Feasibility of Known RNA Polymerase Inhibitors as Anti-SARS-CoV-2 Drugs

 ,
,  , ,
, , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results and Discussion
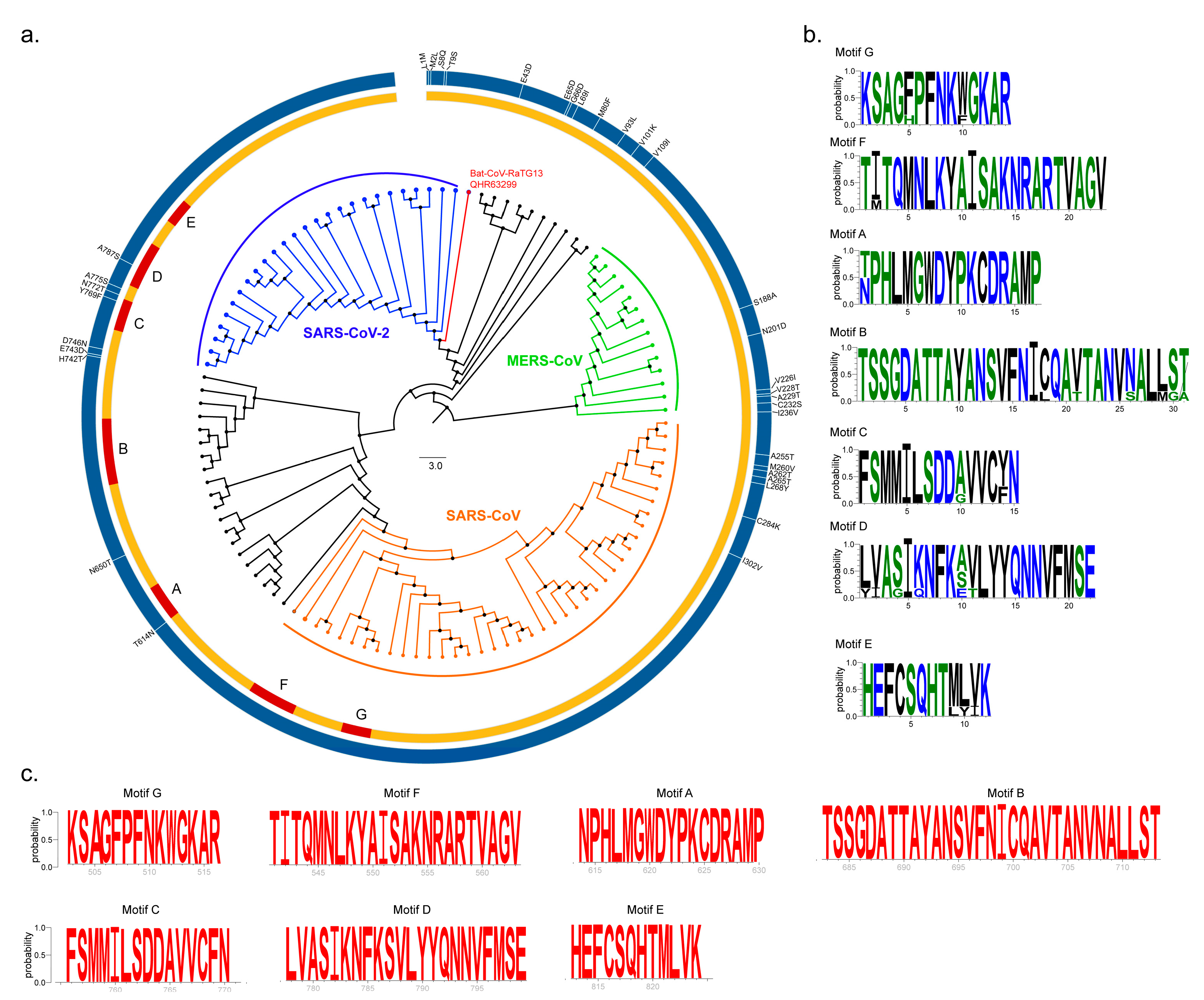
2.1. Sequence Conservation Among SARS-CoV, MERS-CoV and SARS-CoV-2 nsp12 Proteins
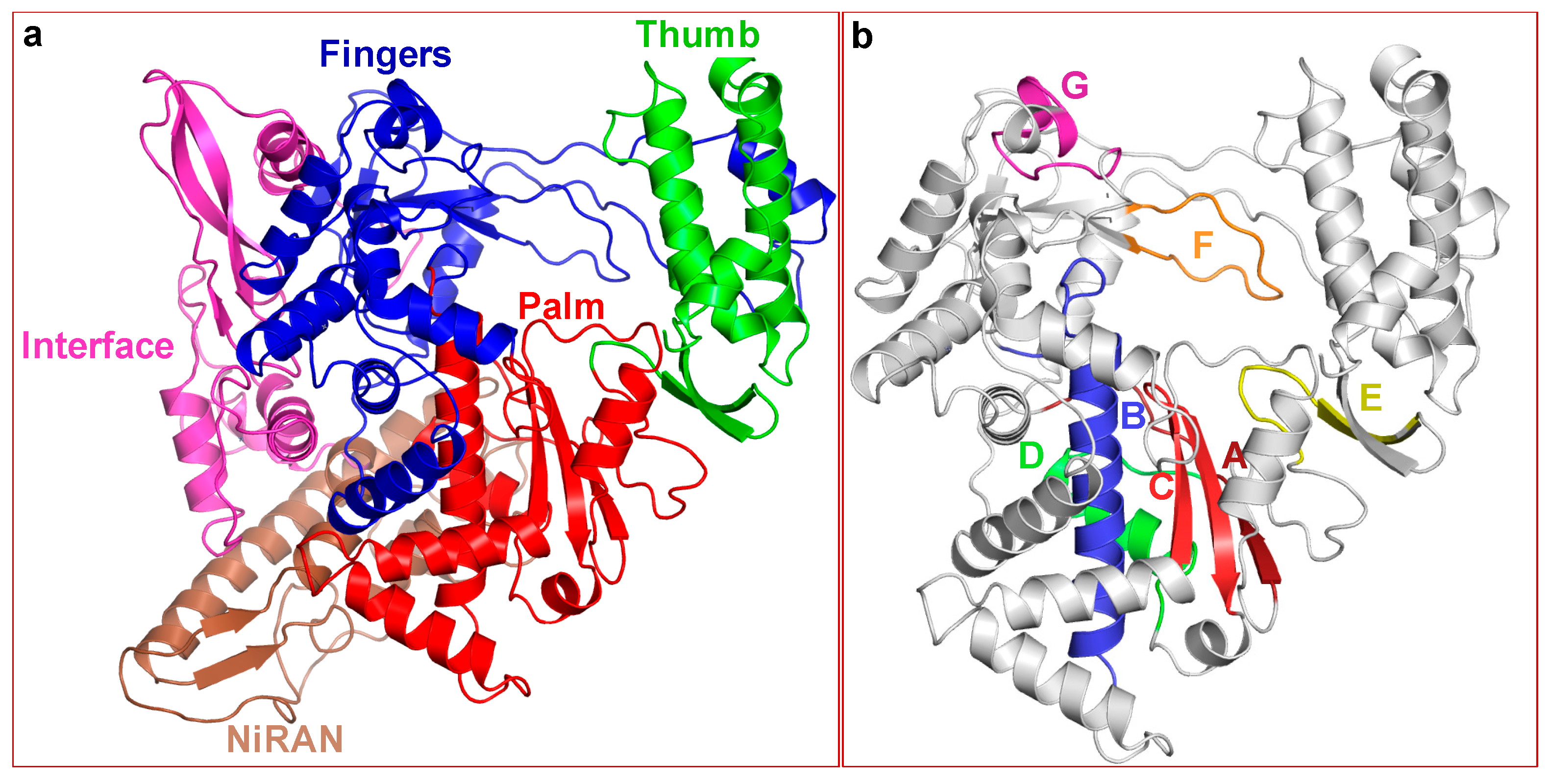
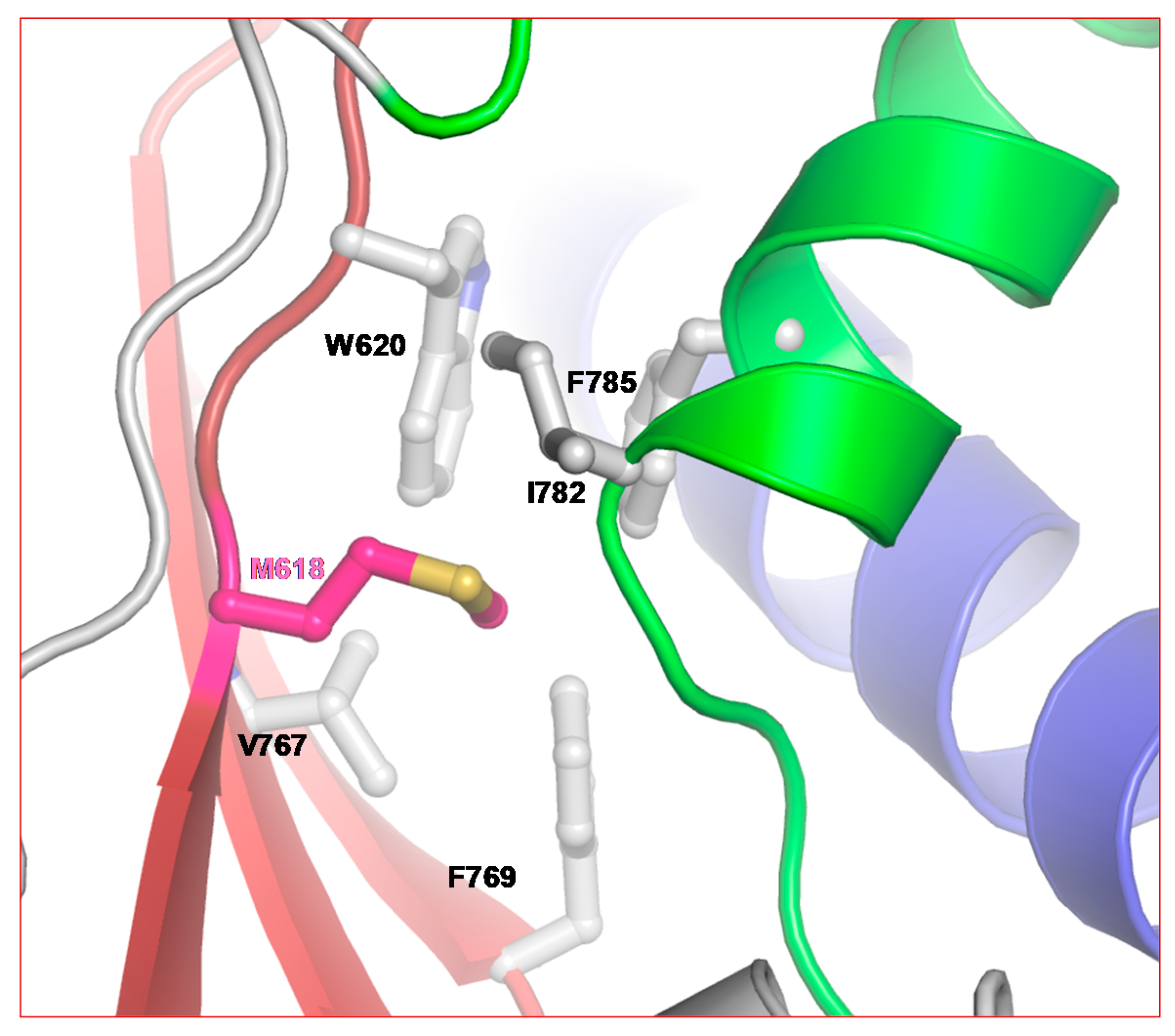
2.2. Structure of SARS-CoV-2 nsp12
2.3. Nucleoside RNA Polymerase Inhibitors
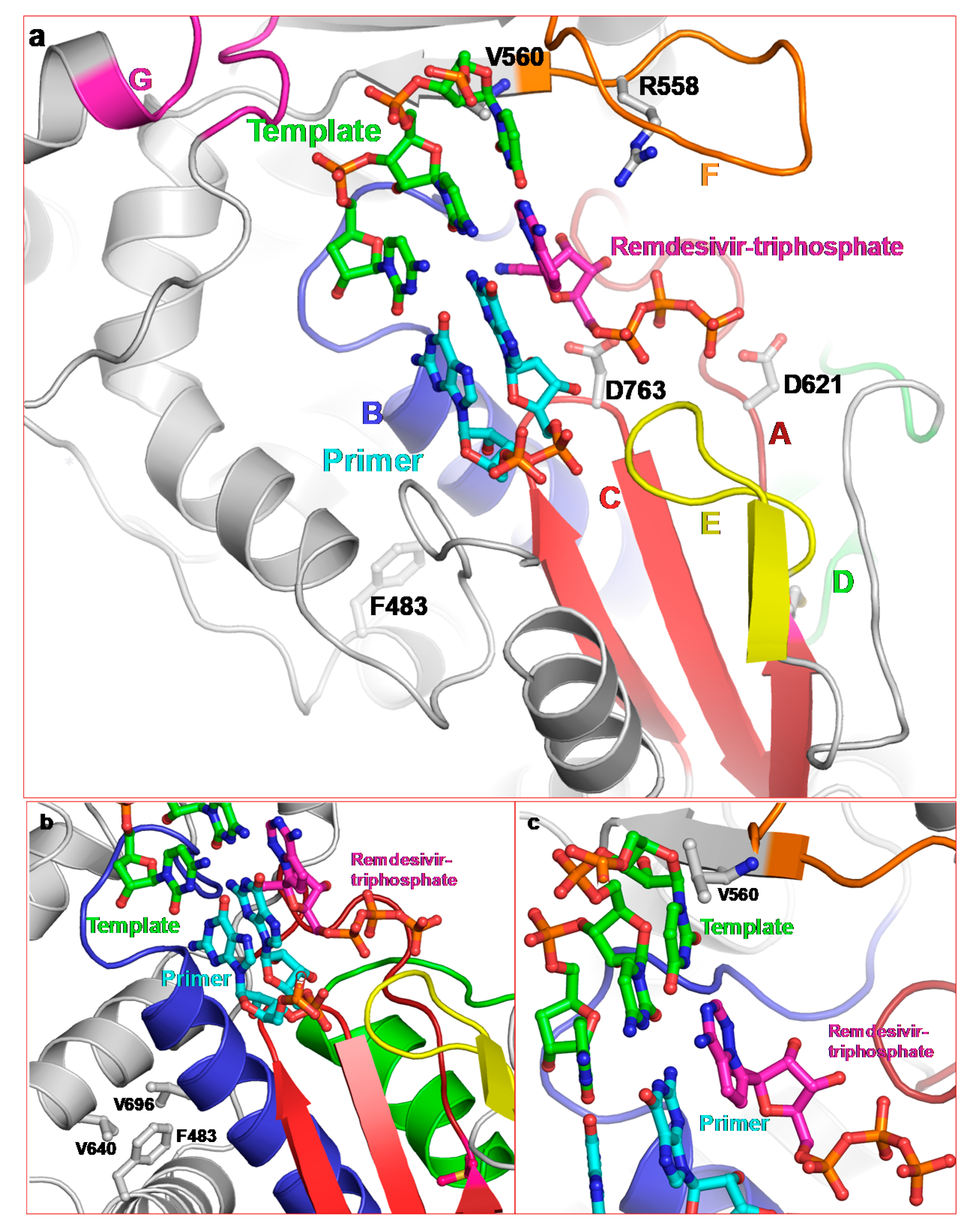
2.3.1. Remdesivir
2.3.2. 5-fluorouracil (5-FU)
2.3.3. Ribavirin
2.3.4. Favipiravir (T-705)
3. Materials and Methods
3.1. Sequence Retrieval and Phylogenetic Analysis
3.2. Molecular Modeling
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Poutanen, S.M.; Low, D.E.; Henry, B.; Finkelstein, S.; Rose, D.; Green, K.; Tellier, R.; Draker, R.; Adachi, D.; Ayers, M.; et al. Identification of severe acute respiratory syndrome in Canada. N. Engl. J. Med. 2003, 348, 1995–2005. [Google Scholar] [CrossRef] [PubMed]
