FCoV Viral Sequences of Systemically Infected Healthy Cats Lack Gene Mutations Previously Linked to the Development of FIP

 , ,
, ,  , and
, and
Abstract
1. Introduction
2. Results
2.1. Molecular Cloning of Viral Sequences
2.2. Sequencing of ORFs 3abc and 7b
2.2.1. Swarm Composition and Mutational Frequencies between Challenge Stock and Tissue/Fecal Virus
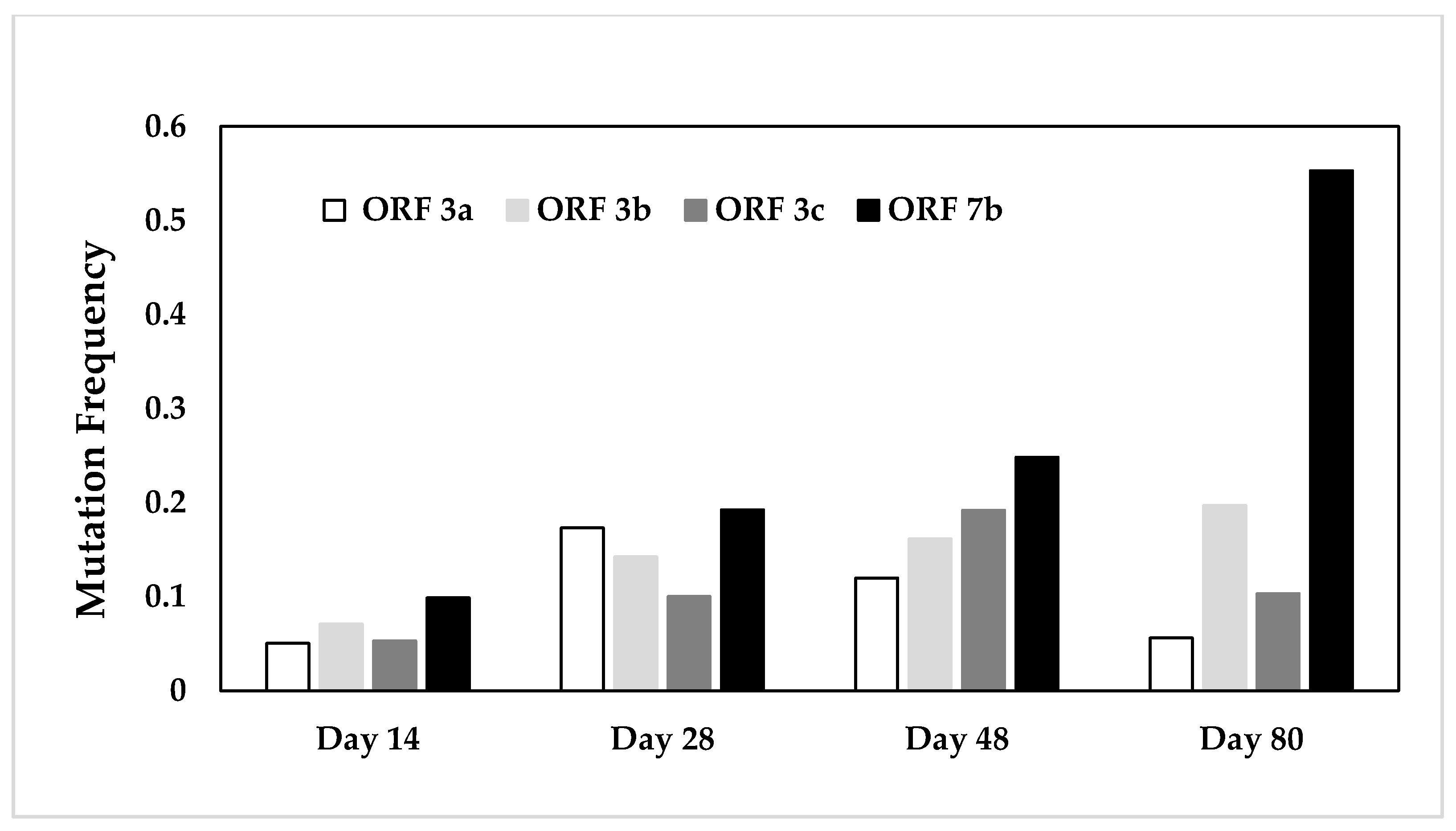
2.2.2. Comparative Assessment of Mutation Frequencies in Genes 3a, 3b, 3c, and 7b
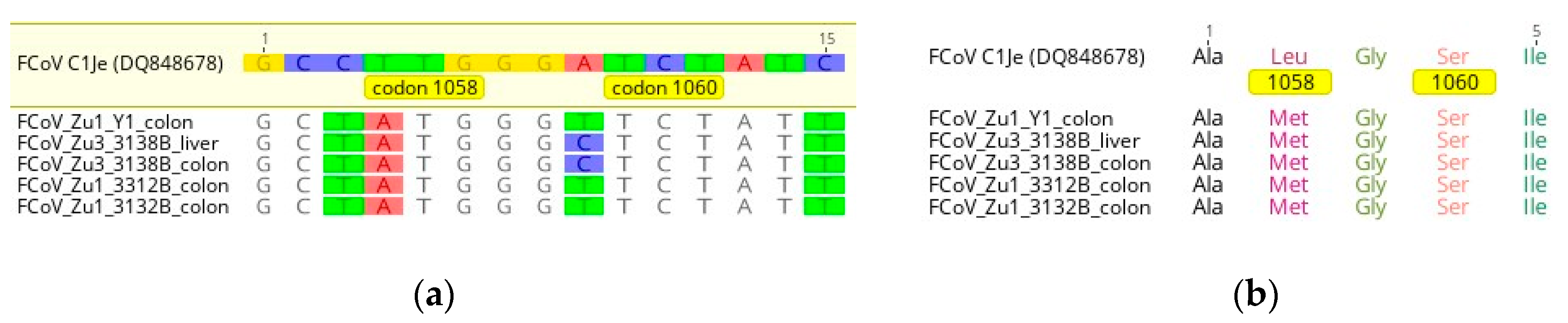
2.2.3. Mutations at the Amino Acid Level
2.2.4. Phylogenetic Analysis of Sequenced Genes and Identification of a New Recombination Site
2.2.5. Evolution Patterns of the Challenge Viruses in Different Cats
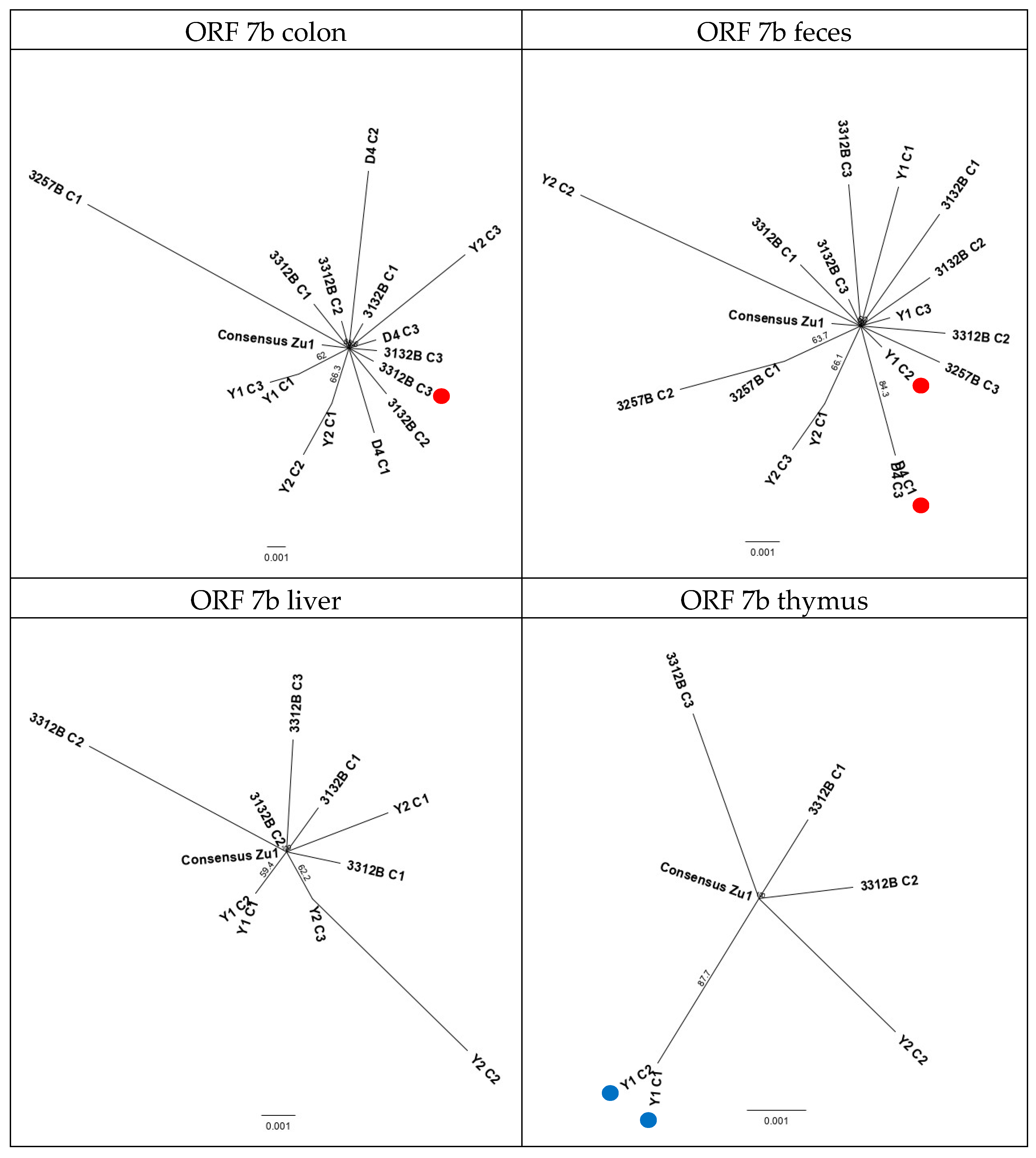
2.2.6. Evolution Patterns of the Challenge Virus FCoV Zu1 in the Different Organs
2.3. Sequencing of the Spike (S) Gene
3. Discussion
4. Materials and Methods
4.1. Sample Characteristics
4.2. Viral RNA Isolation from Tissues and Feces
4.3. Quantitative FCoV Real-Time Reverse Transcription PCR (RT-qPCR)
4.4. One-Step RT-PCR and Nested PCR for Specific Gene Targets
4.5. Purification of PCR Products
4.6. Cloning and Sequencing
4.7. Phylogenetic Analysis
4.8. Statistics
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- de Vries, A.A.; Horzinek, M.C.; Rottier, P.J.; de Groot, R.J. The Genome Organization of the Nidovirales: Similarities and Differences between Arteri-, Toro-, and Coronaviruses. Semin. Virol. 1997, 8, 33–47. [Google Scholar] [CrossRef]
- Denison, M.R.; Graham, R.L.; Donaldson, E.F.; Eckerle, L.D.; Baric, R.S. Coronaviruses. RNA Boil. 2011, 8, 270–279. [Google Scholar] [CrossRef] [PubMed]
- Lauring, A.S.; Andino, R. Quasispecies Theory and the Behavior of RNA Viruses. PLoS Pathog. 2010, 6, e1001005. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, N.C. An overview of feline enteric coronavirus and infectious peritonitis virus infections. Feline Pract. 1995, 23, 7–20. [Google Scholar]
- Pedersen, N.C. A review of feline infectious peritonitis virus infection: 1963–2008. J. Feline Med. Surg. 2009, 11, 225–258. [Google Scholar] [CrossRef]
- Kipar, A.; Meli, M. Feline Infectious Peritonitis. Vet. Pathol. 2014, 51, 505–526. [Google Scholar] [CrossRef]
- Addie, D.; Belák, S.; Boucraut-Baralon, C.; Egberink, H.; Frymus, T.; Gruffydd-Jones, T.; Hartmann, K.; Hosie, M.J.; Lloret, A.; Lutz, H.; et al. Feline Infectious Peritonitis: ABCD Guidelines on Prevention and Management. J. Feline Med. Surg. 2009, 11, 594–604. [Google Scholar] [CrossRef]
