Costello, S.P.; Soo, W.; Bryant, R.V.; Jairath, V.; Hart, A.L.; Andrews, J.M. Systematic review with meta-analysis: Faecal microbiota transplantation for the induction of remission for active ulcerative colitis. Aliment. Pharmacol. Ther. 2017, 46, 213–224. [Google Scholar] [CrossRef]
- Caldeira, L.D.F.; Borba, H.H.; Tonin, F.S.; Wiens, A.; Fernandez-Llimos, F.; Pontarolo, R. Fecal microbiota transplantation in inflammatory bowel disease patients: A systematic review and meta-analysis. PLoS ONE 2020, 15, e0238910. [Google Scholar] [CrossRef] [PubMed]
- Kong, L.; Lloyd-Price, J.; Vatanen, T.; Seksik, P.; Beaugerie, L.; Simon, T.; Vlamakis, H.; Sokol, H.; Xavier, R.J. Linking Strain Engraftment in Fecal Microbiota Transplantation With Maintenance of Remission in Crohn’s Disease. Gastroenterology 2020, 159, 2193–2202.e5. [Google Scholar] [CrossRef] [PubMed]
- Guzzo, G.L.; Andrews, J.M.; Weyrich, L.S. The Neglected Gut Microbiome: Fungi, Protozoa, and Bacteriophages in Inflammatory Bowel Disease. Inflamm. Bowel Dis. 2022, 28, 1112–1122. [Google Scholar] [CrossRef] [PubMed]
- Halwachs, B.; Madhusudhan, N.; Krause, R.; Nilsson, H.; Moissl-Eichinger, C.; Högenauer, C.; Thallinger, G.G.; Gorkiewicz, G. Critical Issues in Mycobiota Analysis. Front. Microbiol. 2017, 8, 180. [Google Scholar] [CrossRef]
- Nash, A.K.; Auchtung, T.A.; Wong, M.C.; Smith, D.P.; Gesell, J.R.; Ross, M.C.; Stewart, C.J.; Metcalf, G.A.; Muzny, D.M.; Gibbs, R.A.; et al. The gut mycobiome of the Human Microbiome Project healthy cohort. Microbiome 2017, 5, 153. [Google Scholar] [CrossRef] [PubMed]
- Sender, R.; Fuchs, S.; Milo, R. Revised Estimates for the Number of Human and Bacteria Cells in the Body. PLoS Biol. 2016, 14, e1002533. [Google Scholar] [CrossRef] [PubMed]
- Sarrabayrouse, G.; Elias, A.; Yáñez, F.; Mayorga, L.; Varela, E.; Bartoli, C.; Casellas, F.; Borruel, N.; de Guise, C.H.; Machiels, K.; et al. Fungal and Bacterial Loads: Noninvasive Inflammatory Bowel Disease Biomarkers for the Clinical Setting. mSystems 2021, 6, e01277-20. [Google Scholar] [CrossRef] [PubMed]
- Richard, M.L.; Sokol, H. The gut mycobiota: Insights into analysis, environmental interactions and role in gastrointestinal diseases. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 1–345. [Google Scholar] [CrossRef]
- Miyoshi, J.; Sofia, M.A.; Pierre, J.F. The evidence for fungus in Crohn’s disease pathogenesis. Clin. J. Gastroenterol. 2018, 11, 449–456. [Google Scholar] [CrossRef]
- Van Schaik, F.D.M.; Oldenburg, B.; Hart, A.R.; Siersema, P.D.; Lindgren, S.; Grip, O.; Teucher, B.; Kaaks, R.; Bergmann, M.M.; Boeing, H.; et al. Serological markers predict inflammatory bowel disease years before the diagnosis. Gut 2012, 62, 683–688. [Google Scholar] [CrossRef]
- Findley, K.; Oh, J.; Yang, J.; Conlan, S.; Deming, C.; Meyer, J.A.; Schoenfeld, D.; Nomicos, E.; Park, M.; Kong, H.H.; et al. Topographic diversity of fungal and bacterial communities in human skin. Nature 2013, 498, 367–370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Standaert-Vitse, A.; Sendid, B.; Joossens, M.; François, N.; Khoury, P.V.-E.; Branche, J.; Van Kruiningen, H.; Jouault, T.; Rutgeerts, P.; Gower-Rousseau, C.; et al. Candida albicans Colonization and ASCA in Familial Crohn’s Disease. Am. J. Gastroenterol. 2009, 104, 1745–1753. [Google Scholar] [CrossRef]
