
Genotyping and Molecular Diagnosis of Hepatitis A Virus in Human Clinical Samples Using Multiplex PCR-Based Next-Generation Sequencing

 , , , , ,
, , , , ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Ethics Approval and Participation Consent
2.2. Study Population and Sample Collection
2.3. Reverse Transcription-PCR (RT-PCR) Assay
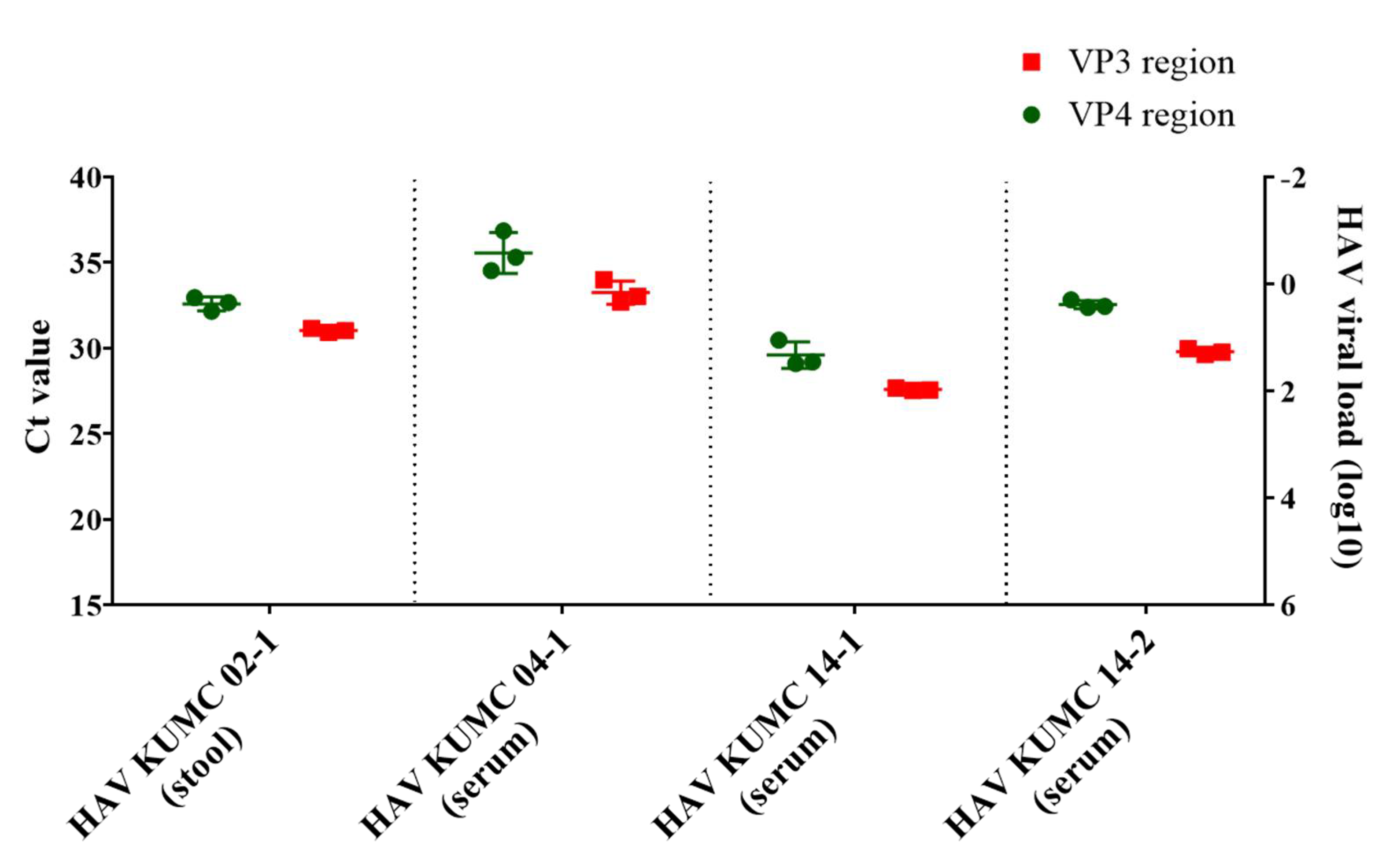
2.4. Quantitative PCR (qPCR) Assay
2.5. Multiplex PCR-Based Next-Generation Sequencing
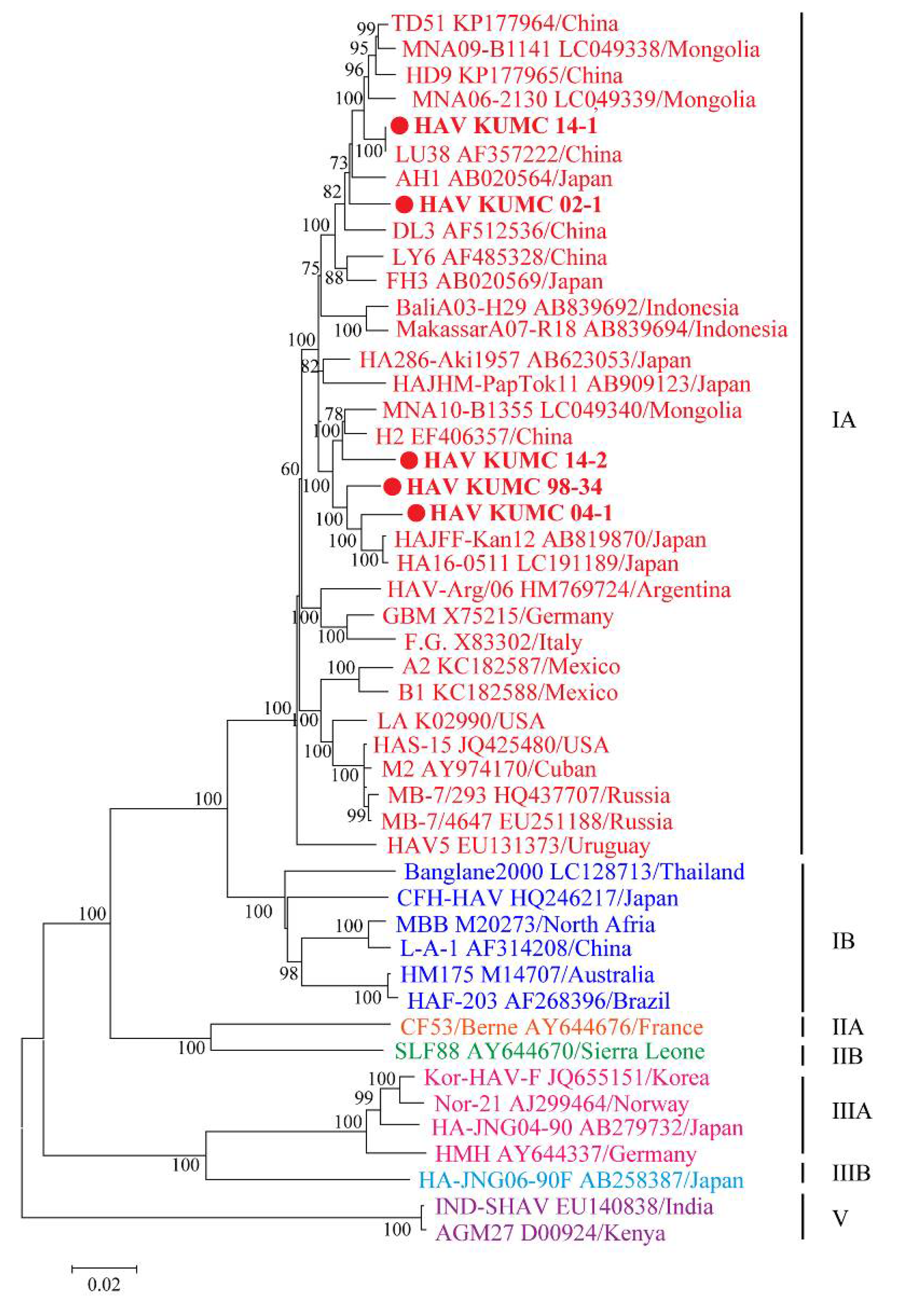
2.6. Phylogenetic Analysis
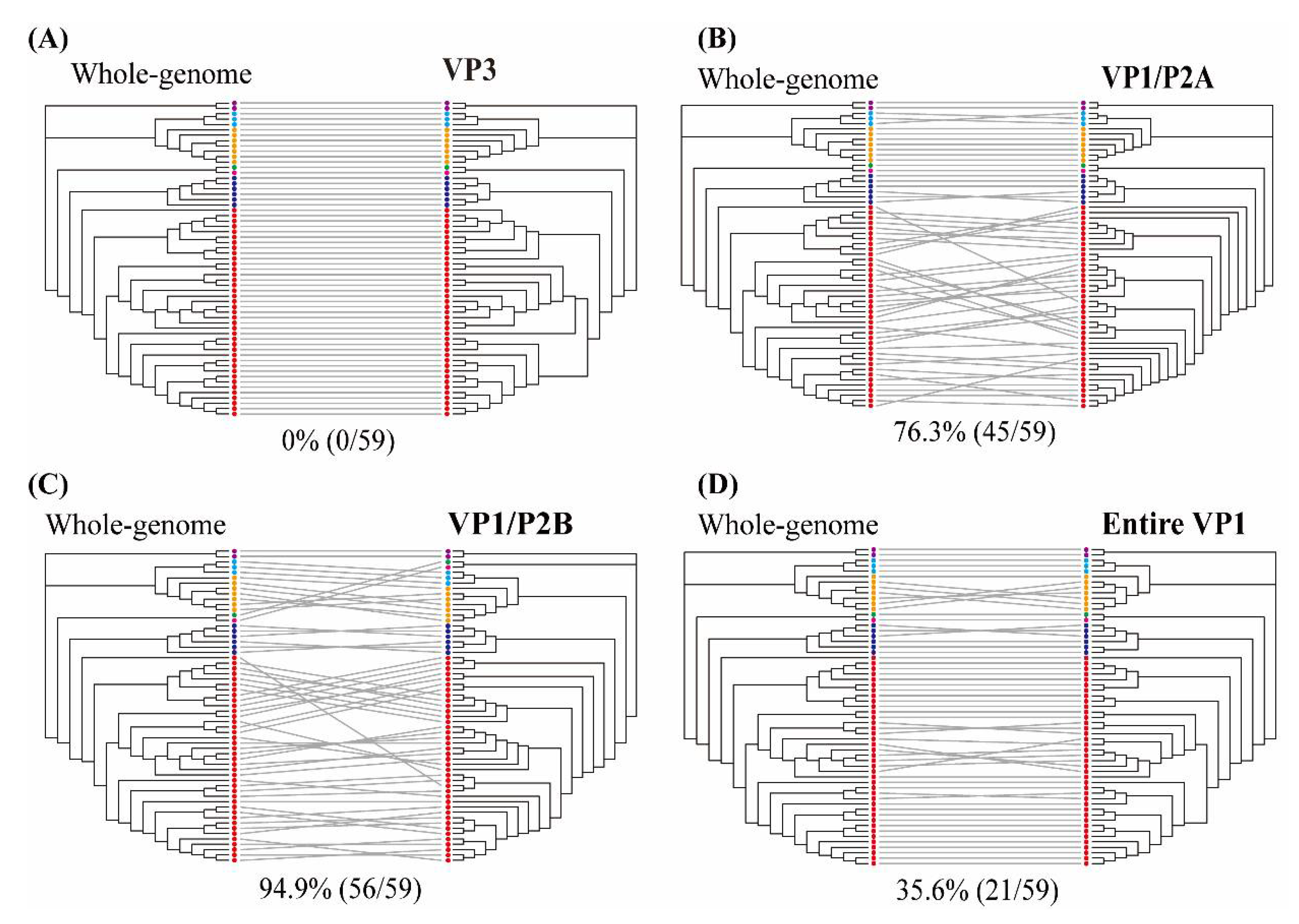
2.7. Tanglegram Analysis for Comparing Different Phylogenies
3. Results
3.1. Clinical Symptoms of HAV-Infected Patients
3.2. Molecular Diagnosis and Quantification of HAV Genomes
3.3. Whole-Genome Sequencing and Genetic Analysis of HAV by Multiplex PCR-Based NGS
3.4. Sequence Similarity of HAV Genomes at the Genotype Level
3.5. Phylogenetic Analysis of Whole-Genome and Partial-Genome Sequences of HAV
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Das, A. An economic analysis of different strategies of immunization against hepatitis A virus in developed countries. Hepatology 1999, 29, 548–552. [Google Scholar] [CrossRef] [PubMed]
- Barzaga, N.G. Hepatitis A shifting epidemiology in South-East Asia and China. Vaccine 2000, 18, S61–S64. [Google Scholar] [CrossRef]
- Kim, J.H.; Yeon, J.E.; Baik, S.K.; Kim, Y.S.; Kim, H.S.; Park, S.H.; Lee, M.S.; Sohn, J.H.; Lee, J.W.; Choi, S.K.; et al. Genotypic shift of the hepatitis A virus and its clinical impact on acute hepatitis A in Korea: A nationwide multicenter study. Scand. J. Infect. Dis. 2013, 45, 811–818. [Google Scholar] [CrossRef] [PubMed]
- Lemon, S.M.; Ott, J.J.; Van Damme, P.; Shouval, D. Type A viral hepatitis: A summary and update on the molecular virology, epidemiology, pathogenesis and prevention. J. Hepatol. 2017, 68, 167–184. [Google Scholar] [CrossRef] [PubMed]
- Robertson, B.H.; Jansen, R.W.; Khanna, B.; Totsuka, A.; Nainan, O.V.; Siegl, G.; Widell, A.; Margolis, H.S.; Isomura, S.; Ito, K.; et al. Genetic Relatedness of Hepatitis-a Virus-Strains Recovered from Different Geographical Regions. J. Gen. Virol. 1992, 73, 1365–1377. [Google Scholar] [CrossRef]
- Aguirre, S.; Malirat, V.; Scodeller, E.; Mattion, N. First full-length genomic sequence of a hepatitis A virus isolated in Argentina shows recombination between subgenotypes IA and IB. Virus Res. 2011, 155, 316–324. [Google Scholar] [CrossRef] [PubMed]
- Jansen, R.W.; Siegl, G.; Lemon, S.M. Molecular epidemiology of human hepatitis A virus defined by an antigen-capture polymerase chain reaction method. Proc. Natl. Acad. Sci. USA 1990, 87, 2867–2871. [Google Scholar] [CrossRef]