- Addie, D.D.; Toth, S.; Murray, G.D.; Jarrett, O. Risk of feline infectious peritonitis in cats naturally infected with feline coronavirus. Am. J. Vet. Res. 1995, 56, 429–434. [Google Scholar]
- Vennema, H.; Polanda, A.; Foleya, J.; Pedersen, N.C. Feline Infectious Peritonitis Viruses Arise by Mutation from Endemic Feline Enteric Coronaviruses. Virology 1998, 243, 150–157. [Google Scholar] [CrossRef]
- Pedersen, N.C.; Boyle, J.F.; Floyd, K.; Fudge, A.; Barker, J. An enteric coronavirus infection of cats and its relationship to feline infectious peritonitis. Am. J. Vet. Res. 1981, 42, 368–377. [Google Scholar]
- Dewerchin, H.L.; Cornelissen, E.; Nauwynck, H.J. Replication of feline coronaviruses in peripheral blood monocytes. Arch. Virol. 2005, 150, 2483–2500. [Google Scholar] [CrossRef] [PubMed]
- Stoddart, C.A.; Scott, F.W. Intrinsic resistance of feline peritoneal macrophages to coronavirus infection correlates with in vivo virulence. J. Virol. 1989, 63, 436–440. [Google Scholar] [CrossRef] [PubMed]
- Gunn-Moore, D.A.; Gruffydd-Jones, T.J.; Harbour, D. Detection of feline coronaviruses by culture and reverse transcriptase-polymerase chain reaction of blood samples from healthy cats and cats with clinical feline infectious peritonitis. Vet. Microbiol. 1998, 62, 193–205. [Google Scholar] [CrossRef]
- Meli, M.; Kipar, A.; Müller, C.; Jenal, K.; Gönczi, E.; Borel, N.; Gunn-Moore, D.; Chalmers, S.; Lin, F.; Reinacher, M.; et al. High viral loads despite absence of clinical and pathological findings in cats experimentally infected with feline coronavirus (FCoV) type I and in naturally FCoV-infected cats. J. Feline Med. Surg. 2004, 6, 69–81. [Google Scholar] [CrossRef] [PubMed]
- Motokawa, K.; Hohdatsu, T.; Hashimoto, H.; Koyama, H. Comparison of the Amino Acid Sequence and Phylogenetic Analysis of the Peplomer, Integral Membrane and Nucleocapsid Proteins of Feline, Canine and Porcine Coronaviruses. Microbiol. Immunol. 1996, 40, 425–433. [Google Scholar] [CrossRef]
- Shiba, N.; Maeda, K.; Kato, H.; Mochizuki, M.; Iwata, H. Differentiation of feline coronavirus type I and II infections by virus neutralization test. Vet. Microbiol. 2007, 124, 348–352. [Google Scholar] [CrossRef]
- Herrewegh, A.A.P.M.; Smeenk, I.; Horzinek, M.C.; Rottier, P.J.M.; de Groot, R. Feline Coronavirus Type II Strains 79-1683 and 79-1146 Originate from a Double Recombination between Feline Coronavirus Type I and Canine Coronavirus. J. Virol. 1998, 72, 4508–4514. [Google Scholar] [CrossRef]