- Yan, L.; Yang, C.; Tang, J. Disruption of the intestinal mucosal barrier in Candida albicans infections. Microbiol. Res. 2013, 168, 389–395. [Google Scholar] [CrossRef]
- Sartor, R.B.; Wu, G.D. Roles for Intestinal Bacteria, Viruses, and Fungi in Pathogenesis of Inflammatory Bowel Diseases and Therapeutic Approaches. Gastroenterology 2017, 152, 327–339.e4. [Google Scholar] [CrossRef] [PubMed]
- Hoarau, G.; Mukherjee, P.K.; Gower, C.; Hager, C.; Chandra, J.; Retuerto, M.A.; Neut, C.; Vermeire, S.; Clemente, J.; Colombel, J.F.; et al. Bacteriome and Mycobiome Interactions Underscore Microbial Dysbiosis in Familial Crohn’s Disease. mBio 2016, 7, e01250-16. [Google Scholar] [CrossRef] [PubMed]
- Garcia, L.S. Dientamoeba fragilis, One of the Neglected Intestinal Protozoa. J. Clin. Microbiol. 2016, 54, 2243–2250. [Google Scholar] [CrossRef] [PubMed]
- Wawrzyniak, I.; Poirier, P.; Viscogliosi, E.; Dionigia, M.; Texier, C.; Delbac, F.; El Alaoui, H. Blastocystis, an unrecognized parasite: An overview of pathogenesis and diagnosis. Ther. Adv. Infect. Dis. 2013, 1, 167–178. [Google Scholar] [CrossRef]
- Nieves-Ramírez, M.E.; Partida-Rodríguez, O.; Laforest-Lapointe, I.; Reynolds, L.A.; Brown, E.M.; Valdez-Salazar, A.; Morán-Silva, P.; Rojas-Velázquez, L.; Morien, E.; Parfrey, L.W.; et al. Asymptomatic Intestinal Colonization with Protist Blastocystis Is Strongly Associated with Distinct Microbiome Ecological Patterns. mSystems 2018, 3, e00007-18. [Google Scholar] [CrossRef]
- De Boer, M.D.; Schuurs, T.A.; Vermeer, M.; Ruijs, G.J.; van der Zanden, A.G.M.; Weel, J.F.; van Coppenraet, L.E.B. Distribution and relevance of Dientamoeba fragilis and Blastocystis species in gastroenteritis: Results from a case-control study. Eur. J. Clin. Microbiol. 2019, 39, 197–203. [Google Scholar] [CrossRef]
- Chabé, M.; Lokmer, A.; Ségurel, L. Gut Protozoa: Friends or Foes of the Human Gut Microbiota? Trends Parasitol. 2017, 33, 925–934. [Google Scholar] [CrossRef]
- Rossen, N.G.; Bart, A.; Verhaar, N.; Van Nood, E.; Kootte, R.; De Groot, P.F.; D’Haens, G.R.; Ponsioen, C.Y.; Van Gool, T. Low prevalence of Blastocystis sp. in active ulcerative colitis patients. Eur. J. Clin. Microbiol. 2015, 34, 1039–1044. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petersen, A.M.; Stensvold, C.R.; Mirsepasi, H.; Engberg, J.; Friis-Møller, A.; Porsbo, L.J.; Hammerum, A.M.; Nordgaard-Lassen, I.; Nielsen, H.V.; Krogfelt, K.A. Active ulcerative colitis associated with low prevalence of Blastocystis and Dientamoeba fragilis infection. Scand. J. Gastroenterol. 2013, 48, 638–639. [Google Scholar] [CrossRef] [PubMed]
- Tito, R.Y.; Chaffron, S.; Caenepeel, C.; Lima-Mendez, G.; Wang, J.; Vieira-Silva, S.; Falony, G.; Hildebrand, F.; Darzi, Y.; Rymenans, L.; et al. Population-level analysis of Blastocystis subtype prevalence and variation in the human gut microbiota. Gut 2019, 68, 1180–1189. [Google Scholar] [CrossRef] [PubMed]
