
Transcriptional Shift and Metabolic Adaptations during Leishmania Quiescence Using Stationary Phase and Drug Pressure as Models

 , ,
, ,
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Leishmania Clinical Isolate and Cell Culture
2.2. Development of an EGFP Clonal Line
2.3. Promastigotes and Amastigotes Enrichment with Ficoll and Percoll Gradients
2.4. Monitoring rEGFP Expression and Cell Viability by Flow Cytometry
2.5. RNA and Library Preparation
2.6. RNAseq Data Analysis
2.7. Transcriptomics Functional Enrichment and Network Analysis
2.8. Metabolomics
3. Results
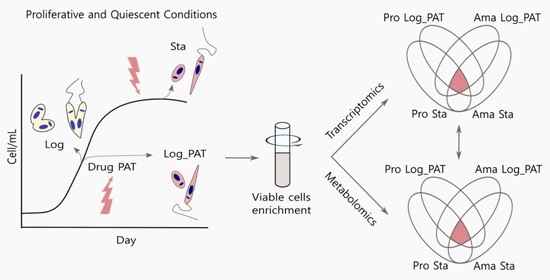
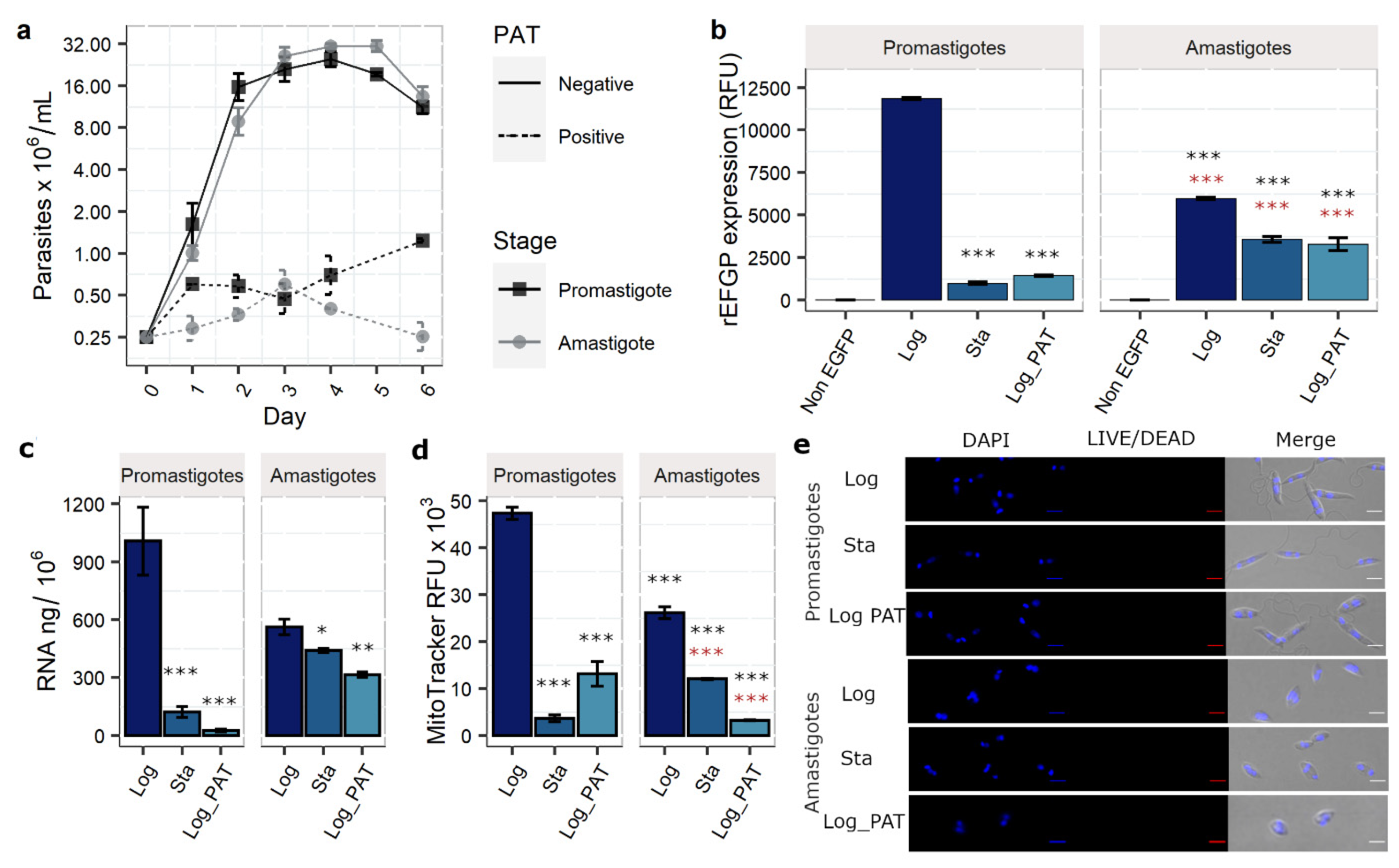
3.1. PAT Drug Pressure and Stationary Phase as Models of Quiescence
3.2. Evaluation of Additional Quiescence Indicators: Total RNA Content and Mitochondrial Activity
3.3. Evaluation of the mRNAs Transcriptome Size and Global Shift in the Levels of mRNAs
3.4. Evaluation of the Transcriptome Composition
3.5. GO and GSEA Analysis
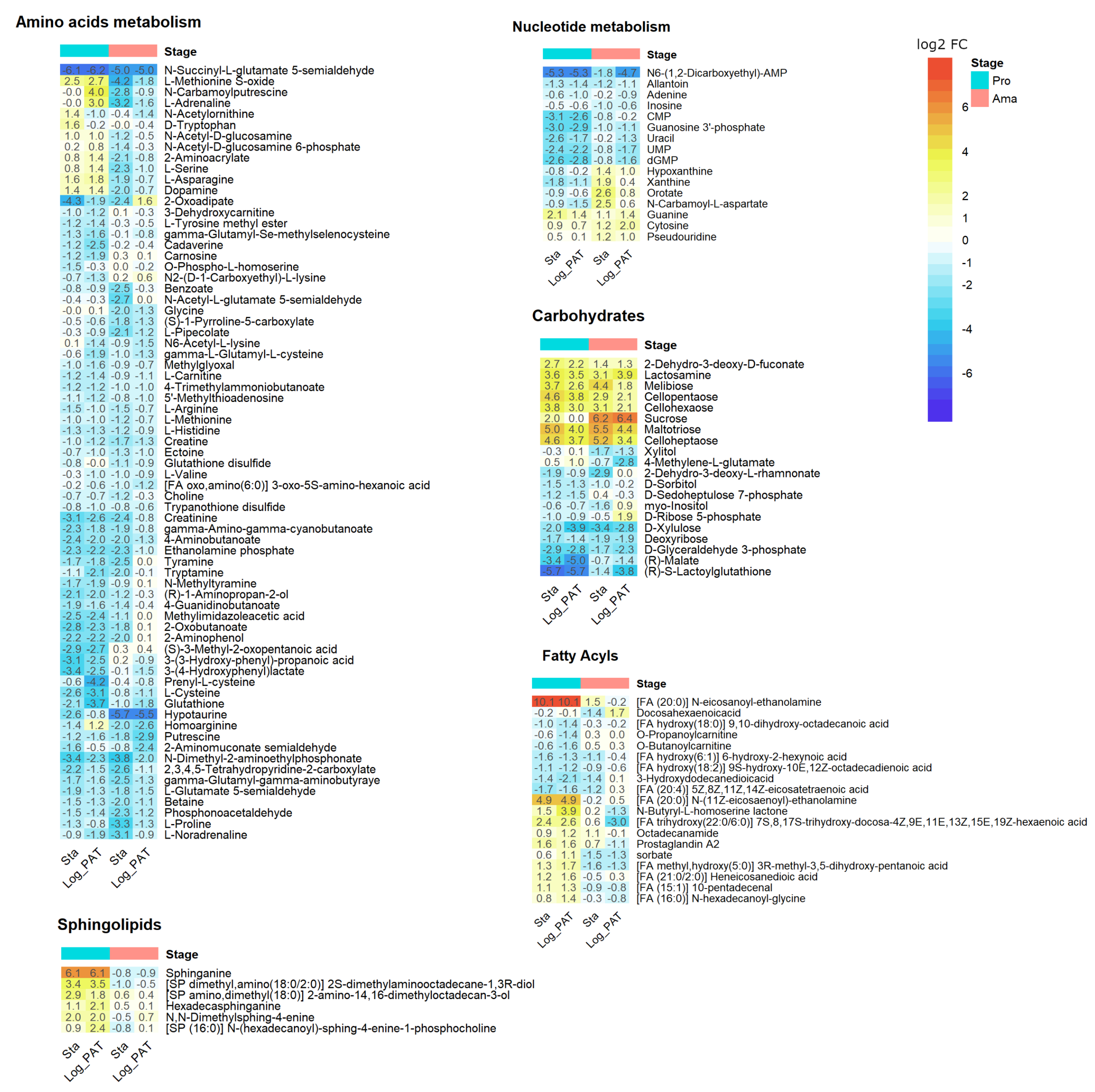
3.6. Metabolomics
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- da Silva, R.; Sacks, D.L. Metacyclogenesis is a major determinant of Leishmania promastigote virulence and attenuation. Infect. Immun. 1987, 55, 2802–2806. [Google Scholar] [CrossRef]
- Lopez, M.M.; Soto, M.; Iborra, S.; Sancho, D. Leishmania Hijacks Myeloid Cells for Immune Escape. Front. Microbiol. 2018, 9, 883. [Google Scholar] [CrossRef]
- Saunders, E.; Ng, W.W.; Kloehn, J.; Chambers, J.M.; Ng, M.; McConville, M.J. Induction of a Stringent Metabolic Response in Intracellular Stages of Leishmania mexicana Leads to Increased Dependence on Mitochondrial Metabolism. PLoS Pathog. 2014, 10, e1003888. [Google Scholar] [CrossRef] [PubMed]
- Mandell, M.A.; Beverley, S.M. Continual renewal and replication of persistent Leishmania major parasites in concomitantly immune hosts. Proc. Natl. Acad. Sci. USA 2017, 31, E801–E810. [Google Scholar] [CrossRef] [PubMed]
- Kloehn, J.; Saunders, E.; O’Callaghan, S.; Dagley, M.J.; McConville, M.J. Characterization of Metabolically Quiescent Leishmania Parasites in Murine Lesions Using Heavy Water Labeling. PLoS Pathog. 2015, 11, e1004683. [Google Scholar] [CrossRef]
- Jara, M.; Maes, I.; Imamura, H.; Domagalska, M.A.; Dujardin, J.C.; Arevalo, J. Tracking of quiescence in Leishmania by quantifying the expression of GFP in the ribosomal DNA locus. Sci. Rep. 2019, 9, 18951. [Google Scholar] [CrossRef]
- Barrett, M.P.; Kyle, D.E.; Sibley, L.D.; Radke, J.B.; Tarleton, R.L. Protozoan persister-like cells and drug treatment failure. Nat. Rev. Genet. 2019, 17, 607–620. [Google Scholar] [CrossRef]