- Chang, H.-W.; de Groot, R.J.; Egberink, H.F.; Rottier, P.J. Feline infectious peritonitis: Insights into feline coronavirus pathobiogenesis and epidemiology based on genetic analysis of the viral 3c gene. J. Gen. Virol. 2009, 91, 415–420. [Google Scholar] [CrossRef]
- Balint, A.; Farsang, A.; Zádori, Z.; Belák, S. Comparative In Vivo Analysis of Recombinant Type II Feline Coronaviruses with Truncated and Completed ORF3 Region. PLoS ONE 2014, 9, e88758. [Google Scholar] [CrossRef]
- Pedersen, N.C.; Liu, H.; Scarlett, J.; Leutenegger, C.M.; Golovko, L.; Kennedy, H.; Mustaffa-Kamal, F. Feline infectious peritonitis: Role of the feline coronavirus 3c gene in intestinal tropism and pathogenicity based upon isolates from resident and adopted shelter cats. Virus Res. 2012, 165, 17–28. [Google Scholar] [CrossRef]
- Pedersen, N.C.; Liu, H.; Dodd, K.A.; Pesavento, P.A. Significance of Coronavirus Mutants in Feces and Diseased Tissues of Cats Suffering from Feline Infectious Peritonitis. Viruses 2009, 1, 166–184. [Google Scholar] [CrossRef] [PubMed]
- Bank-Wolf, B.R.; Stallkamp, I.; Wiese, S.; Moritz, A.; Tekes, G.; Thiel, H.-J. Mutations of 3c and spike protein genes correlate with the occurrence of feline infectious peritonitis. Vet. Microbiol. 2014, 173, 177–188. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.-T.; Su, B.-L.; Hsieh, L.-E.; Chueh, L.-L. An outbreak of feline infectious peritonitis in a Taiwanese shelter: Epidemiologic and molecular evidence for horizontal transmission of a novel type II feline coronavirus. Vet. Res. 2013, 44, 57. [Google Scholar] [CrossRef] [PubMed]
- Dedeurwaerder, A.; Desmarets, L.M.; Olyslaegers, D.A.; Vermeulen, B.L.; Dewerchin, H.L.; Nauwynck, H.J. The role of accessory proteins in the replication of feline infectious peritonitis virus in peripheral blood monocytes. Vet. Microbiol. 2013, 162, 447–455. [Google Scholar] [CrossRef] [PubMed]
- Takano, T.; Tomiyama, Y.; Katoh, Y.; Nakamura, M.; Satoh, R.; Hohdatsu, T. Mutation of neutralizing/antibody-dependent enhancing epitope on spike protein and 7b gene of feline infectious peritonitis virus: Influences of viral replication in monocytes/macrophages and virulence in cats. Virus Res. 2011, 156, 72–80. [Google Scholar] [CrossRef]