- Imhann, F.; Van Der Velde, K.J.; Barbieri, R.; Alberts, R.; Voskuil, M.D.; Vila, A.V.; Collij, V.; Spekhorst, L.M.; Van Der Sloot, K.W.J.; Peters, V.; et al. The 1000IBD project: Multi-omics data of 1000 inflammatory bowel disease patients; data release 1. BMC Gastroenterol. 2019, 19, 1–10. [Google Scholar] [CrossRef]
- Schirmer, M.; Smeekens, S.P.; Vlamakis, H.; Jaeger, M.; Oosting, M.; Franzosa, E.A.; Ter Horst, R.; Jansen, T.; Jacobs, L.; Bonder, M.J.; et al. Linking the Human Gut Microbiome to Inflammatory Cytokine Production Capacity. Cell 2016, 167, 1125–1136.e8. [Google Scholar] [CrossRef]
- Sokol, H.; Landman, C.; Seksik, P.; Berard, L.; Montil, M.; Nion-Larmurier, I.; Bourrier, A.; Le Gall, G.; Lalande, V.; De Rougemont, A.; et al. Fecal microbiota transplantation to maintain remission in Crohn’s disease: A pilot randomized controlled study. Microbiome 2020, 8, 12. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef]
- Xie, C.; Goi, C.L.W.; Huson, D.H.; Little, P.F.R.; Williams, R.B.H. RiboTagger: Fast and unbiased 16S/18S profiling using whole community shotgun metagenomic or metatranscriptome surveys. BMC Bioinform. 2016, 17, 277–282. [Google Scholar] [CrossRef]
- Lind, A.L.; Pollard, K.S. Accurate and sensitive detection of microbial eukaryotes from whole metagenome shotgun sequencing. Microbiome 2021, 9, 1–18. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2020. [Google Scholar]
- McMurdie, P.J.; Holmes, S. phyloseq: An R package for reproducible interactive analysis and graphics of microbiome census data. PLoS ONE 2013, 8, e61217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2016. [Google Scholar]
- DeWitt, P.; Bennett, T. qwraps2: Quick Wraps 2,0.5.2; CRAN. 2021. Available online: https://cran.r-project.org/web/packages/qwraps2/index.html (accessed on 14 August 2022).
- Little, R.J.A.; Rubin, D.B. Statistical Analysis with Missing Data; John Wiley & Sons, Inc.: New York, NY, USA, 2002. [Google Scholar]
- McCullagh, P.; Nelder, J.A. Generalized Linear Models; CRC Press LLC: Boca Raton, FL, USA, 1989. [Google Scholar]
- Zeileis, A.; Kleiber, C.; Jackman, S. Regression Models for Count Data in R. J. Stat. Softw. 2008, 27, 1–25. [Google Scholar] [CrossRef]
- Jackman, S. pscl: Classes and Methods for R Developed in the Political Science Computational Laboratory; United States Studies Centre, University of Sydney: Sydney, NSW, Australia, 2020. [Google Scholar]
- Zeileis, A.; Köll, S.; Graham, N. Various Versatile Variances: An Object-Oriented Implementation of Clustered Covariances in R. J. Stat. Softw. 2020, 95, 1–36. [Google Scholar] [CrossRef]
- McMurdie, P.J.; Holmes, S. Waste Not, Want Not: Why Rarefying Microbiome Data Is Inadmissible. PLoS Comput. Biol. 2014, 10, e1003531. [Google Scholar] [CrossRef]
- Cameron, E.S.; Schmidt, P.J.; Tremblay, B.J.-M.; Emelko, M.B.; Müller, K.M. Enhancing diversity analysis by repeatedly rarefying next generation sequencing data describing microbial communities. Sci. Rep. 2021, 11, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Zhang, Y.-R.; Yu, Y.-B. The important role of fungi in inflammatory bowel diseases. Scand. J. Gastroenterol. 2021, 56, 1312–1322. [Google Scholar] [CrossRef] [PubMed]