- Ostyn, B.; Gidwani, K.; Khanal, B.; Picado, A.; Chappuis, F.; Singh, S.P.; Rijal, S.; Sundar, S.; Boelaert, M. Incidence of Symptomatic and Asymptomatic Leishmania donovani Infections in High-Endemic Foci in India and Nepal: A Prospective Study. PLoS Neglected Trop. Dis. 2011, 5, e1284. [Google Scholar] [CrossRef]
- Prajapati, V.K.; Sharma, S.; Rai, M.; Ostyn, B.; Salotra, P.; Vanaerschot, M.; Dujardin, J.-C.; Sundar, S. In vitro Susceptibility of Leishmania donovani to Miltefosine in Indian Visceral Leishmaniasis. Am. J. Trop. Med. Hyg. 2013, 89, 750–754. [Google Scholar] [CrossRef]
- Baptista, C.; Miranda, L.D.F.C.; Madeira, M.D.F.; Leon, L.L.P.; Conceição-Silva, F.; Schubach, A.D.O. In Vitro Sensitivity of Paired Leishmania (Viannia) braziliensis Samples Isolated before Meglumine Antimoniate Treatment and after Treatment Failure or Reactivation of Cutaneous Leishmaniasis. Dis. Markers 2015, 2015, 1–6. [Google Scholar] [CrossRef]
- Yanagida, M. Cellular quiescence: Are controlling genes conserved? Trends Cell Biol. 2009, 19, 705–715. [Google Scholar] [CrossRef]
- Rittershaus, E.S.; Baek, S.-H.; Sassetti, C.M. The Normalcy of Dormancy: Common Themes in Microbial Quiescence. Cell Host Microbe 2013, 13, 643–651. [Google Scholar] [CrossRef]
- Aragon, A.D.; Rodriguez, A.L.; Meirelles, O.; Roy, S.; Davidson, G.S.; Tapia, P.H.; Allen, C.; Joe, R.; Benn, D.; Werner-Washburne, M. Characterization of Differentiated Quiescent and Nonquiescent Cells in Yeast Stationary-Phase Cultures. Mol. Biol. Cell 2008, 19, 1271–1280. [Google Scholar] [CrossRef]
- Laporte, D.; Lebaudy, A.; Sahin, A.; Pinson, B.; Ceschin, J.; Daignan-Fornier, B.; Sagot, I. Metabolic status rather than cell cycle signals control quiescence entry and exit. J. Cell Biol. 2011, 192, 949–957. [Google Scholar] [CrossRef]
- Witkowski, B.; Lelièvre, J.; Barragán, M.J.L.; Laurent, V.; Su, X.-Z.; Berry, A.; Benoit-Vical, F. Increased Tolerance to Artemisinin in Plasmodium falciparum Is Mediated by a Quiescence Mechanism. Antimicrob. Agents Chemother. 2010, 54, 1872–1877. [Google Scholar] [CrossRef]
- Leung, V.; Lévesque, C.M. A Stress-Inducible Quorum-Sensing Peptide Mediates the Formation of Persister Cells with Noninherited Multidrug Tolerance. J. Bacteriol. 2012, 194, 2265–2274. [Google Scholar] [CrossRef]
- Valcourt, J.; Lemons, J.M.; Haley, E.M.; Kojima, M.; Demuren, O.O.; Coller, H.A. Staying alive: Metabolic adaptations to quiescence. Cell Cycle 2012, 11, 1680–1696. [Google Scholar] [CrossRef]
- Tegazzini, D.; Díaz, R.; Aguilar, F.; Peña, I.; Presa, J.L.; Yardley, V.; Martin, J.J.; Coteron, J.M.; Croft, S.L.; Cantizani, J. A Replicative In Vitro Assay for Drug Discovery against Leishmania donovani. Antimicrob. Agents Chemother. 2016, 60, 3524–3532. [Google Scholar] [CrossRef]