- Hutin, Y.J.; Pool, V.; Cramer, E.H.; Nainan, O.V.; Weth, J.; Williams, I.T.; Goldstein, S.T.; Gensheimer, K.F.; Bell, B.P.; Shapiro, C.N.; et al. A multistate, foodborne outbreak of hepatitis A. National Hepatitis A Investigation Team. N. Engl. J. Med. 1999, 340, 595–602. [Google Scholar] [CrossRef]
- Nainan, O.V.; Armstrong, G.L.; Han, X.H.; Williams, I.; Bell, B.P.; Margolis, H.S. Hepatitis a molecular epidemiology in the United States, 1996–1997: Sources of infection and implications of vaccination policy. J. Infect. Dis. 2005, 191, 957–963. [Google Scholar] [CrossRef]
- Apaire-Marchais, V.; Robertson, B.H.; Aubineau-Ferre, V.; Le Roux, M.G.; Leveque, F.; Schwartzbrod, L.; Billaudel, S. Direct sequencing of hepatitis A virus strains isolated during an epidemic in France. Appl. Environ. Microbiol. 1995, 61, 3977–3980. [Google Scholar] [CrossRef]
- Costa-Mattioli, M.; Cristina, J.; Romero, H.; Perez-Bercof, R.; Casane, D.; Colina, R.; Garcia, L.; Vega, I.; Glikman, G.; Romanowsky, V.; et al. Molecular evolution of hepatitis A virus: A new classification based on the complete VP1 protein. J. Virol. 2002, 76, 9516–9525. [Google Scholar] [CrossRef]
- Nainan, O.V.; Margolis, H.S.; Robertson, B.H.; Balayan, M.; Brinton, M.A. Sequence analysis of a new hepatitis A virus naturally infecting cynomolgus macaques (Macaca fascicularis). J. Gen. Virol. 1991, 72, 1685–1689. [Google Scholar] [CrossRef]
- Yilmaz, H.; Karakullukcu, A.; Turan, N.; Cizmecigil, U.Y.; Yilmaz, A.; Ozkul, A.A.; Aydin, O.; Gunduz, A.; Mete, M.; Zeyrek, F.Y.; et al. Genotypes of hepatitis a virus in Turkey: First report and clinical profile of children infected with sub-genotypes IA and IIIA. BMC Infect. Dis. 2017, 17, 561. [Google Scholar] [CrossRef]
- Probert, W.S.; Gonzalez, C.; Espinosa, A.; Hacker, J.K. Molecular Genotyping of Hepatitis A Virus, CA, USA, 2017–2018. Emerg. Infect. Dis. 2019, 25, 1594–1596. [Google Scholar] [CrossRef]
- Bosch, A.; Sanchez, G.; Le Guyader, F.; Vanaclocha, H.; Haugarreau, L.; Pinto, R.M. Human enteric viruses in Coquina clams associated with a large hepatitis A outbreak. Water Sci. Technol. 2001, 43, 61–65. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, G.; Pinto, R.M.; Vanaclocha, H.; Bosch, A. Molecular characterization of hepatitis A virus isolates from a transcontinental shellfish-borne outbreak. J. Clin. Microbiol. 2002, 40, 4148–4155. [Google Scholar] [CrossRef]
- Frank, C.; Walter, J.; Muehlen, M.; Jansen, A.; van Treeck, U.; Hauri, A.M.; Zoellner, I.; Rakha, M.; Hoehne, M.; Hamouda, O.; et al. Major outbreak of hepatitis A associated with orange juice among tourists, Egypt, 2004. Emerg. Infect. Dis. 2007, 13, 156–158. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, C.; Thomas, H.L.; Balogun, K.; Tedder, R.; Pebody, R.; Ramsay, M.; Ngui, S.L. A possible outbreak of hepatitis A associated with semi-dried tomatoes, England, July–November 2011. Eurosurveillance 2012, 17, 14–17. [Google Scholar] [CrossRef]