- Sokol, H.; Leducq, V.; Aschard, H.; Pham, H.P.; Jegou, S.; Landman, C.; Cohen, D.; Liguori, G.; Bourrier, A.; Nion-Larmurier, I.; et al. Fungal microbiota dysbiosis in IBD. Gut 2017, 66, 1039–1048. [Google Scholar] [CrossRef]
- Liguori, G.; Lamas, B.; Richard, M.L.; Brandi, G.; Da Costa, G.; Hoffmann, T.W.; Di Simone, M.P.; Calabrese, C.; Poggioli, G.; Langella, P.; et al. Fungal Dysbiosis in Mucosa-associated Microbiota of Crohn’s Disease Patients. J. Crohns Colitis 2016, 10, 296–305. [Google Scholar] [CrossRef]
- Jain, U.; Heul, A.M.V.; Xiong, S.; Gregory, M.H.; Demers, E.G.; Kern, J.T.; Lai, C.-W.; Muegge, B.D.; Barisas, D.A.G.; Leal-Ekman, J.S.; et al. Debaryomyces is enriched in Crohn’s disease intestinal tissue and impairs healing in mice. Science 2021, 371, 1154–1159. [Google Scholar] [CrossRef]
- David, L.A.; Maurice, C.F.; Carmody, R.N.; Gootenberg, D.B.; Button, J.E.; Wolfe, B.E.; Ling, A.V.; Devlin, A.S.; Varma, Y.; Fischbach, M.A.; et al. Diet rapidly and reproducibly alters the human gut microbiome. Nature 2014, 505, 559–563. [Google Scholar] [CrossRef]
- Hoffmann, C.; Dollive, S.; Grunberg, S.; Chen, J.; Li, H.; Wu, G.D.; Lewis, J.D.; Bushman, F.D. Archaea and Fungi of the Human Gut Microbiome: Correlations with Diet and Bacterial Residents. PLoS ONE 2013, 8, e66019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ali, T.; Bronze, M.S.; Kaitha, S.; Mahmood, S.; Ftaisi, A.; Stone, J. Clinical use of anti-TNF therapy and increased risk of infections. Drug Health Patient Saf. 2013, 5, 79–99. [Google Scholar] [CrossRef] [PubMed]
- Toruner, M.; Loftus, E.; Harmsen, W.S.; Zinsmeister, A.R.; Orenstein, R.; Sandborn, W.J.; Colombel, J.; Egan, L.J. Risk Factors for Opportunistic Infections in Patients With Inflammatory Bowel Disease. Gastroenterology 2008, 134, 929–936. [Google Scholar] [CrossRef]
- Samonis, G.; Gikas, A.; Anaissie, E.J.; Vrenzos, G.; Maraki, S.; Tselentis, Y.; Bodey, G.P. Prospective evaluation of effects of broad-spectrum antibiotics on gastrointestinal yeast colonization of humans. Antimicrob. Agents Chemother. 1993, 37, 51–53. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, C.; Adame, E.C.; Attaluri, A.; Valestin, J.; Rao, S.S.C. Dysmotility and proton pump inhibitor use are independent risk factors for small intestinal bacterial and/or fungal overgrowth. Aliment. Pharmacol. Ther. 2013, 37, 1103–1111. [Google Scholar] [CrossRef] [PubMed]
- Erdogan, A.; Rao, S.S.C. Small Intestinal Fungal Overgrowth. Curr. Gastroenterol. Rep. 2015, 17, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, P.; Lang, S.; Zeng, S.; Duan, Y.; Zhang, X.; Wang, Y.; Bondareva, M.; Kruglov, A.; Fouts, D.E.; Stärkel, P.; et al. Dynamic Changes of the Fungal Microbiome in Alcohol Use Disorder. Front. Physiol. 2021, 12, 699253. [Google Scholar] [CrossRef]
- García-Gamboa, R.; Kirchmayr, M.R.; Gradilla-Hernández, M.S.; Pérez-Brocal, V.; Moya, A.; González-Avila, M. The intestinal mycobiota and its relationship with overweight, obesity and nutritional aspects. J. Hum. Nutr. Diet. 2021, 34, 645–655. [Google Scholar] [CrossRef]