- Jara, M.; Berg, M.; Caljon, G.; De Muylder, G.; Cuypers, B.; Castillo, D.; Maes, I.; Orozco, M.D.C.; Vanaerschot, M.; Dujardin, J.-C.; et al. Macromolecular biosynthetic parameters and metabolic profile in different life stages of Leishmania braziliensis: Amastigotes as a functionally less active stage. PLoS ONE 2017, 12, e0180532. [Google Scholar] [CrossRef]
- Ducharme, O.; Simon, S.; Ginouves, M.; Prévot, G.; Couppie, P.; Demar, M.; Blaizot, R. Leishmania naiffi and lainsoni in French Guiana: Clinical features and phylogenetic variability. PLoS Neglected Trop. Dis. 2020, 14, e0008380. [Google Scholar] [CrossRef]
- Martinez, E.; Le Pont, F.; Mollinedo, S.; Cupolillo, E. A first case of cutaneous leishmaniasis due to Leishmania (Viannia) lainsoni in Bolivia. Trans. R. Soc. Trop. Med. Hyg. 2001, 95, 375–377. [Google Scholar] [CrossRef]
- Lucas, C.M.; Franke, E.D.; Cachay, M.I.; Tejada, A.; Carrizales, D.; Kreutzer, R.D. Leishmania (Viannia) lainsoni: First isolation in Peru. Am. J. Trop. Med. Hyg. 1994, 51, 533–537. [Google Scholar] [CrossRef]
- Van der Auwera, G.; Dujardin, J.-C. Species Typing in Dermal Leishmaniasis. Clin. Microbiol. Rev. 2015, 28, 265–294. [Google Scholar] [CrossRef]
- Bolhassani, A.; Taheri, T.; Taslimi, Y.; Zamanilui, S.; Zahedifard, F.; Seyed, N.; Torkashvand, F.; Vaziri, B.; Rafati, S. Fluorescent Leishmania species: Development of stable GFP expression and its application for in vitro and in vivo studies. Exp. Parasitol. 2011, 127, 637–645. [Google Scholar] [CrossRef] [PubMed]
- Adaui, V.; Schnorbusch, K.; Zimic, M.; Gutiérrez, A.; Decuypere, S.; Vanaerschot, M.; de Doncker, S.; Maes, I.; Llanos-Cuentas, A.; Chappuis, F.; et al. Comparison of gene expression patterns among Leishmania braziliensis clinical isolates showing a different in vitro susceptibility to pentavalent antimony. Parasitology 2011, 138, 183–193. [Google Scholar] [CrossRef]
- Späth, G.F.; Beverley, S. A Lipophosphoglycan-Independent Method for Isolation of Infective Leishmania Metacyclic Promastigotes by Density Gradient Centrifugation. Exp. Parasitol. 2001, 99, 97–103. [Google Scholar] [CrossRef] [PubMed]
- La Fuente, S.G.-D.; Camacho, E.; Peiro-Pastor, R.; Rastrojo, A.; Carrasco-Ramiro, F.; Aguado, B.; Requena, J.M. Complete and de novo assembly of the Leishmania braziliensis (M2904) genome. Memórias Inst. Oswaldo Cruz 2019, 114, e180438. [Google Scholar] [CrossRef]
- Raudvere, U.; Kolberg, L.; Kuzmin, I.; Arak, T.; Adler, P.; Peterson, H.; Vilo, J. g:Profiler: A web server for functional enrichment analysis and conversions of gene lists (2019 update). Nucleic Acids Res. 2019, 47, W191–W198. [Google Scholar] [CrossRef]