- Tsang, K.W.; Ho, P.L.; Ooi, G.C.; Yee, W.K.; Wang, T.; Chan-Yeung, M.; Lam, W.K.; Seto, W.H.; Yam, L.Y.; Cheung, T.M.; et al. A cluster of cases of severe acute respiratory syndrome in Hong Kong. N. Engl. J. Med. 2003, 348, 1977–1985. [Google Scholar] [CrossRef] [PubMed]
- Peiris, J.S.; Lai, S.T.; Poon, L.L.; Guan, Y.; Yam, L.Y.; Lim, W.; Nicholls, J.; Yee, W.K.; Yan, W.W.; Cheung, M.T.; et al. Coronavirus as a possible cause of severe acute respiratory syndrome. Lancet 2003, 361, 1319–1325. [Google Scholar] [CrossRef]
- Lee, N.; Hui, D.; Wu, A.; Chan, P.; Cameron, P.; Joynt, G.M.; Ahuja, A.; Yung, M.Y.; Leung, C.B.; To, K.F.; et al. A major outbreak of severe acute respiratory syndrome in Hong Kong. N. Engl. J. Med. 2003, 348, 1986–1994. [Google Scholar] [CrossRef] [PubMed]
- Rota, P.A.; Oberste, M.S.; Monroe, S.S.; Nix, W.A.; Campagnoli, R.; Icenogle, J.P.; Penaranda, S.; Bankamp, B.; Maher, K.; Chen, M.H.; et al. Characterization of a novel coronavirus associated with severe acute respiratory syndrome. Science 2003, 300, 1394–1399. [Google Scholar] [CrossRef] [PubMed]