- Petrignani, M.; Harms, M.; Verhoef, L.; van Hunen, R.; Swaan, C.; van Steenbergen, J.; Boxman, I.; Sala, R.P.I.; Ober, H.J.; Vennema, H.; et al. Update: A food-borne outbreak of hepatitis A in the Netherlands related to semi-dried tomatoes in oil, January–February 2010. Eurosurveillance 2010, 15, 19572. [Google Scholar] [CrossRef] [PubMed]
- Donnan, E.J.; Fielding, J.E.; Gregory, J.E.; Lalor, K.; Rowe, S.; Goldsmith, P.; Antoniou, M.; Fullerton, K.E.; Knope, K.; Copland, J.G.; et al. A Multistate Outbreak of Hepatitis A Associated With Semidried Tomatoes in Australia, 2009. Clin. Infect. Dis. 2012, 54, 775–781. [Google Scholar] [CrossRef] [PubMed]
- Lowe, C.F.; Merrick, L.; Harrigan, P.R.; Mazzulli, T.; Sherlock, C.H.; Ritchie, G. Implementation of Next-Generation Sequencing for Hepatitis B Virus Resistance Testing and Genotyping in a Clinical Microbiology Laboratory. J. Clin. Microbiol. 2016, 54, 127–133. [Google Scholar] [CrossRef] [PubMed]
- Buermans, H.P.; den Dunnen, J.T. Next generation sequencing technology: Advances and applications. Biochim. Biophys. Acta 2014, 1842, 1932–1941. [Google Scholar] [CrossRef]
- Gu, W.; Miller, S.; Chiu, C.Y. Clinical Metagenomic Next-Generation Sequencing for Pathogen Detection. Annu. Rev. Pathol.-Mech. 2019, 14, 319–338. [Google Scholar] [CrossRef] [PubMed]
- Xiao, M.F.; Liu, X.Q.; Ji, J.K.; Li, M.; Li, J.D.; Yang, L.; Sun, W.Y.; Ren, P.D.; Yang, G.F.; Zhao, J.C.; et al. Multiple approaches for massively parallel sequencing of SARS-CoV-2 genomes directly from clinical samples. Genome Med. 2020, 12, 57. [Google Scholar] [CrossRef]
- Batista, F.M.; Stapleton, T.; Lowther, J.A.; Fonseca, V.G.; Shaw, R.; Pond, C.; Walker, D.I.; van Aerle, R.; Martinez-Urtaza, J. Whole Genome Sequencing of Hepatitis A Virus Using a PCR-Free Single-Molecule Nanopore Sequencing Approach. Front. Microbiol. 2020, 11, 874. [Google Scholar] [CrossRef]
- Jiang, X.; Wang, M.; Wang, K.; Estes, M.K.J.V. Sequence and genomic organization of Norwalk virus. Virology 1993, 195, 51–61. [Google Scholar] [CrossRef]
- Kim, W.K.; Kim, J.A.; Song, D.H.; Lee, D.; Kim, Y.C.; Lee, S.Y.; Lee, S.H.; No, J.S.; Kim, J.H.; Kho, J.H.; et al. Phylogeographic analysis of hemorrhagic fever with renal syndrome patients using multiplex PCR-based next generation sequencing. Sci. Rep. 2016, 6, 26017. [Google Scholar] [CrossRef]
- Kim, W.K.; No, J.S.; Lee, S.H.; Song, D.H.; Lee, D.; Kim, J.A.; Gu, S.H.; Park, S.; Jeong, S.T.; Kim, H.C.; et al. Multiplex PCR-Based Next-Generation Sequencing and Global Diversity of Seoul Virus in Humans and Rats. Emerg. Infect. Dis. 2018, 24, 249–257. [Google Scholar] [CrossRef]
- Deng, X.; Gu, W.; Federman, S.; Du Plessis, L.; Pybus, O.G.; Faria, N.R.; Wang, C.; Yu, G.; Bushnell, B.; Pan, C.-Y. Genomic surveillance reveals multiple introductions of SARS-CoV-2 into Northern California. Science 2020, 369, 582–587. [Google Scholar] [CrossRef] [PubMed]