- Supek, F.; Bošnjak, M.; Škunca, N.; Smuc, T. REVIGO Summarizes and Visualizes Long Lists of Gene Ontology Terms. PLoS ONE 2011, 6, e21800. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A Software Environment for Integrated Models of Biomolecular Interaction Networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Luo, W.; Friedman, M.S.; Shedden, K.; Hankenson, K.D.; Woolf, P.J. GAGE: Generally applicable gene set enrichment for pathway analysis. BMC Bioinform. 2009, 10, 161. [Google Scholar] [CrossRef]
- Creek, D.; Jankevics, A.; Burgess, K.; Breitling, R.; Barrett, M.P. IDEOM: An Excel interface for analysis of LC–MS-based metabolomics data. Bioinformatics 2012, 28, 1048–1049. [Google Scholar] [CrossRef] [PubMed]
- De Livera, A.M.; Olshansky, M.; Speed, T.P. Statistical Analysis of Metabolomics Data. Springer Protoc. Handb. 2013, 1055, 291–307. [Google Scholar] [CrossRef]
- Yardley, V.; Ortuño, N.; Llanos-Cuentas, A.; Chappuis, F.; De Doncker, S.; Ramirez, L.; Croft, S.; Arevalo, J.; Adaui, V.; Bermudez, H.; et al. American Tegumentary Leishmaniasis: Is Antimonial Treatment Outcome Related to Parasite Drug Susceptibility? J. Infect. Dis. 2006, 194, 1168–1175. [Google Scholar] [CrossRef]
- Gengenbacher, M.; Rao, S.P.S.; Pethe, K.; Dick, T. Nutrient-starved, non-replicating Mycobacterium tuberculosis requires respiration, ATP synthase and isocitrate lyase for maintenance of ATP homeostasis and viability. Microbiology 2009, 156, 81–87. [Google Scholar] [CrossRef]
- Stallings, C.L.; Stephanou, N.C.; Chu, L.; Hochschild, A.; Nickels, B.E.; Glickman, M.S. CarD Is an Essential Regulator of rRNA Transcription Required for Mycobacterium tuberculosis Persistence. Cell 2009, 138, 146–159. [Google Scholar] [CrossRef]
- Marguerat, S.; Schmidt, A.; Codlin, S.; Chen, W.; Aebersold, R.; Bähler, J. Quantitative Analysis of Fission Yeast Transcriptomes and Proteomes in Proliferating and Quiescent Cells. Cell 2012, 151, 671–683. [Google Scholar] [CrossRef]
- Xu, Q.; Zhang, X. The Influence of the Global Gene Expression Shift on Downstream Analyses. PLoS ONE 2016, 11, e0153903. [Google Scholar] [CrossRef]
- Lun, A.T.; Calero-Nieto, F.J.; Haim-Vilmovsky, L.; Göttgens, B.; Marioni, J.C. Assessing the reliability of spike-in normalization for analyses of single-cell RNA sequencing data. Genome Res. 2017, 27, 1795–1806. [Google Scholar] [CrossRef]
- Coate, J.E.; Doyle, J.J. Variation in transcriptome size: Are we getting the message? Chromosoma 2014, 124, 27–43. [Google Scholar] [CrossRef] [PubMed]
- Boehm, E.; Gildenberg, M.; Washington, M. The Many Roles of PCNA in Eukaryotic DNA Replication. Hydrolys. Peptide Bonds 2016, 39, 231–254. [Google Scholar] [CrossRef]