- Booth, C.M.; Matukas, L.M.; Tomlinson, G.A.; Rachlis, A.R.; Rose, D.B.; Dwosh, H.A.; Walmsley, S.L.; Mazzulli, T.; Avendano, M.; Derkach, P.; et al. Clinical features and short-term outcomes of 144 patients with SARS in the greater toronto area. JAMA J. Am. Med. Assoc. 2003, 289, 2801–2809. [Google Scholar] [CrossRef]
- Frieman, M.; Basu, D.; Matthews, K.; Taylor, J.; Jones, G.; Pickles, R.; Baric, R.; Engel, D.A. Yeast based small molecule screen for inhibitors of SARS-cov. PLoS ONE 2011, 6, e28479. [Google Scholar] [CrossRef]
- Hsu, L.Y.; Lee, C.C.; Green, J.A.; Ang, B.; Paton, N.I.; Lee, L.; Villacian, J.S.; Lim, P.L.; Earnest, A.; Leo, Y.S. Severe acute respiratory syndrome (SARS) in Singapore: Clinical features of index patient and initial contacts. Emerg. Infect. Dis. 2003, 9, 713–717. [Google Scholar] [CrossRef]
- Word Health Organization. MERS Situation Update, November 2019. 2019. Available online: http://applications.emro.who.int/docs/EMRPUB-CSR-241-2019-EN.pdf?ua=1 (accessed on 24 February 2020).