- Chiapponi, C.; Pavoni, E.; Bertasi, B.; Baioni, L.; Scaltriti, E.; Chiesa, E.; Cianti, L.; Losio, M.N.; Pongolini, S. Isolation and Genomic Sequence of Hepatitis A Virus from Mixed Frozen Berries in Italy. Food Environ. Virol. 2014, 6, 202–206. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Martinez, M.; Diez-Valcarce, M.; Hernandez, M.; Rodriguez-Lazaro, D. Design and Application of Nucleic Acid Standards for Quantitative Detection of Enteric Viruses by Real-Time PCR. Food Environ. Virol. 2011, 3, 92–98. [Google Scholar] [CrossRef] [PubMed]
- Coudray-Meunier, C.; Fraisse, A.; Mokhtari, C.; Martin-Latil, S.; Roque-Afonso, A.-M.; Perelle, S.J.B.M. Hepatitis A virus subgenotyping based on RT-qPCR assays. BMC Microbiol. 2014, 14, 296. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [PubMed]
- Galili, T. dendextend: An R package for visualizing, adjusting and comparing trees of hierarchical clustering. Bioinformatics 2015, 31, 3718–3720. [Google Scholar] [CrossRef] [PubMed]
- Su, Q.; Zhang, Z.; Zhang, Y.; Cui, Z.; Chang, S.; Zhao, P.J.V.M. Complete genome analysis of avian hepatitis E virus from chicken with hepatic rupture hemorrhage syndrome. Vet. Microbiol. 2020, 242, 108577. [Google Scholar] [CrossRef]
- Lim, K.L.; Hewitt, J.; Sitabkhan, A.; Eden, J.-S.; Lun, J.; Levy, A.; Merif, J.; Smith, D.; Rawlinson, W.D.; White, P.A. A multi-site study of norovirus molecular epidemiology in Australia and New Zealand, 2013–2014. PLoS ONE 2016, 11, e0145254. [Google Scholar] [CrossRef] [PubMed]
- Ruchusatsawat, K.; Wongpiyabovorn, J.; Kawidam, C.; Thiemsing, L.; Sangkitporn, S.; Yoshizaki, S.; Tatsumi, M.; Takeda, N.; Ishii, K. An Outbreak of Acute Hepatitis Caused by Genotype IB Hepatitis A Viruses Contaminating the Water Supply in Thailand. Intervirology 2016, 59, 197–203. [Google Scholar] [CrossRef]
- Takahashi, M.; Nishizawa, T.; Gotanda, Y.; Tsuda, F.; Komatsu, F.; Kawabata, T.; Hasegawa, K.; Altankhuu, M.; Chimedregzen, U.; Narantuya, L.; et al. High prevalence of antibodies to hepatitis A and E viruses and viremia of hepatitis B, C, and D viruses among apparently healthy populations in Mongolia. Clin. Vaccine Immunol. 2004, 11, 392–398. [Google Scholar] [CrossRef]
- Song, Y.J.; Jeong, H.J.; Kim, Y.J.; Lee, S.W.; Lee, J.B.; Park, S.Y.; Song, C.S.; Park, H.M.; Choi, I.S. Analysis of complete genome sequences of swine hepatitis E virus and possible risk factors for transmission of HEV to humans in Korea. J. Med. Virol. 2010, 82, 583–591. [Google Scholar] [CrossRef]
- Tominaga, A.; Kanda, T.; Akiike, T.; Komoda, H.; Ito, K.; Abe, A.; Aruga, A.; Kaneda, S.; Saito, M.; Kiyohara, T.; et al. Hepatitis A outbreak associated with a revolving sushi bar in Chiba, Japan: Application of molecular epidemiology. Hepatol. Res. 2012, 42, 828–834. [Google Scholar] [CrossRef]