- Choe, K.N.; Moldovan, G.-L. Forging Ahead through Darkness: PCNA, Still the Principal Conductor at the Replication Fork. Mol. Cell 2017, 65, 380–392. [Google Scholar] [CrossRef] [PubMed]
- Martens, S.; Fracchiolla, D. Activation and targeting of ATG8 protein lipidation. Cell Discov. 2020, 6, 1–11. [Google Scholar] [CrossRef]
- Derrick, S.; Large, P.J. Activities of the enzymes of the Ehrlich pathway and formation of branched-chain alcohols in Saccharomyces cerevisiae and Candida utilis grown in continuous culture on valine or ammonium as sole nitrogen source. J. Gen. Microbiol. 1993, 139, 2783–2792. [Google Scholar] [CrossRef]
- Kovářová, J.; Pountain, A.W.; Wildridge, D.; Weidt, S.; Bringaud, F.; Burchmore, R.J.S.; Achcar, F.; Barrett, M.P. Deletion of transketolase triggers a stringent metabolic response in promastigotes and loss of virulence in amastigotes of Leishmania mexicana. PLoS Pathog. 2018, 14, e1006953. [Google Scholar] [CrossRef] [PubMed]
- Ralton, J.E.; Naderer, T.; Piraino, H.L.; Bashtannyk, T.A.; Callaghan, J.M.; McConville, M.J. Evidence That Intracellular β1-2 Mannan Is a Virulence Factor in Leishmania Parasites. J. Biol. Chem. 2003, 278, 40757–40763. [Google Scholar] [CrossRef]
- McKnight, J.N.; Breeden, L.L.; Tsukiyama, T. A molecular off switch for transcriptional quiescence. Cell Cycle 2015, 14, 3667–3668. [Google Scholar] [CrossRef]
- McKnight, J.N.; Tsukiyama, T. The conserved HDAC Rpd3 drives transcriptional quiescence in S. cerevisiae. Genom. Data 2015, 6, 245–248. [Google Scholar] [CrossRef]
- Grünebast, J.; Lorenzen, S.; Zummack, J.; Clos, J. Life Cycle Stage-Specific Accessibility of Leishmania donovani Chromatin at Transcription Start Regions. mSystems 2021, 6, e0062821. [Google Scholar] [CrossRef]
- Gala, H.P.; Saha, D.; Venugopal, N.; Aloysius, A.; Dhawan, J. RNA polymerase II pausing regulates a quiescence-dependent transcriptional program, priming cells for cell cycle reentry. bioRxiv 2018, 250910. [Google Scholar] [CrossRef]
- Kim, N.; Jinks-Robertson, S. Transcription as a source of genome instability. Nat. Rev. Genet. 2012, 13, 204–214. [Google Scholar] [CrossRef]
- Roche, B.; Arcangioli, B.; Martienssen, R. Transcriptional reprogramming in cellular quiescence. RNA Biol. 2017, 14, 843–853. [Google Scholar] [CrossRef]
- Young, C.P.; Hillyer, C.; Hokamp, K.; Fitzpatrick, D.J.; Konstantinov, N.K.; Welty, J.S.; Ness, S.; Werner-Washburne, M.; Fleming, A.B.; Osley, M.A. Distinct histone methylation and transcription profiles are established during the development of cellular quiescence in yeast. BMC Genom. 2017, 18, 107. [Google Scholar] [CrossRef]
- Piel, L.; Rajan, K.S.; Bussotti, G.; Varet, H.; Legendre, R.; Proux, C.; Douché, T.; Giai-Gianetto, Q.; Chaze, T.; Vojtkova, B.; et al. Post-transcriptional regulation of Leishmania fitness gain. bioRxiv 2021. [Google Scholar] [CrossRef]