- Kelesidis, T.; Pothoulakis, C. Efficacy and safety of the probiotic Saccharomyces boulardii for the prevention and therapy of gastrointestinal disorders. Ther. Adv. Gastroenterol. 2011, 5, 111–125. [Google Scholar] [CrossRef]
- Kara, I.; Yıldırım, F.; Özgen, Ö.; Erganiş, S.; Aydoğdu, M.; Dizbay, M.; Gürsel, G.; Kalkanci, A. Saccharomyces cerevisiae fungemia after probiotic treatment in an intensive care unit patient. J. Mycol. Med. 2018, 28, 218–221. [Google Scholar] [CrossRef]
- Martin, I.W.; Tonner, R.; Trivedi, J.; Miller, H.; Lee, R.; Liang, X.; Rotello, L.; Isenbergh, E.; Anderson, J.; Perl, T.; et al. Saccharomyces boulardii probiotic-associated fungemia: Questioning the safety of this preventive probiotic’s use. Diagn. Microbiol. Infect. Dis. 2017, 87, 286–288. [Google Scholar] [CrossRef] [PubMed]
- Santino, I.; Alari, A.; Bono, S.; Teti, E.; Marangi, M.; Bernardini, A.; Magrini, L.; Di Somma, S.; Teggi, A. Saccharomyces Cerevisiae Fungemia, a Possible Consequence of the Treatment of Clostridium Difficile Colitis with a Probioticum. Int. J. Immunopathol. Pharmacol. 2014, 27, 143–146. [Google Scholar] [CrossRef] [PubMed]
- Sivananthan, K.; Petersen, A.M. Review of Saccharomyces boulardii as a treatment option in IBD. Immunopharmacol. Immunotoxicol. 2018, 40, 465–475. [Google Scholar] [CrossRef]
- Khatri, I.; Tomar, R.; Ganesan, K.; Prasad, G.S.; Subramanian, S. Complete genome sequence and comparative genomics of the probiotic yeast Saccharomyces boulardii. Sci. Rep. 2017, 7, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Petersen, A.M.; Nielsen, H.V.; Stensvold, C.R.; Engberg, J.H.; Friis-Møller, A.; Nordgaard-Lassen, I.; Wildt, S.; Krogfelt, K.A. Blastocystis and Dientamoeba Fragilis in Active and Inactive Inflammatory Bowel Disease. Gastroenterology 2011, 140, S-329–S-330. [Google Scholar] [CrossRef]
- Brands, M.R.; Van de Vijver, E.; Haisma, S.M.; Heida, A.; van Rheenen, P.F. No association between abdominal pain and Dientamoeba in Dutch and Belgian children. Arch. Dis. Child. 2019, 104, 686–689. [Google Scholar] [CrossRef]
- Holtman, A.G.; Kranenberg, J.J.; Blanker, M.H.; Ott, A.; Leeuwen, Y.L.-V.; Berger, M.Y. Di entamoeba fragilis colonization is not associated with gastrointestinal symptoms in children at primary care level. Fam. Pract. 2016, 34, 25–29. [Google Scholar] [CrossRef]
- Jokelainen, P.; Jensen, B.H.; Andreassen, B.U.; Petersen, A.M.; Röser, D.; Krogfelt, K.A.; Nielsen, H.V.; Stensvold, C.R. Dientamoeba fragilis, a Commensal in Children in Danish Day Care Centers. J. Clin. Microbiol. 2017, 55, 1707–1713. [Google Scholar] [CrossRef]
- Laforest-Lapointe, I.; Arrieta, M.-C. Microbial Eukaryotes: A Missing Link in Gut Microbiome Studies. mSystems 2018, 3, e00201-17. [Google Scholar] [CrossRef]
- Nagata, N.; Marriott, D.; Harkness, J.; Ellis, J.; Stark, D. Current treatment options for Dientamoeba fragilis infections. Int. J. Parasitol. Drugs Drug Resist. 2012, 2, 204–215. [Google Scholar] [CrossRef]