- Wang, H.; Zheng, H.; Cao, J.; Zhou, W.; Yi, Y.; Jia, Z.; Bi, S. Genetic diversity of hepatitis A virus in China: VP3-VP1-2A genes and evidence of quasispecies distribution in the isolates. PLoS ONE 2013, 8, e74752. [Google Scholar]
- Wang, H.; Wang, X.Y.; Zheng, H.H.; Cao, J.Y.; Zhou, W.T.; Bi, S.L. Evolution and genetic characterization of hepatitis A virus isolates in China. Int. J. Infect. Dis. 2015, 33, 156–158. [Google Scholar] [CrossRef]
- No, J.S.; Kim, W.K.; Cho, S.; Lee, S.H.; Kim, J.A.; Lee, D.; Song, D.H.; Gu, S.H.; Jeong, S.T.; Wiley, M.R.; et al. Comparison of targeted next-generation sequencing for whole- genome sequencing of Hantaan orthohantavirus in Apodemus agrarius lung tissues. Sci. Rep. 2019, 9, 16631. [Google Scholar] [CrossRef]
- Bartsch, C.; Hoper, D.; Made, D.; Johne, R. Analysis of frozen strawberries involved in a large norovirus gastroenteritis outbreak using next generation sequencing and digital PCR. Food Microbiol. 2018, 76, 390–395. [Google Scholar] [CrossRef]
- Lee, G.Y.; Kim, W.K.; Park, K.; Lee, S.H.; Hwang, J.; No, J.S.; Cho, S.; Lee, D.; Song, D.H.; Gu, S.H.; et al. Phylogeographic diversity and hybrid zone of Hantaan orthohantavirus collected in Gangwon Province, Republic of Korea. PLoS Negl. Trop. Dis. 2020, 14, e0008714. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.R.; Lee, S.G.; Kang, L.H.; Jheong, W.H.; Paik, S.Y. Full-length genomic sequence of subgenotype IIIA hepatitis A virus isolate in Republic of Korea. BioMed Res. Int. 2013, 2013, 426034. [Google Scholar] [CrossRef]
- Endo, K.; Takahashi, M.; Masuko, K.; Inoue, K.; Akahane, Y.; Okamoto, H. Full-length sequences of subgenotype IIIA and IIIB hepatitis A virus isolates: Characterization of genotype IIIHAV genornes. Virus Res. 2007, 126, 116–127. [Google Scholar] [CrossRef] [PubMed]
- Lin, K.Y.; Fang, C.T. Public Health Responses to Person-to-Person Hepatitis A Outbreaks. J. Infect. Dis. 2021, 223, 359–361. [Google Scholar] [CrossRef] [PubMed]
- Foster, M.A.; Hofmeister, M.G.; Kupronis, B.A.; Lin, Y.L.; Xia, G.L.; Yin, S.M.; Teshale, E. Increase in Hepatitis A Virus Infections—United States, 2013–2018. Morb. Mortal. Wkly. Rep. 2019, 68, 413–415. [Google Scholar] [CrossRef]
- Sfetcu, O.; Irvine, N.; Ngui, S.L.; Emerson, C.; McCaughey, C.; Donaghy, P. Hepatitis A outbreak predominantly affecting men who have sex with men in Northern Ireland, October 2008 to July 2009. Eurosurveillance 2011, 16, 11–16. [Google Scholar] [CrossRef]
- Freidl, G.S.; Sonder, G.J.; Bovee, L.P.; Friesema, I.H.; van Rijckevorsel, G.G.; Ruijs, W.L.; van Schie, F.; Siedenburg, E.C.; Yang, J.; Vennema, H. Hepatitis A outbreak among men who have sex with men (MSM) predominantly linked with the EuroPride, the Netherlands, July 2016 to February 2017. Eurosurveillance 2017, 22, 2–6. [Google Scholar] [CrossRef] [PubMed]