- Cuypers, B.; Meysman, P.; Erb, I.; Bittremieux, W.; Valkenborg, D.; Baggerman, G.; Mertens, I.; Sundar, S.; Khanal, B.; Notredame, C.; et al. Four layer multi-omics reveals molecular responses to aneuploidy in Leishmania. bioRxiv 2021. [Google Scholar] [CrossRef]
- Hansen, M.; Rubinsztein, D.C.; Walker, D.W. Autophagy as a promoter of longevity: Insights from model organisms. Nat. Rev. Mol. Cell Biol. 2018, 19, 579–593. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.; Gresham, D. Cellular quiescence in budding yeast. Yeast 2021, 38, 12–29. [Google Scholar] [CrossRef]
- Johansen, T.; Lamark, T. Selective autophagy mediated by autophagic adapter proteins. Autophagy 2011, 7, 279–296. [Google Scholar] [CrossRef] [PubMed]
- Khaminets, A.; Behl, C.; Dikic, I. Ubiquitin-Dependent and Independent Signals in Selective Autophagy. Trends Cell Biol. 2016, 26, 6–16. [Google Scholar] [CrossRef]
- Besteiro, S.; Williams, R.; Morrison, L.S.; Coombs, G.H.; Mottram, J. Endosome Sorting and Autophagy Are Essential for Differentiation and Virulence of Leishmania major. J. Biol. Chem. 2006, 281, 11384–11396. [Google Scholar] [CrossRef]
- Eapen, V.V.; Waterman, D.P.; Bernard, A.; Schiffmann, N.; Sayas, E.; Kamber, R.; Lemos, B.; Memisoglu, G.; Ang, J.; Mazella, A.; et al. A pathway of targeted autophagy is induced by DNA damage in budding yeast. Proc. Natl. Acad. Sci. USA 2017, 114, E1158–E1167. [Google Scholar] [CrossRef]
- Cebollero, E.; Reggiori, F. Regulation of autophagy in yeast Saccharomyces cerevisiae. Biochim. Biophys. Acta (BBA)-Bioenerg. 2009, 1793, 1413–1421. [Google Scholar] [CrossRef] [PubMed]
- Pimenta, P.F.; Saraiva, E.M.; Sacks, D.L. The comparative fine structure and surface glycoconjugate expression of three life stages of Leishmania major. Exp. Parasitol. 1991, 72, 191–204. [Google Scholar] [CrossRef]
- Eisnard, A.; Shio, M.T.; Eolivier, M. Impact of Leishmania metalloprotease GP63 on macrophage signaling. Front. Cell. Infect. Microbiol. 2012, 2, 72. [Google Scholar] [CrossRef] [PubMed]
- Hsiao, C.-H.C.; Yao, C.; Storlie, P.; Donelson, J.E.; Wilson, M.E. The major surface protease (MSP or GP63) in the intracellular amastigote stage of Leishmania chagasi. Mol. Biochem. Parasitol. 2008, 157, 148–159. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Yao, C.; Leidal, K.G.; Brittingham, A.; Tarr, D.E.; Donelson, J.E.; Wilson, M.E. Biosynthesis of the major surface protease GP63 of Leishmania chagasi. Mol. Biochem. Parasitol. 2002, 121, 119–128. [Google Scholar] [CrossRef]