- Gralinski, L.E.; Menachery, V.D. Return of the coronavirus: 2019-nCOV. Viruses 2020, 12, 135. [Google Scholar] [CrossRef] [PubMed]
- Word Health Organization. Coronavirus Disease 2019 (covid-19) Situation Report—35. 2020. Available online: https://www.who.int/docs/default-source/coronaviruse/situation-reports/20200224-sitrep-35-covid-19.pdf?sfvrsn=1ac4218d_2 (accessed on 24 February 2020).
- Rothan, H.A.; Byrareddy, S.N. The epidemiology and pathogenesis of coronavirus disease (COVID-19) outbreak. J. Autoimmun. 2020, 109, 102433. [Google Scholar] [CrossRef]
- Adedeji, A.O.; Sarafianos, S.G. Antiviral drugs specific for coronaviruses in preclinical development. Curr. Opin. Virol. 2014, 8, 45–53. [Google Scholar] [CrossRef]
- Adams, M.J.; Carstens, E.B. Ratification vote on taxonomic proposals to the international committee on taxonomy of viruses (2012). Arch. Virol. 2012, 157, 1411–1422. [Google Scholar] [CrossRef]
- de Groot, R.J.; Baker, S.C.; Baric, R.S.; Brown, C.S.; Drosten, C.; Enjuanes, L.; Fouchier, R.A.; Galiano, M.; Gorbalenya, A.E.; Memish, Z.A.; et al. Middle east respiratory syndrome coronavirus (MERS-CoV): Announcement of the coronavirus study group. J. Virol. 2013, 87, 7790–7792. [Google Scholar] [CrossRef]
- Chan, J.F.; Li, K.S.; To, K.K.; Cheng, V.C.; Chen, H.; Yuen, K.Y. Is the discovery of the novel human betacoronavirus 2c EMC/2012 (hCoV-EMC) the beginning of another SARS-like pandemic? J. Infect. 2012, 65, 477–489. [Google Scholar] [CrossRef]
- Letko, M.; Marzi, A.; Munster, V. Functional assessment of cell entry and receptor usage for SARS-cov-2 and other lineage B betacoronaviruses. Nat. Microbiol. 2020, 5, 562–569. [Google Scholar] [CrossRef]
- Gorbalenya, A.E.; Enjuanes, L.; Ziebuhr, J.; Snijder, E.J. Nidovirales: Evolving the largest RNA virus genome. Virus Res. 2006, 117, 17–37. [Google Scholar] [CrossRef]
- Gorbalenya, A.E.; Snijder, E.J.; Spaan, W.J. Severe acute respiratory syndrome coronavirus phylogeny: Toward consensus. J. Virol. 2004, 78, 7863–7866. [Google Scholar] [CrossRef]
- Chan, J.F.; Lau, S.K.; To, K.K.; Cheng, V.C.; Woo, P.C.; Yuen, K.Y. Middle east respiratory syndrome coronavirus: Another zoonotic betacoronavirus causing SARS-like disease. Clin. Microbiol. Rev. 2015, 28, 465–522. [Google Scholar] [CrossRef]
- Lau, S.K.; Woo, P.C.; Yip, C.C.; Fan, R.Y.; Huang, Y.; Wang, M.; Guo, R.; Lam, C.S.; Tsang, A.K.; Lai, K.K.; et al. Isolation and characterization of a novel betacoronavirus subgroup a coronavirus, rabbit coronavirus HKU14, from domestic rabbits. J. Virol. 2012, 86, 5481–5496. [Google Scholar] [CrossRef]
- Marra, M.A.; Jones, S.J.; Astell, C.R.; Holt, R.A.; Brooks-Wilson, A.; Butterfield, Y.S.; Khattra, J.; Asano, J.K.; Barber, S.A.; Chan, S.Y.; et al. The genome sequence of the SARS-associated coronavirus. Science 2003, 300, 1399–1404. [Google Scholar] [CrossRef]
- Herold, J.; Siddell, S.; Ziebuhr, J. Characterization of coronavirus RNA polymerase gene products. Methods Enzymol. 1996, 275, 68–89. [Google Scholar] [PubMed]
- Thiel, V.; Ivanov, K.A.; Putics, A.; Hertzig, T.; Schelle, B.; Bayer, S.; Weissbrich, B.; Snijder, E.J.; Rabenau, H.; Doerr, H.W.; et al. Mechanisms and enzymes involved in SARS coronavirus genome expression. J. Gen. Virol. 2003, 84, 2305–2315. [Google Scholar] [CrossRef] [PubMed]
- Snijder, E.J.; Bredenbeek, P.J.; Dobbe, J.C.; Thiel, V.; Ziebuhr, J.; Poon, L.L.; Guan, Y.; Rozanov, M.; Spaan, W.J.; Gorbalenya, A.E. Unique and conserved features of genome and proteome of SARS-coronavirus, an early split-off from the coronavirus group 2 lineage. J. Mol. Biol. 2003, 331, 991–1004. [Google Scholar] [CrossRef]
- Ziebuhr, J. The coronavirus replicase. Curr. Top Microbiol. Immunol. 2005, 287, 57–94. [Google Scholar]
- Ziebuhr, J. Molecular biology of severe acute respiratory syndrome coronavirus. Curr. Opin. Microbiol. 2004, 7, 412–419. [Google Scholar] [CrossRef]
- Putics, A.; Filipowicz, W.; Hall, J.; Gorbalenya, A.E.; Ziebuhr, J. ADP-ribose-1″-monophosphatase: A conserved coronavirus enzyme that is dispensable for viral replication in tissue culture. J. Virol. 2005, 79, 12721–12731. [Google Scholar] [CrossRef]
- Perlman, S.; Netland, J. Coronaviruses post-SARS: Update on replication and pathogenesis. Nat. Rev. Microbiol. 2009, 7, 439–450. [Google Scholar] [CrossRef]