- Haijema, B.J.; Volders, H.; Rottier, P.J.M. Live, Attenuated Coronavirus Vaccines through the Directed Deletion of Group-Specific Genes Provide Protection against Feline Infectious Peritonitis. J. Virol. 2004, 78, 3863–3871. [Google Scholar] [CrossRef] [PubMed]
- Herrewegh, A.A.; Vennema, H.; Horzinek, M.C.; Rottier, P.J.; de Groot, R.J. The Molecular Genetics of Feline Coronaviruses: Comparative Sequence Analysis of the ORF7a/7b Transcription Unit of Different Biotypes. Virology 1995, 212, 622–631. [Google Scholar] [CrossRef]
- Kennedy, M.A.; Boedeker, N.; Gibbs, P.; Kania, S.A. Deletions in the 7a ORF of feline coronavirus associated with an epidemic of feline infectious peritonitis. Vet. Microbiol. 2001, 81, 227–234. [Google Scholar] [CrossRef]
- Lin, C.-N.; Su, B.-L.; Wang, C.-H.; Hsieh, M.-W.; Chueh, T.-J.; Chueh, L.-L. Genetic diversity and correlation with feline infectious peritonitis of feline coronavirus type I and II: A 5-year study in Taiwan. Vet Microbiol. 2009, 136, 233–239. [Google Scholar] [CrossRef]
- Rottier, P.J.M.; Nakamura, K.; Schellen, P.; Volders, H.; Haijema, B.J. Acquisition of Macrophage Tropism during the Pathogenesis of Feline Infectious Peritonitis Is Determined by Mutations in the Feline Coronavirus Spike Protein. J. Virol. 2005, 79, 14122–14130. [Google Scholar] [CrossRef]
- Battilani, M.; Balboni, A.; Bassani, M.; Scagliarini, A.; Paltrinieri, S.; Prosperi, S. Sequence analysis of the nucleocapsid gene of feline coronaviruses circulating in Italy. New Microbiol. 2010, 33, 387–392. [Google Scholar] [PubMed]
- Belouzard, S.; Millet, J.K.; Licitra, B.N.; Whittaker, G.R. Mechanisms of Coronavirus Cell Entry Mediated by the Viral Spike Protein. Viruses 2012, 4, 1011–1033. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.-W.; Egberink, H.F.; Halpin, R.; Spiro, D.J.; Rottier, P.J. Spike Protein Fusion Peptide and Feline Coronavirus Virulence. Emerg. Infect. Dis. 2012, 18, 1089–1095. [Google Scholar] [CrossRef] [PubMed]
- Lewis, C.S.; Porter, E.; Matthews, D.A.; Kipar, A.; Tasker, S.; Helps, C.; Siddell, S.G. Genotyping coronaviruses associated with feline infectious peritonitis. J. Gen. Virol. 2015, 96, 1358–1368. [Google Scholar] [CrossRef] [PubMed]
- Porter, E.L.; Tasker, S.; Day, M.J.; Harley, R.; Kipar, A.; Siddell, S.G.; Helps, C. Amino acid changes in the spike protein of feline coronavirus correlate with systemic spread of virus from the intestine and not with feline infectious peritonitis. Vet. Res. 2014, 45, 49. [Google Scholar] [CrossRef]