- Roberts, T.; Stark, D.; Harkness, J.; Ellis, J. Update on the pathogenic potential and treatment options for Blastocystis sp. Gut Pathog. 2014, 6, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haifer, C.; Kelly, C.R.; Paramsothy, S.; Andresen, D.; Papanicolas, E.L.; McKew, G.L.; Borody, T.J.; Kamm, M.; Costello, S.P.; Andrews, J.M.; et al. Australian consensus statements for the regulation, production and use of faecal microbiota transplantation in clinical practice. Gut 2020, 69, 801–810. [Google Scholar] [CrossRef] [PubMed]
- Allaband, C.; McDonald, D.; Vázquez-Baeza, Y.; Minich, J.J.; Tripathi, A.; Brenner, D.A.; Loomba, R.; Smarr, L.; Sandborn, W.J.; Schnabl, B.; et al. Microbiome 101: Studying, Analyzing, and Interpreting Gut Microbiome Data for Clinicians. Clin. Gastroenterol. Hepatol. 2018, 17, 218–230. [Google Scholar] [CrossRef] [PubMed]
- Gaulke, C.A.; Sharpton, T.J. The influence of ethnicity and geography on human gut microbiome composition. Nat. Med. 2018, 24, 1495–1496. [Google Scholar] [CrossRef]
- Kabwe, M.H.; Vikram, S.; Mulaudzi, K.; Jansson, J.K.; Makhalanyane, T.P. The gut mycobiota of rural and urban individuals is shaped by geography. BMC Microbiol. 2020, 20, 1–12. [Google Scholar] [CrossRef]
- Martino, C.; Dilmore, A.H.; Burcham, Z.M.; Metcalf, J.L.; Jeste, D.; Knight, R. Microbiota succession throughout life from the cradle to the grave. Nat. Rev. Genet. 2022; Online ahead of print. [Google Scholar] [CrossRef]
- Strati, F.; Di Paola, M.; Stefanini, I.; Albanese, D.; Rizzetto, L.; Lionetti, P.; Calabrò, A.; Jousson, O.; Donati, C.; Cavalieri, D.; et al. Age and Gender Affect the Composition of Fungal Population of the Human Gastrointestinal Tract. Front. Microbiol. 2016, 7, 1227. [Google Scholar] [CrossRef]
- Ahmad, H.F.; Castro Mejia, J.L.; Krych, L.; Khakimov, B.; Kot, W.; Bechshøft, R.L.; Reitelseder, S.; Højfeldt, G.W.; Engelsen, S.B.; Holm, L.; et al. Gut Mycobiome Dysbiosis Is Linked to Hypertriglyceridemia among Home Dwelling Elderly Danes. bioRxiv 2020. [Google Scholar] [CrossRef]
- Nagpal, R.; Neth, B.J.; Wang, S.; Mishra, S.P.; Craft, S.; Yadav, H. Gut mycobiome and its interaction with diet, gut bacteria and alzheimer’s disease markers in subjects with mild cognitive impairment: A pilot study. eBioMedicine 2020, 59, 102950. [Google Scholar] [CrossRef]
- Eisenhofer, R.; Minich, J.J.; Marotz, C.; Cooper, A.; Knight, R.; Weyrich, L.S. Contamination in Low Microbial Biomass Microbiome Studies: Issues and Recommendations. Trends Microbiol. 2019, 27, 105–117. [Google Scholar] [CrossRef]
- Hallen-Adams, H.E.; Suhr, M.J. Fungi in the healthy human gastrointestinal tract. Virulence 2016, 8, 352–358. [Google Scholar] [CrossRef] [PubMed]
- Underhill, D.M.; Iliev, I.D. The mycobiota: Interactions between commensal fungi and the host immune system. Nat. Rev. Immunol. 2014, 14, 405–416. [Google Scholar] [CrossRef] [PubMed]
- Olm, M.R.; West, P.T.; Brooks, B.; Firek, B.A.; Baker, R.; Morowitz, M.J.; Banfield, J.F. Genome-resolved metagenomics of eukaryotic populations during early colonization of premature infants and in hospital rooms. Microbiome 2019, 7, 26. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
{kind=link}
{kind=link}