- Navas-Martin, S.R.; Weiss, S. Coronavirus replication and pathogenesis: Implications for the recent outbreak of severe acute respiratory syndrome (SARS), and the challenge for vaccine development. J. Neurovirol. 2004, 10, 75–85. [Google Scholar] [CrossRef]
- Sawicki, S.G.; Sawicki, D.L.; Siddell, S.G. A contemporary view of coronavirus transcription. J. Virol. 2007, 81, 20–29. [Google Scholar] [CrossRef]
- Harcourt, B.H.; Jukneliene, D.; Kanjanahaluethai, A.; Bechill, J.; Severson, K.M.; Smith, C.M.; Rota, P.A.; Baker, S.C. Identification of severe acute respiratory syndrome coronavirus replicase products and characterization of papain-like protease activity. J. Virol. 2004, 78, 13600–13612. [Google Scholar] [CrossRef]
- Ziebuhr, J.; Snijder, E.J.; Gorbalenya, A.E. Virus-encoded proteinases and proteolytic processing in the nidovirales. J. Gen. Virol. 2000, 81, 853–879. [Google Scholar] [PubMed]
- Fan, K.; Wei, P.; Feng, Q.; Chen, S.; Huang, C.; Ma, L.; Lai, B.; Pei, J.; Liu, Y.; Chen, J.; et al. Biosynthesis, purification, and substrate specificity of severe acute respiratory syndrome coronavirus 3C-like proteinase. J. Biol. Chem. 2004, 279, 1637–1642. [Google Scholar] [PubMed]
- Anand, K.; Ziebuhr, J.; Wadhwani, P.; Mesters, J.R.; Hilgenfeld, R. Coronavirus main proteinase (3CLpro) structure: Basis for design of anti-SARS drugs. Science 2003, 300, 1763–1767. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Yang, M.; Ding, Y.; Liu, Y.; Lou, Z.; Zhou, Z.; Sun, L.; Mo, L.; Ye, S.; Pang, H.; et al. The crystal structures of severe acute respiratory syndrome virus main protease and its complex with an inhibitor. Proc. Natl. Acad. Sci. USA 2003, 100, 13190–13195. [Google Scholar]
- Bernini, A.; Spiga, O.; Venditti, V.; Prischi, F.; Bracci, L.; Huang, J.; Tanner, J.A.; Niccolai, N. Tertiary structure prediction of SARS coronavirus helicase. Biochem. Biophys. Res. Commun. 2006, 343, 1101–1104. [Google Scholar] [CrossRef]
- Minskaia, E.; Hertzig, T.; Gorbalenya, A.E.; Campanacci, V.; Cambillau, C.; Canard, B.; Ziebuhr, J. Discovery of an RNA virus 3’- > 5’ exoribonuclease that is critically involved in coronavirus RNA synthesis. Proc. Natl. Acad. Sci. USA 2006, 103, 5108–5113. [Google Scholar]
- Ricagno, S.; Egloff, M.P.; Ulferts, R.; Coutard, B.; Nurizzo, D.; Campanacci, V.; Cambillau, C.; Ziebuhr, J.; Canard, B. Crystal structure and mechanistic determinants of SARS coronavirus nonstructural protein 15 define an endoribonuclease family. Proc. Natl. Acad. Sci. USA 2006, 103, 11892–11897. [Google Scholar]
- Bhardwaj, K.; Sun, J.; Holzenburg, A.; Guarino, L.A.; Kao, C.C. RNA recognition and cleavage by the SARS coronavirus endoribonuclease. J. Mol. Biol. 2006, 361, 243–256. [Google Scholar]
- Te Velthuis, A.J.; Arnold, J.J.; Cameron, C.E.; van den Worm, S.H.; Snijder, E.J. The RNA polymerase activity of SARS-coronavirus nsp12 is primer dependent. Nucleic Acids Res. 2010, 38, 203–214. [Google Scholar]
- Lehmann, K.C.; Gulyaeva, A.; Zevenhoven-Dobbe, J.C.; Janssen, G.M.; Ruben, M.; Overkleeft, H.S.; van Veelen, P.A.; Samborskiy, D.V.; Kravchenko, A.A.; Leontovich, A.M.; et al. Discovery of an essential nucleotidylating activity associated with a newly delineated conserved domain in the RNA polymerase-containing protein of all nidoviruses. Nucleic Acids Res. 2015, 43, 8416–8434. [Google Scholar]
- Kirchdoerfer, R.N.; Ward, A.B. Structure of the SARS-CoV nsp12 polymerase bound to nsp7 and nsp8 co-factors. Nat. Commun. 2019, 10, 2342. [Google Scholar]
- Gao, Y.; Yan, L.; Huang, T.; Liu, F.; Yao Zhao, Y.; Cao, L.; Wang, T.; Sun, Q.; Ming, Z.; Zhang, L.; et al. Structure of RNA-dependent RNA polymerase from 2019-nCoV, a major antiviral drug target. BioRxiv 2020, 1–24. [Google Scholar] [CrossRef]
- Falzarano, D.; de Wit, E.; Rasmussen, A.L.; Feldmann, F.; Okumura, A.; Scott, D.P.; Brining, D.; Bushmaker, T.; Martellaro, C.; Baseler, L.; et al. Treatment with interferon-alpha2b and ribavirin improves outcome in MERS-CoV-infected rhesus macaques. Nat. Med. 2013, 19, 1313–1317. [Google Scholar]
- Sexton, N.R.; Smith, E.C.; Blanc, H.; Vignuzzi, M.; Peersen, O.B.; Denison, M.R. Homology-based identification of a mutation in the coronavirus RNA-dependent RNA polymerase that confers resistance to multiple mutagens. J. Virol. 2016, 90, 7415–7428. [Google Scholar]