- Licitra, B.N.; Millet, J.K.; Regan, A.D.; Hamilton, B.S.; Rinaldi, V.; Duhamel, G.E.; Whittaker, G.R. Mutation in Spike Protein Cleavage Site and Pathogenesis of Feline Coronavirus. Emerg. Infect. Dis. 2013, 19, 1066–1073. [Google Scholar] [CrossRef]
- Tekes, G.; Spies, D.; Bank-Wolf, B.; Thiel, V.; Thiel, H.-J. A Reverse Genetics Approach to Study Feline Infectious Peritonitis. J. Virol. 2012, 86, 6994–6998. [Google Scholar] [CrossRef]
- Thiel, V.; Thiel, H.-J.; Tekes, G. Tackling feline infectious peritonitis via reverse genetics. Bioengineered 2014, 5, 396–400. [Google Scholar] [CrossRef]
- Dye, C.; Siddell, S.G. Genomic RNA sequence of feline coronavirus strain FCoV C1Je. J. Feline Med. Surg. 2007, 9, 202–213. [Google Scholar] [CrossRef]
- Hora, A.S.; Tonietti, P.O.; Taniwaki, S.A.; Asano, K.M.; Maiorka, P.; Richtzenhain, L.J.; Brandão, P.E. Feline Coronavirus 3c Protein: A Candidate for a Virulence Marker? BioMed Res. Int. 2016, 1–9. [Google Scholar] [CrossRef]
- McKay, L.A.; Meachem, M.; Snead, E.; Brannen, T.; Mutlow, N.; Ruelle, L.; Davies, J.L.; van der Meer, F. Prevalence and mutation analysis of the spike protein in feline enteric coronavirus and feline infectious peritonitis detected in household and shelter cats in western Canada. Can. J. Vet. Res. 2020, 84, 18–23. [Google Scholar] [PubMed]
- Oguma, K.; Ohno, M.; Yoshida, M.; Sentsui, H. Mutation of the S and 3c genes in genomes of feline coronaviruses. J. Vet. Med. Sci. 2018, 80, 1094–1100. [Google Scholar] [CrossRef] [PubMed]
- Sangl, L.; Matiasek, K.; Felten, S.; Gründl, S.; Bergmann, M.; Balzer, H.-J.; Pantchev, N.; Leutenegger, C.M.; Hartmann, K. Detection of feline coronavirus mutations in paraffin-embedded tissues in cats with feline infectious peritonitis and controls. J. Feline Med. Surg. 2018, 21, 133–142. [Google Scholar] [CrossRef] [PubMed]
- Woo, P.C.Y.; Lau, S.K.P.; Huang, Y.; Yuen, K.-Y. Coronavirus Diversity, Phylogeny, and Interspecies Jumping. Exp. Boil. Med. 2009, 234, 1117–1127. [Google Scholar] [CrossRef]
- Addie, D.D.; Schaap, I.A.T.; Nicolson, L.; Jarrett, O. Persistence, and transmission of natural type I feline coronavirus infection. J. Gen. Virol. 2003, 84, 2735–2744. [Google Scholar] [CrossRef]
- Más, A.; Lopez-Galindez, C.; Cacho, I.; Gómez, J.; Martinez, M.A. Unfinished Stories on Viral Quasispecies and Darwinian Views of Evolution. J. Mol. Boil. 2010, 397, 865–877. [Google Scholar] [CrossRef]