| Cohort Characteristics | IBD Group (n = 355) | Control Group (n = 471) | |
|---|---|---|---|
| Sex | Female | 214 (60.28%) | 265 (56.26%) |
| Male | 141 (39.72%) | 200 (42.46%) | |
| Unspecified | 0 (0.00%) | 6 (1.27%) | |
| Age (years) | Median (IQR) | 45.00 (34.25, 59.00) | 23.00 (21.00, 27.00) |
| 18–40 | 145 (41.43%) | 408 (87.74%) | |
| 41–60 | 132 (37.71%) | 31 (6.67%) | |
| 61–80 | 69 (19.71%) | 26 (5.59%) | |
| 81+ | 4 (1.14%) | 0 (0.00%) | |
| Missing | 5 | 6 | |
| BMI | Median (IQR) | 24.80 (21.70, 28.10) | 22.30 (20.72, 24.39) |
| Missing | 0 | 14 | |
| Smoking Status | Current | 78 (22.35%) | 60 (14.12%) |
| Past | 146 (41.83%) | 65 (15.29%) | |
| Never | 125 (35.82%) | 300 (70.59%) | |
| Missing | 6 | 46 | |
| Diagnosis | CD | 206 (58.03%) | NA |
| UC | 126 (35.49%) | ||
| IBDU | 23 (6.48%) | ||
| Model | Coefficient | Blastocystis spp. 1 | Saccharomyces cerevisiae | ||||
|---|---|---|---|---|---|---|---|
| Estimate | Std. Error | p-Value | Estimate | Std. Error | p-Value | ||
| Count | IBD | −0.34 | 0.39 | 0.38 | 1.05 | 0.34 | <0.01 |
| Sex—male | 0.23 | 0.22 | 0.29 | −1.15 | 0.41 | 0.01 | |
| Age | 0.01 | 0.01 | 0.35 | −0.03 | 0.01 | <0.01 | |
| BMI—underweight | −0.85 | 0.40 | 0.04 | −2.19 | 0.61 | <0.01 | |
| BMI—overweight | −0.81 | 0.28 | <0.01 | −0.69 | 0.45 | 0.13 | |
| BMI—obese | −1.03 | 0.43 | 0.02 | 0.27 | 0.48 | 0.58 | |
| Smoking—past | −0.15 | 0.33 | 0.64 | 0.52 | 0.50 | 0.30 | |
| Smoking—current | −0.44 | 0.38 | 0.25 | −0.68 | 0.38 | 0.08 | |
| Zero | IBD | 1.84 | 0.37 | <0.01 | −1.40 | 0.44 | <0.01 |
| Sex—male | 0.32 | 0.24 | 0.18 | −0.47 | 0.35 | 0.18 | |
| Age | −0.02 | 0.01 | 0.02 | 0.01 | 0.01 | 0.37 | |
| BMI—underweight | −0.03 | 0.68 | 0.97 | −0.06 | 1.27 | 0.96 | |
| BMI—overweight | 0.46 | 0.34 | 0.17 | 0.01 | 0.43 | 0.98 | |
| BMI—obese | 0.88 | 0.78 | 0.26 | −0.53 | 0.50 | 0.29 | |
| Smoking—past | 0.19 | 0.31 | 0.55 | 0.48 | 0.45 | 0.29 | |
| Smoking—current | 0.66 | 0.39 | 0.10 | −0.42 | 0.39 | 0.27 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guzzo, G.L.; Mittinty, M.N.; Llamas, B.; Andrews, J.M.; Weyrich, L.S. Individuals with Inflammatory Bowel Disease Have an Altered Gut Microbiome Composition of Fungi and Protozoa. Microorganisms 2022, 10, 1910. https://doi.org/10.3390/microorganisms10101910
Guzzo GL, Mittinty MN, Llamas B, Andrews JM, Weyrich LS. Individuals with Inflammatory Bowel Disease Have an Altered Gut Microbiome Composition of Fungi and Protozoa. Microorganisms. 2022; 10(10):1910. https://doi.org/10.3390/microorganisms10101910
Chicago/Turabian StyleGuzzo, Gina L., Murthy N. Mittinty, Bastien Llamas, Jane M. Andrews, and Laura S. Weyrich. 2022. "Individuals with Inflammatory Bowel Disease Have an Altered Gut Microbiome Composition of Fungi and Protozoa" Microorganisms 10, no. 10: 1910. https://doi.org/10.3390/microorganisms10101910
APA StyleGuzzo, G. L., Mittinty, M. N., Llamas, B., Andrews, J. M., & Weyrich, L. S. (2022). Individuals with Inflammatory Bowel Disease Have an Altered Gut Microbiome Composition of Fungi and Protozoa. Microorganisms, 10(10), 1910. https://doi.org/10.3390/microorganisms10101910