- Dyall, J.; Coleman, C.M.; Hart, B.J.; Venkataraman, T.; Holbrook, M.R.; Kindrachuk, J.; Johnson, R.F.; Olinger, G.G., Jr.; Jahrling, P.B.; Laidlaw, M.; et al. Repurposing of clinically developed drugs for treatment of middle east respiratory syndrome coronavirus infection. Antimicrob. Agents Chemother. 2014, 58, 4885–4893. [Google Scholar]
- Wang, M.; Cao, R.; Zhang, L.; Yang, X.; Liu, J.; Xu, M.; Shi, Z.; Hu, Z.; Zhong, W.; Xiao, G. Remdesivir and chloroquine effectively inhibit the recently emerged novel coronavirus (2019-nCoV) in vitro. Cell Res. 2020, 30, 269–271. [Google Scholar]
- Li, G.; De Clercq, E. Therapeutic options for the 2019 novel coronavirus (2019-nCoV). Nat. Rev. Drug Discov. 2020, 19, 149–150. [Google Scholar]
- Gordon, C.J.; Tchesnokov, E.P.; Feng, J.Y.; Porter, D.P.; Gotte, M. The antiviral compound remdesivir potently inhibits RNA-dependent RNA polymerase from middle east respiratory syndrome coronavirus. J. Biol. Chem. 2020, 295, 4773–4779. [Google Scholar]
- Tchesnokov, E.P.; Feng, J.Y.; Porter, D.P.; Gotte, M. Mechanism of inhibition of Ebola virus RNA-dependent RNA polymerase by remdesivir. Viruses 2019, 11, 326. [Google Scholar]
- de Wit, E.; Feldmann, F.; Cronin, J.; Jordan, R.; Okumura, A.; Thomas, T.; Scott, D.; Cihlar, T.; Feldmann, H. Prophylactic and therapeutic remdesivir (gs-5734) treatment in the rhesus macaque model of MERS-CoV infection. Proc. Natl. Acad. Sci. USA 2020, 117, 6771–6776. [Google Scholar]
- Singh, K.; Modak, M.J. A unified DNA- and dNTP-binding mode for DNA polymerases. Trends Biochem. Sci. 1998, 23, 277–281. [Google Scholar] [CrossRef]
- Peersen, O.B. Picornaviral polymerase structure, function, and fidelity modulation. Virus Res. 2017, 234, 4–20. [Google Scholar] [CrossRef] [PubMed]
- Dong, L.; Hu, S.; Gao, J. Discovering drugs to treat coronavirus disease 2019 (COVID-19). Drug Discov. Ther. 2020, 14, 58–60. [Google Scholar] [CrossRef] [PubMed]
- Grein, J.; Ohmagari, N.; Shin, D.; Diaz, G.; Asperges, E.; Castagna, A.; Feldt, T.; Green, G.; Green, M.L.; Lescure, F.X.; et al. Compassionate use of remdesivir for patients with severe COVID-19. N. Engl. J. Med. 2020. [Google Scholar] [CrossRef] [PubMed]
- Agostini, M.L.; Andres, E.L.; Sims, A.C.; Graham, R.L.; Sheahan, T.P.; Lu, X.; Smith, E.C.; Case, J.B.; Feng, J.Y.; Jordan, R.; et al. Coronavirus susceptibility to the antiviral remdesivir (GS-5734) is mediated by the viral polymerase and the proofreading exoribonuclease. mBio 2018, 9, e00221-18. [Google Scholar] [CrossRef]
- Ferrer-Orta, C.; Arias, A.; Perez-Luque, R.; Escarmis, C.; Domingo, E.; Verdaguer, N. Sequential structures provide insights into the fidelity of RNA replication. Proc. Natl. Acad. Sci. USA 2007, 104, 9463–9468. [Google Scholar]
- Ferrer-Orta, C.; Sierra, M.; Agudo, R.; de la Higuera, I.; Arias, A.; Perez-Luque, R.; Escarmis, C.; Domingo, E.; Verdaguer, N. Structure of foot-and-mouth disease virus mutant polymerases with reduced sensitivity to ribavirin. J. Virol. 2010, 84, 6188–6199. [Google Scholar]
- de Castro, S.; Ferrer-Orta, C.; Mills, A.; Fernandez-Cureses, G.; Gago, F.; Verdaguer, N.; Camarasa, M.J. (F)uridylylated peptides linked to vpg1 of Foot-and- Mouth disease virus (FMDV): Design, synthesis and X-ray crystallography of the complexes with FMDV RNA-dependent RNA polymerase. Molecules 2019, 24, 2360. [Google Scholar]
- Weckbecker, G. Biochemical pharmacology and analysis of fluoropyrimidines alone and in combination with modulators. Pharmacol. Ther. 1991, 50, 367–424. [Google Scholar]
- Rosenberg, A.J.; Rademaker, A.; Hochster, H.S.; Ryan, T.; Hensing, T.; Shankaran, V.; Baddi, L.; Mahalingam, D.; Mulcahy, M.F.; Benson, A.B., III. Docetaxel, oxaliplatin, and 5-fluorouracil (DOF) in metastatic and unresectable gastric/gastroesophageal junction adenocarcinoma: A phase II study with long-term follow-up. Oncologist 2019, 24, 1039–1642. [Google Scholar]
- Sierra, S.; Davila, M.; Lowenstein, P.R.; Domingo, E. Response of foot-and-mouth disease virus to increased mutagenesis: Influence of viral load and fitness in loss of infectivity. J. Virol. 2000, 74, 8316–8323. [Google Scholar] [CrossRef]
- Airaksinen, A.; Pariente, N.; Menendez-Arias, L.; Domingo, E. Curing of foot-and-mouth disease virus from persistently infected cells by ribavirin involves enhanced mutagenesis. Virology 2003, 311, 339–349. [Google Scholar] [CrossRef]
- Agudo, R.; Arias, A.; Domingo, E. 5-fluorouracil in lethal mutagenesis of foot-and-mouth disease virus. Future Med. Chem. 2009, 1, 529–539. [Google Scholar] [CrossRef]