- Battilani, M.; Coradin, T.; Scagliarini, A.; Ciulli, S.; Ostanello, F.; Prosperi, S.; Morganti, L. Quasispecies composition and phylogenetic analysis of feline coronaviruses (FCoVs) in naturally infected cats. FEMS Immunol. Med. Microbiol. 2003, 39, 141–147. [Google Scholar] [CrossRef]
- Vennema, H.; Poland, A.; Hawkins, K.F.; Pedersen, N.C. A comparison of the genomes of FECVs and FIPVs and what they tell us about the relationships between feline coronaviruses and their evolution. Feline Pract. 1995, 23, 40–44. [Google Scholar]
- Steinhauer, D.A.; Holland, J.J. Direct method for quantitation of extreme polymerase error frequencies at selected single base sites in viral RNA. J. Virol. 1986, 57, 219–228. [Google Scholar] [CrossRef]
- Lin, C.-N.; Chang, R.-Y.; Su, B.-L.; Chueh, L.-L. Full genome analysis of a novel type II feline coronavirus NTU156. Virus Genes 2012, 46, 316–322. [Google Scholar] [CrossRef]
- Terada, Y.; Matsui, N.; Noguchi, K.; Kuwata, R.; Shimoda, H.; Soma, T.; Mochizuki, M.; Maeda, K. Emergence of Pathogenic Coronaviruses in Cats by Homologous Recombination between Feline and Canine Coronaviruses. PLoS ONE 2014, 9, e106534. [Google Scholar] [CrossRef] [PubMed]
- Lai, M.M. Recombination in large RNA viruses: Coronaviruses. Semin. Virol. 1996, 7, 381–388. [Google Scholar] [CrossRef]
- Shirato, K.; Chang, H.-W.; Rottier, P.J. Differential susceptibility of macrophages to serotype II feline coronaviruses correlates with differences in the viral spike protein. Virus Res. 2018, 255, 14–23. [Google Scholar] [CrossRef] [PubMed]
- de Haan, C.A.M.; Haijema, B.J.; Schellen, P.; Schreur, P.W.; Lintelo, E.T.; Vennema, H.; Rottier, P.J. Cleavage of Group 1 Coronavirus Spike Proteins: How Furin Cleavage Is Traded Off against Heparan Sulfate Binding upon Cell Culture Adaptation. J. Virol. 2008, 82, 6078–6083. [Google Scholar] [CrossRef]
- Leutenegger, C.M.; Boretti, F.S.; Mislin, C.N.; Flynn, J.N.; Schroff, M.; Habel, A.; Junghans, C.; Koenig-Merediz, S.A.; Sigrist, B.; Aubert, A.; et al. Immunization of Cats against Feline Immunodeficiency Virus (FIV) Infection by Using Minimalistic Immunogenic Defined Gene Expression Vector Vaccines Expressing FIV gp140 Alone or with Feline Interleukin-12 (IL-12), IL-16, or a CpG Motif. J. Virol. 2000, 74, 10447–10457. [Google Scholar] [CrossRef][Green Version]
- Gut, M.; Leutenegger, C.M.; Huder, J.B.; Pedersen, N.C.; Lutz, H. One-tube fluorogenic reverse transcription-polymerase chain reaction for the quantitation of feline coronaviruses. J. Virol. Methods 1999, 77, 37–46. [Google Scholar] [CrossRef]