- Yao, C.; Donelson, J.E.; Wilson, M.E. The major surface protease (MSP or GP63) of Leishmania sp. Biosynthesis, regulation of expression, and function. Mol. Biochem. Parasitol. 2003, 132, 1–16. [Google Scholar] [CrossRef]
- Matte, C.; Casgrain, P.-A.; Séguin, O.; Moradin, N.; Hong, W.J.; Descoteaux, A. Leishmania major Promastigotes Evade LC3-Associated Phagocytosis through the Action of GP63. PLoS Pathog. 2016, 12, e1005690. [Google Scholar] [CrossRef] [PubMed]
- Vieira, T.D.S.; Duque, G.A.; Ory, K.; Gontijo, C.M.; Soares, R.P.; Descoteaux, A. Leishmania braziliensis: Strain-Specific Modulation of Phagosome Maturation. Front. Cell. Infect. Microbiol. 2019, 9, 319. [Google Scholar] [CrossRef]
- Kulkarni, M.M.; Jones, E.A.; McMaster, W.R.; McGwire, B.S. Fibronectin Binding and Proteolytic Degradation by Leishmania and Effects on Macrophage Activation. Infect. Immun. 2008, 76, 1738–1747. [Google Scholar] [CrossRef]
- Neto, A.L.C.; Brito, A.N.A.L.M.; Rezende, A.M.; Magalhães, F.B.; Neto, O.P.D.M. In silico characterization of multiple genes encoding the GP63 virulence protein from Leishmania braziliensis: Identification of sources of variation and putative roles in immune evasion. BMC Genom. 2019, 20, 118. [Google Scholar] [CrossRef]
- De Souza Leao, S.; Lang, T.; Prina, E.; Hellio, R.; Antoine, J.C. Intracellular Leishmania amazonensis amastigotes internalize and degrade MHC class II molecules of their host cells. J. Cell Sci. 1995, 108 Pt 10, 3219–3231. [Google Scholar] [CrossRef] [PubMed]
- De Paiva, R.M.C.; Grazielle-Silva, V.; Cardoso, M.S.; Nakagaki, B.N.; Mendonça-Neto, R.P.; Canavaci, A.; Melo, N.S.; Martinelli, P.M.; Fernandes, A.P.; DaRocha, W.D.; et al. Amastin Knockdown in Leishmania braziliensis Affects Parasite-Macrophage Interaction and Results in Impaired Viability of Intracellular Amastigotes. PLoS Pathog. 2015, 11, e1005296. [Google Scholar] [CrossRef] [PubMed]
- Kangussu-Marcolino, M.M.; de Paiva, R.M.C.; Araújo, P.R.; de Mendonça-Neto, R.P.; Lemos, L.; Bartholomeu, D.C.; Mortara, R.A.; Darocha, W.D.; Teixeira, S.M.R. Distinct genomic organization, mRNA expression and cellular localization of members of two amastin sub-families present in Trypanosoma cruzi. BMC Microbiol. 2013, 13, 10. [Google Scholar] [CrossRef] [PubMed]
- Alcolea, P.J.; Alonso, A.; Degayón, M.A.; Moreno-Paz, M.; Jiménez, M.; Molina, R.; Larraga, V. In vitro infectivity and differential gene expression of Leishmania infantum metacyclic promastigotes: Negative selection with peanut agglutinin in culture versus isolation from the stomodeal valve of Phlebotomus perniciosus. BMC Genom. 2016, 17, 375. [Google Scholar] [CrossRef]
- Biswas, B.; Laha, B.; Chaudhury, A.; Ghosh, M. Characterization of a novel amastin-like surface protein (ALSP) of Leishmania donovani, a probable lipase. bioRxiv 2021. [Google Scholar] [CrossRef]