- Pariente, N.; Sierra, S.; Lowenstein, P.R.; Domingo, E. Efficient virus extinction by combinations of a mutagen and antiviral inhibitors. J. Virol. 2001, 75, 9723–9730. [Google Scholar] [CrossRef]
- Agudo, R.; Arias, A.; Pariente, N.; Perales, C.; Escarmis, C.; Jorge, A.; Marina, A.; Domingo, E. Molecular characterization of a dual inhibitory and mutagenic activity of 5-fluorouridine triphosphate on viral RNA synthesis. Implications for lethal mutagenesis. J. Mol. Biol. 2008, 382, 652–666. [Google Scholar] [CrossRef]
- Beaucourt, S.; Borderia, A.V.; Coffey, L.L.; Gnadig, N.F.; Sanz-Ramos, M.; Beeharry, Y.; Vignuzzi, M. Isolation of fidelity variants of RNA viruses and characterization of virus mutation frequency. J. Vis. Exp. 2011, e2953. [Google Scholar] [CrossRef] [PubMed]
- Smith, E.C.; Blanc, H.; Surdel, M.C.; Vignuzzi, M.; Denison, M.R. Coronaviruses lacking exoribonuclease activity are susceptible to lethal mutagenesis: Evidence for proofreading and potential therapeutics. PLoS Pathog. 2013, 9, e1003565. [Google Scholar] [CrossRef] [PubMed]
- de la Higuera, I.; Ferrer-Orta, C.; de Avila, A.I.; Perales, C.; Sierra, M.; Singh, K.; Sarafianos, S.G.; Dehouck, Y.; Bastolla, U.; Verdaguer, N.; et al. Molecular and functional bases of selection against a mutation bias in an RNA virus. Genome Biol. Evol. 2017, 9, 1212–1228. [Google Scholar] [CrossRef] [PubMed]
- Campagnola, G.; McDonald, S.; Beaucourt, S.; Vignuzzi, M.; Peersen, O.B. Structure-function relationships underlying the replication fidelity of viral RNA-dependent RNA polymerases. J. Virol. 2015, 89, 275–286. [Google Scholar] [CrossRef]
- Contreras, A.M.; Hiasa, Y.; He, W.; Terella, A.; Schmidt, E.V.; Chung, R.T. Viral RNA mutations are region specific and increased by ribavirin in a full-length hepatitis C virus replication system. J. Virol. 2002, 76, 8505–8517. [Google Scholar] [CrossRef]
- Crotty, S.; Cameron, C.E.; Andino, R. RNA virus error catastrophe: Direct molecular test by using ribavirin. Proc. Natl. Acad. Sci. USA 2001, 98, 6895–6900. [Google Scholar] [CrossRef]
- Crotty, S.; Maag, D.; Arnold, J.J.; Zhong, W.; Lau, J.Y.; Hong, Z.; Andino, R.; Cameron, C.E. The broad-spectrum antiviral ribonucleoside ribavirin is an RNA virus mutagen. Nat. Med. 2000, 6, 1375–1379. [Google Scholar] [CrossRef]
- Day, C.W.; Smee, D.F.; Julander, J.G.; Yamshchikov, V.F.; Sidwell, R.W.; Morrey, J.D. Error-prone replication of West Nile virus caused by ribavirin. Antivir. Res. 2005, 67, 38–45. [Google Scholar] [CrossRef]
- Pariente, N.; Sierra, S.; Airaksinen, A. Action of mutagenic agents and antiviral inhibitors on foot-and-mouth disease virus. Virus Res. 2005, 107, 183–193. [Google Scholar] [CrossRef]
- Asahina, Y.; Izumi, N.; Enomoto, N.; Uchihara, M.; Kurosaki, M.; Onuki, Y.; Nishimura, Y.; Ueda, K.; Tsuchiya, K.; Nakanishi, H.; et al. Mutagenic effects of ribavirin and response to interferon/ribavirin combination therapy in chronic hepatitis C. J. Hepatol. 2005, 43, 623–629. [Google Scholar] [CrossRef]
- Martin, P.; Jensen, D.M. Ribavirin in the treatment of chronic hepatitis C. J. Gastroenterol. Hepatol. 2008, 23, 844–855. [Google Scholar] [CrossRef]
- Choi, J.H.; Jeong, K.; Kim, S.M.; Ko, M.K.; You, S.H.; Lyoo, Y.S.; Kim, B.; Ku, J.M.; Park, J.H. Synergistic effect of ribavirin and vaccine for protection during early infection stage of foot-and-mouth disease. J. Vet. Sci. 2018, 19, 788–797. [Google Scholar] [CrossRef]
- De Palma, A.M.; Purstinger, G.; Wimmer, E.; Patick, A.K.; Andries, K.; Rombaut, B.; De Clercq, E.; Neyts, J. Potential use of antiviral agents in polio eradication. Emerg. Infect. Dis. 2008, 14, 545–551. [Google Scholar] [CrossRef]
- Omrani, A.S.; Saad, M.M.; Baig, K.; Bahloul, A.; Abdul-Matin, M.; Alaidaroos, A.Y.; Almakhlafi, G.A.; Albarrak, M.M.; Memish, Z.A.; Albarrak, A.M. Ribavirin and interferon alfa-2a for severe middle east respiratory syndrome coronavirus infection: A retrospective cohort study. Lancet Infect. Dis. 2014, 14, 1090–1095. [Google Scholar] [CrossRef]
- Khalid, M.; Al Rabiah, F.; Khan, B.; Al Mobeireek, A.; Butt, T.S.; Al Mutairy, E. Ribavirin and interferon-alpha2b as primary and preventive treatment for middle east respiratory syndrome coronavirus: A preliminary report of two cases. Antivir. Ther. 2015, 20, 87–91. [Google Scholar] [CrossRef]