- Saitou, N.; Nei, M. The neighbor-joining method: A new method for reconstructing phylogenetic trees. Mol. Boil. Evol. 1987, 4, 406–425. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cat ID | Challenge Virus Strain (Origin) | Timepoint of Euthanasia (Days p.i.) | Virus Load * in Different Organs | Virus Load * in Feces | |||
|---|---|---|---|---|---|---|---|
| Colon | Liver | Thymus | Other | ||||
| 3132B | FCoV Zu1 (feces) | 14 | 14.9 | 31.2 | 40.3 | 26.4 | |
| 3138B | FCoV Zu3 (feces) | 28 | 14.2 | 23.9 | 42.3 | 20.5 | |
| 3312B | FCoV Zu1 (feces) | 28 | 17.0 | 32.2 | 37.3 | 25.3 | |
| D4 | FCoV Zu1 (gut homogenate) | 48 | 28.4 | 27.9 | negative | 39.4 a | 28.8 |
| Y1 | FCoV Zu1 (gut homogenate) | 48 | 24.2 | 29.7 | 35.4 | 30.3 | |
| Y2 | FCoV Zu1 (gut homogenate) | 48 | 27.0 | 38.0 | 39.0 | 28.4 | |
| 3257B | FCoV Zu1 (gut homogenate) | 80 | 27.7 | 37.9 | negative | 39.3 b | 22.9 |
| Challenge Stock Virus | Gene | Number (Percentage) of Sequences Identical to Consensus (1) | Number (Percentage) of SNPs | ||
|---|---|---|---|---|---|
| Challenge Stock | Tissue/Fecal FCoVs | Challenge Stock (2) | Tissue/Fecal FCoVs (3) | ||
| FCoV Zu1 | ORF | 7/23 | 13/63 | 28/25668 | 91/70308 |
| 3abc | (30.4%) | (20.6%) | (0.109%) | (0.129%) | |
| FCoV Zu1 | ORF | 12/23 | 7/46 | 19/14559 | 75/29118 |
| 7b | (52.2%) | (15.2%) | (0.130%) | (0.256%) | |
| FCoV Zu3 | ORF | 5/21 | 2/9 | 39/22932 | 16/9828 |
| 3abc | (23.8%) | (22.2%) | (0.170%) | (0.163%) | |
| FCoV Zu3 | ORF | 9/21 | 2/8 | 15/13041 | 9/4968 |
| 7b | (42.9%) | (25%) | (0.115%) | (0.181%) | |
| Cat ID/Challenge Virus Stock | Days p.i. | ORF 3a | ORF 3b | ORF 3c | ORF 7b | ||||
|---|---|---|---|---|---|---|---|---|---|
| # SNPs/Nts (1) | # AA/SNPs (2) | # SNPs/Nts (1) | # AA/SNPs (2) | # SNPs/Nts (1) | # AA/SNPs (2) | # SNPs/Nts (1) | # AA/ SNPs (2) | ||
| 3132B | 14 | 1/1980 | 0/1 | 2/2820 | 0/2 | 4/7530 | 4/4 | 5/5064 | 4/5 |
| (0.051%) | (0.071%) | (0.053%) | (0.099%) | ||||||
| 3138B | 28 | 4/1917 | 3/4 | 5/3213 | 1/5 | 9/6426 | 9/9 | 7/4347 | 4/7 |
| (0.209%) | (0.156%) | (0.140%) | (0.161%) | ||||||
| 3312B | 28 | 3/2178 | 1/3 | 4/3102 | 2/4 | 5/8283 | 3/5 | 17/7596 | 13/17 |
| (0.138%) | (0.129%) | (0.060%) | (0.224%) | ||||||
| D4 | 48 | – | – | 4/3102 | 0/4 | 13/8283 | 11/13 | 11/4431 | 6/11 |
| (0.129%) | (0.157%) | (0.248%) | |||||||