- Foster, M.A.; Hofmeister, M.G.; Albertson, J.P.; Brown, K.B.; Burakoff, A.W.; Gandhi, A.P.; Glenn-Finer, R.E.; Gounder, P.; Ho, P.Y.; Kavanaugh, T.; et al. Hepatitis A Virus Infections Among Men Who Have Sex with Men—Eight US States, 2017–2018. Morb. Mortal. Wkly. Rep. 2021, 70, 875–878. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, R.; Faber, M.; Dudareva, S.; Ingiliz, P.; Jessen, H.; Koch, J.; Marcus, U.; Michaelis, K.; Rieck, T.; Ruscher, C.; et al. Hepatitis A outbreak among MSM in Berlin due to low vaccination coverage: Epidemiology, management, and successful interventions. Int. J. Infect. Dis. 2021, 103, 146–153. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.J.; Lin, K.Y.; Hung, C.C.; Chang, S.C. Hepatitis A Outbreak Among Men Who Have Sex With Men in a Country of Low Endemicity of Hepatitis A Infection. J. Infect. Dis. 2017, 215, 1339–1340. [Google Scholar] [CrossRef]
- Chen, H.F.; Wang, W.M.; Wang, S.L.; Hu, Y. Near-Complete Genome Sequence of a Hepatitis A Subgenotype IB Virus Isolated from Frozen Raspberries. Microbiol. Resour. Ann. 2019, 8, e00522-19. [Google Scholar] [CrossRef]
- Hebeler-Barbosa, F.; Wolf, I.R.; Valente, G.T.; Mello, F.C.D.; Lampe, E.; Pardini, M.I.D.C.; Grotto, R.M.T. A New Method for Next-Generation Sequencing of the Full Hepatitis B Virus Genome from A Clinical Specimen: Impact for Virus Genotyping. Microorganisms 2020, 8, 1391. [Google Scholar] [CrossRef]
- Endo, K.; Inoue, J.; Takahashi, M.; Mitsui, T.; Masuko, K.; Alkahane, Y.; Okamoto, H. Analysis of the full-length genome of a subgenotype IIIB hepatitis a virus isolate: Primers for broadly reactive PCR and genotypic analysis. J. Med. Virol. 2007, 79, 8–17. [Google Scholar] [CrossRef]
- Vaughan, G.; Forbi, J.C.; Xia, G.L.; Fonseca-Ford, M.; Vazquez, R.; Khudyakov, Y.E.; Montiel, S.; Waterman, S.; Alpuche, C.; Goncalves Rossi, L.M.; et al. Full-length genome characterization and genetic relatedness analysis of hepatitis A virus outbreak strains associated with acute liver failure among children. J. Med. Virol. 2014, 86, 202–208. [Google Scholar] [CrossRef]
- Costa-Mattioli, M.; Ferre, V.; Casane, D.; Perez-Bercoff, R.; Coste-Burel, M.; Imbert-Marcille, B.M.; Andre, E.C.M.; Bressollette-Bodin, C.; Billaudel, S.; Cristina, J. Evidence of recombination in natural populations of hepatitis A virus. Virology 2003, 311, 51–59. [Google Scholar] [CrossRef][Green Version]
- Belalov, I.S.; Isaeva, O.V.; Lukashev, A.N. Recombination in hepatitis A virus: Evidence for reproductive isolation of genotypes. J. Gen. Virol. 2011, 92, 860–872. [Google Scholar] [CrossRef]
- Liu, W.; Zhai, J.; Liu, J.; Xie, Y. Identification of recombination between subgenotypes IA and IB of hepatitis A virus. Virus Genes 2010, 40, 222–224. [Google Scholar] [CrossRef] [PubMed]