- De Virgilio, C. The essence of yeast quiescence. FEMS Microbiol. Rev. 2012, 36, 306–339. [Google Scholar] [CrossRef]
- Huang, X.; Duan, X.; Li, J.; Niu, J.; Yuan, S.; Wang, X.; Lambert, N.; Li, X.; Xu, J.; Gong, Z.; et al. The Synergistic Effect of Exogenous Glutamine and Rifampicin against Mycobacterium Persisters. Front. Microbiol. 2018, 9, 1625. [Google Scholar] [CrossRef]
- Sernee, M.F.; Ralton, J.E.; Nero, T.L.; Sobala, L.; Kloehn, J.; Vieira-Lara, M.; Cobbold, S.A.; Stanton, L.; Pires, D.E.; Hanssen, E.; et al. A Family of Dual-Activity Glycosyltransferase-Phosphorylases Mediates Mannogen Turnover and Virulence in Leishmania Parasites. Cell Host Microbe 2019, 26, 385–399.e9. [Google Scholar] [CrossRef]
- Guo, H.; Novozhilova, N.M.; Bandini, G.; Turco, S.J.; Ferguson, M.A.J.; Beverley, S.M. Genetic metabolic complementation establishes a requirement for GDP-fucose in Leishmania. J. Biol. Chem. 2017, 292, 10696–10708. [Google Scholar] [CrossRef]
- Turnock, D.C.; Izquierdo, L.; Ferguson, M. The de Novo Synthesis of GDP-fucose Is Essential for Flagellar Adhesion and Cell Growth in Trypanosoma brucei. J. Biol. Chem. 2007, 282, 28853–28863. [Google Scholar] [CrossRef] [PubMed]
- de Aquino, G.P.; Gomes, M.A.M.; Salinas, R.K.; Laranjeira-Silva, M.F. Lipid and fatty acid metabolism in trypanosomatids. Microb. Cell 2021, 8, 262–275. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jara, M.; Barrett, M.; Maes, I.; Regnault, C.; Imamura, H.; Domagalska, M.A.; Dujardin, J.-C. Transcriptional Shift and Metabolic Adaptations during Leishmania Quiescence Using Stationary Phase and Drug Pressure as Models. Microorganisms 2022, 10, 97. https://doi.org/10.3390/microorganisms10010097
Jara M, Barrett M, Maes I, Regnault C, Imamura H, Domagalska MA, Dujardin J-C. Transcriptional Shift and Metabolic Adaptations during Leishmania Quiescence Using Stationary Phase and Drug Pressure as Models. Microorganisms. 2022; 10(1):97. https://doi.org/10.3390/microorganisms10010097
Chicago/Turabian StyleJara, Marlene, Michael Barrett, Ilse Maes, Clement Regnault, Hideo Imamura, Malgorzata Anna Domagalska, and Jean-Claude Dujardin. 2022. "Transcriptional Shift and Metabolic Adaptations during Leishmania Quiescence Using Stationary Phase and Drug Pressure as Models" Microorganisms 10, no. 1: 97. https://doi.org/10.3390/microorganisms10010097
APA StyleJara, M., Barrett, M., Maes, I., Regnault, C., Imamura, H., Domagalska, M. A., & Dujardin, J.-C. (2022). Transcriptional Shift and Metabolic Adaptations during Leishmania Quiescence Using Stationary Phase and Drug Pressure as Models. Microorganisms, 10(1), 97. https://doi.org/10.3390/microorganisms10010097