| Y1 | 48 | 1/1782 | 0/1 | 7/2538 | 2/7 | 14/6777 | 9/14 | 11/5697 | 11/11 |
| (0.056%) | (0.276%) | (0.207%) | (0.193%) | ||||||
| Y2 | 48 | 4/2178 | 0/4 | 5/3102 | 2/5 | 13/6777 | 3/13 | 27/6330 | 24/27 |
| (0.184%) | (0.161%) | (0.192%) | (0.427%) | ||||||
| 3257B | 80 | 1/1782 | 1/1 | 5/2538 | 3/5 | 7/6777 | 7/7 | 14/2532 | 11/14 |
| (0.056%) | (0.197%) | (0.103%) | (0.553%) | ||||||
| FCoV Zu1 | n.a. | 4/4554 | 2/4 | 11/6486 | 7/11 | 19/17319 | 13/19 | 19/14559 | 14/19 |
| (0.088%) | (0.170%) | (0.110%) | (0.130%) | ||||||
| FCoV Zu3 | n.a. | 9/4899 | 6/9 | 15/8211 | 11/15 | 23/16422 | 8/23 | 15/13041 | 9/15 |
| (0.184%) | (0.183%) | (0.140%) | (0.115%) | ||||||
| Cat/Virus | Origin | ORF | SNP or Deletion (# Nts) | Effect on Protein |
|---|---|---|---|---|
| Y1 | Feces | 3a | Deletion (1) | Truncation |
| Feces | 7b | Deletion (1) | Truncation | |
| Thymus | 7b | SNP | No start codon | |
| Liver | 3c | SNP | No stop codon | |
| Y2 | Colon | 3c | Deletion (3) | Shortening |
| 3312B | Colon | 7b | Deletion (4) | Truncation |
| D4 | Feces | 7b | Deletion (35) | Truncation |
| 3138B | Liver | 3a | SNP | Truncation |
| FCoV Zu1 challenge stock virus | Gut homogenate | 3a | Deletion (22) | No stop codon |
| 3b | Deletion (22) | Truncation |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lutz, M.; Steiner, A.R.; Cattori, V.; Hofmann-Lehmann, R.; Lutz, H.; Kipar, A.; Meli, M.L. FCoV Viral Sequences of Systemically Infected Healthy Cats Lack Gene Mutations Previously Linked to the Development of FIP. Pathogens 2020, 9, 603. https://doi.org/10.3390/pathogens9080603
Lutz M, Steiner AR, Cattori V, Hofmann-Lehmann R, Lutz H, Kipar A, Meli ML. FCoV Viral Sequences of Systemically Infected Healthy Cats Lack Gene Mutations Previously Linked to the Development of FIP. Pathogens. 2020; 9(8):603. https://doi.org/10.3390/pathogens9080603
Chicago/Turabian StyleLutz, Mirjam, Aline R. Steiner, Valentino Cattori, Regina Hofmann-Lehmann, Hans Lutz, Anja Kipar, and Marina L. Meli. 2020. "FCoV Viral Sequences of Systemically Infected Healthy Cats Lack Gene Mutations Previously Linked to the Development of FIP" Pathogens 9, no. 8: 603. https://doi.org/10.3390/pathogens9080603
APA StyleLutz, M., Steiner, A. R., Cattori, V., Hofmann-Lehmann, R., Lutz, H., Kipar, A., & Meli, M. L. (2020). FCoV Viral Sequences of Systemically Infected Healthy Cats Lack Gene Mutations Previously Linked to the Development of FIP. Pathogens, 9(8), 603. https://doi.org/10.3390/pathogens9080603