- Escalera-Zamudio, M.; Rojas-Anaya, E.; Kolokotronis, S.O.; Taboada, B.; Loza-Rubio, E.; Mendez-Ojeda, M.L.; Arias, C.F.; Osterrieder, N.; Greenwood, A.D. Bats, Primates, and the Evolutionary Origins and Diversification of Mammalian Gammaherpesviruses. Mbio 2016, 7, e01425-16. [Google Scholar] [CrossRef] [PubMed]
- Maddison, W.P. Gene trees in species trees. Syst. Biol. 1997, 46, 523–536. [Google Scholar] [CrossRef]
- Lauber, C.; Seitz, S.; Mattei, S.; Suh, A.; Beck, J.; Herstein, J.; Borold, J.; Salzburger, W.; Kaderali, L.; Briggs, J.A.G.; et al. Deciphering the Origin and Evolution of Hepatitis B Viruses by Means of a Family of Non-enveloped Fish Viruses. Cell Host Microbe 2017, 22, 387–399. [Google Scholar] [CrossRef]
- Geoghegan, J.L.; Duchêne, S.; Holmes, E.C. Comparative analysis estimates the relative frequencies of co-divergence and cross-species transmission within viral families. PLoS Pathog. 2017, 13, e1006215. [Google Scholar] [CrossRef]
- Nainan, O.V.; Xia, G.; Vaughan, G.; Margolis, H.S. Diagnosis of hepatitis a virus infection: A molecular approach. Clin. Microbiol. Rev. 2006, 19, 63–79. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Observation | HAV KUMC 98-34 | HAV KUMC 02-1 | HAV KUMC 04-1 | HAV KUMC 14-1 | HAV KUMC 14-2 |
|---|---|---|---|---|---|
| Year | 1998 | 2002 | 2004 | 2014 | 2014 |
| Age | 28 | 30 | 22 | 38 | 36 |
| Gender | Male | Male | Female | Female | Female |
| Anti-HAV IgM | Positive | Positive | Positive | Positive | Positive |
| Anti-HAV IgG | Negative | Negative | Negative | Negative | Negative |
| ALT (IU/L) | 5431 | 4672 | 10,592 | 2572 | 456 |
| AST (IU/L) | 3447.8 | 2389 | 18,912 | 1872 | 410 |
| ALP (IU/L) | 166 | 207 | 184 | 236 | 232 |
| Total bilirubin (mg/dl) | 4.55 | 8.00 | 3.40 | 3.73 | 2.98 |
| Prothrombin time (sec) | 49 | 74.2 | 38.1 | 89 | 105 |
| Albumin (g/dl) | 3.79 | 3.63 | 4.36 | 3.4 | 3.1 |
| HBsAg | Negative | Negative | Negative | Negative | Negative |
| Anti-HCV | Negative | Negative | Negative | Negative | Negative |
| HAV RNA Copy Number (log10 copies/μL) | Sample | Sample Type | Ct Value | HAV Coverage 1 | Total Reads | Reads Mapped to Reference Sequence 1 | Mapping Reads /Total Reads | Depth of Coverage 2 |
|---|---|---|---|---|---|---|---|---|
| 2.2 | HAV KUMC 14-1 | Serum | 27.6 | 94.6% | 3,072,916 | 2,835,008 | 92.3% | 56,719 |
| 1.4 | HAV KUMC 14-2 | Serum | 29.8 | 94% | 4,391,220 | 2,969,551 | 67.6% | 57,894 |
| 1 | HAV KUMC 02-1 | Stool | 31 | 92.3% | 2,030,476 | 1,382,172 | 68.1% | 27,264 |
| 0.3 | HAV KUMC 04-1 | Serum | 33.3 | 86.1% | 4,328,092 | 861,676 | 19.9% | 16,630 |
| Nucleotide (%) | Amino Acid (%) | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Genotype | Strain | HAV KUMC 98-34 | HAV KUMC 02-1 | HAV KUMC 04-1 | HAV KUMC 14-1 | HAV KUMC 14-2 | HAV KUMC 98-34 | HAV KUMC 02-1 | HAV KUMC 04-1 | HAV KUMC 14-1 | HAV KUMC 14-2 |
| IA | HAV KUMC 98-34 | - | - | - | - | - | - | - | - | - | - |
| HAV KUMC 02-1 | 96.4 | - | - | - | - | 99.1 | - | - | - | - | |
| HAV KUMC 04-1 | 97.4 | 96.2 | - | - | - | 98.7 | 98.7 | - | - | - | |
| HAV KUMC 14-1 | 97.8 | 95.7 | 97.6 | - | - | 99.4 | 98.8 | 98.3 | - | - | |
| HAV KUMC 14-2 | 96.5 | 95.5 | 95.7 | 96 | - | 99.2 | 99.1 | 98.4 | 98.9 | - | |
| LU38 | 97.9 | 96.7 | 97 | 97.1 | 97.4 | 99.1 | 99.3 | 98.5 | 98.8 | 99.1 | |
| H2 | 96.3 | 97.9 | 96 | 95.7 | 95.4 | 99.6 | 99.3 | 98.7 | 99.1 | 99.4 | |
| AH1 | 96.4 | 97.8 | 95.9 | 95.7 | 95.6 | 99.1 | 99.3 | 98.5 | 98.8 | 99 | |
| HAJFF-Kan12 | 98.2 | 96.2 | 98.2 | 99.3 | 96.3 | 99.7 | 99.3 | 98.9 | 99.4 | 99.5 | |
| GBM | 95.7 | 95.5 | 95.1 | 95.1 | 95 | 98.6 | 98.6 | 97.9 | 98.2 | 98.4 | |
| IB | HM-175 | 91.5 | 91.3 | 91.4 | 91.2 | 90.6 | 98.8 | 98.9 | 98.1 | 98.4 | 98.8 |
| MBB | 91.6 | 91.3 | 91.2 | 91.1 | 90.8 | 98.2 | 98.3 | 97.5 | 97.8 | 98.2 | |
| IIA | CF53/Berne | 86.1 | 86.2 | 85.9 | 85.9 | 86 | 96.5 | 96.5 | 95.7 | 96 | 96.3 |
| IIB | SLF88 | 86.5 | 86.3 | 86.3 | 86.4 | 86.1 | 97.1 | 97.3 | 96.5 | 96.7 | 97 |
| IIIA | Kor-HAV-F | 82.9 | 83.4 | 82.8 | 82.7 | 82.5 | 94.2 | 94.6 | 93.6 | 93.8 | 94 |
| IIIB | HAJ85-1 | 83 | 83.2 | 82.8 | 82.8 | 82.8 | 94.4 | 94.5 | 93.8 | 93.9 | 94.1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, G.-Y.; Kim, W.-K.; Cho, S.; Park, K.; Kim, J.; Lee, S.-H.; Lee, J.; Lee, Y.-S.; Kim, J.H.; Byun, K.S.; et al. Genotyping and Molecular Diagnosis of Hepatitis A Virus in Human Clinical Samples Using Multiplex PCR-Based Next-Generation Sequencing. Microorganisms 2022, 10, 100. https://doi.org/10.3390/microorganisms10010100
Lee G-Y, Kim W-K, Cho S, Park K, Kim J, Lee S-H, Lee J, Lee Y-S, Kim JH, Byun KS, et al. Genotyping and Molecular Diagnosis of Hepatitis A Virus in Human Clinical Samples Using Multiplex PCR-Based Next-Generation Sequencing. Microorganisms. 2022; 10(1):100. https://doi.org/10.3390/microorganisms10010100
Chicago/Turabian StyleLee, Geum-Young, Won-Keun Kim, Seungchan Cho, Kyungmin Park, Jongwoo Kim, Seung-Ho Lee, Jingyeong Lee, Young-Sun Lee, Ji Hoon Kim, Kwan Soo Byun, and et al. 2022. "Genotyping and Molecular Diagnosis of Hepatitis A Virus in Human Clinical Samples Using Multiplex PCR-Based Next-Generation Sequencing" Microorganisms 10, no. 1: 100. https://doi.org/10.3390/microorganisms10010100
APA StyleLee, G.-Y., Kim, W.-K., Cho, S., Park, K., Kim, J., Lee, S.-H., Lee, J., Lee, Y.-S., Kim, J. H., Byun, K. S., & Song, J.-W. (2022). Genotyping and Molecular Diagnosis of Hepatitis A Virus in Human Clinical Samples Using Multiplex PCR-Based Next-Generation Sequencing. Microorganisms, 10(1), 100. https://doi.org/10.3390/microorganisms10010100