- Falzarano, D.; de Wit, E.; Martellaro, C.; Callison, J.; Munster, V.J.; Feldmann, H. Inhibition of novel beta coronavirus replication by a combination of interferon-alpha2b and ribavirin. Sci. Rep. 2013, 3, 1686. [Google Scholar] [CrossRef]
- Pfeiffer, J.K.; Kirkegaard, K. A single mutation in poliovirus RNA-dependent RNA polymerase confers resistance to mutagenic nucleotide analogs via increased fidelity. Proc. Natl. Acad. Sci. USA 2003, 100, 7289–7294. [Google Scholar] [CrossRef]
- Sierra, M.; Airaksinen, A.; Gonzalez-Lopez, C.; Agudo, R.; Arias, A.; Domingo, E. Foot-and-Mouth disease virus mutant with decreased sensitivity to ribavirin: Implications for error catastrophe. J. Virol. 2007, 81, 2012–2024. [Google Scholar] [CrossRef]
- Kempf, B.J.; Peersen, O.B.; Barton, D.J. Poliovirus polymerase Leu420 facilitates RNA recombination and ribavirin resistance. J. Virol. 2016, 90, 8410–8421. [Google Scholar] [CrossRef]
- Sadeghipour, S.; Bek, E.J.; McMinn, P.C. Ribavirin-resistant mutants of human enterovirus 71 express a high replication fidelity phenotype during growth in cell culture. J. Virol. 2013, 87, 1759–1769. [Google Scholar] [CrossRef]
- Shao, R.X.; Zhang, L.; Peng, L.F.; Sun, E.; Chung, W.J.; Jang, J.Y.; Tsai, W.L.; Hyppolite, G.; Chung, R.T. Suppressor of cytokine signaling 3 suppresses hepatitis C virus replication in an mTOR-dependent manner. J. Virol. 2010, 84, 6060–6069. [Google Scholar] [CrossRef]
- Kumthip, K.; Chusri, P.; Jilg, N.; Zhao, L.; Fusco, D.N.; Zhao, H.; Goto, K.; Cheng, D.; Schaefer, E.A.; Zhang, L.; et al. Hepatitis C virus NS5A disrupts STAT1 phosphorylation and suppresses type I interferon signaling. J. Virol. 2012, 86, 8581–8591. [Google Scholar] [CrossRef]
- Pfeiffer, J.K.; Kirkegaard, K. Ribavirin resistance in hepatitis C virus replicon-containing cell lines conferred by changes in the cell line or mutations in the replicon RNA. J. Virol. 2005, 79, 2346–2355. [Google Scholar] [CrossRef]
- Young, K.C.; Lindsay, K.L.; Lee, K.J.; Liu, W.C.; He, J.W.; Milstein, S.L.; Lai, M.M. Identification of a ribavirin-resistant NS5B mutation of hepatitis C virus during ribavirin monotherapy. Hepatology 2003, 38, 869–878. [Google Scholar] [CrossRef]
- Gong, P.; Peersen, O.B. Structural basis for active site closure by the poliovirus RNA-dependent RNA polymerase. Proc. Natl. Acad. Sci. USA 2010, 107, 22505–22510. [Google Scholar] [CrossRef]
- Mosley, R.T.; Edwards, T.E.; Murakami, E.; Lam, A.M.; Grice, R.L.; Du, J.; Sofia, M.J.; Furman, P.A.; Otto, M.J. Structure of hepatitis C virus polymerase in complex with primer-template RNA. J. Virol. 2012, 86, 6503–6511. [Google Scholar] [CrossRef] [PubMed]
- Pfeiffer, J.K.; Kirkegaard, K. Increased fidelity reduces poliovirus fitness and virulence under selective pressure in mice. PLoS Pathog. 2005, 1, e11. [Google Scholar] [CrossRef]
- Arias, A.; Arnold, J.J.; Sierra, M.; Smidansky, E.D.; Domingo, E.; Cameron, C.E. Determinants of RNA-dependent RNA polymerase (in)fidelity revealed by kinetic analysis of the polymerase encoded by a foot-and-mouth disease virus mutant with reduced sensitivity to ribavirin. J. Virol. 2008, 82, 12346–12355. [Google Scholar] [CrossRef]
- Furuta, Y.; Komeno, T.; Nakamura, T. Favipiravir (T-705), a broad spectrum inhibitor of viral RNA polymerase. Proc. Jpn. Acad Ser. B Phys. Biol. Sci. 2017, 93, 449–463. [Google Scholar] [CrossRef]
- Furuta, Y.; Takahashi, K.; Fukuda, Y.; Kuno, M.; Kamiyama, T.; Kozaki, K.; Nomura, N.; Egawa, H.; Minami, S.; Watanabe, Y.; et al. In vitro and in vivo activities of anti-influenza virus compound T-705. Antimicrob. Agents Chemother. 2002, 46, 977–981. [Google Scholar] [CrossRef]
- Oestereich, L.; Ludtke, A.; Wurr, S.; Rieger, T.; Munoz-Fontela, C.; Gunther, S. Successful treatment of advanced ebola virus infection with T-705 (favipiravir) in a small animal model. Antivir. Res. 2014, 105, 17–21. [Google Scholar] [CrossRef]
- Oestereich, L.; Rieger, T.; Neumann, M.; Bernreuther, C.; Lehmann, M.; Krasemann, S.; Wurr, S.; Emmerich, P.; de Lamballerie, X.; Olschlager, S.; et al. Evaluation of antiviral efficacy of ribavirin, arbidol, and T-705 (favipiravir) in a mouse model for Crimean-Congo hemorrhagic fever. PLoS Negl. Trop. Dis. 2014, 8, e2804. [Google Scholar] [CrossRef]
- Oestereich, L.; Rieger, T.; Ludtke, A.; Ruibal, P.; Wurr, S.; Pallasch, E.; Bockholt, S.; Krasemann, S.; Munoz-Fontela, C.; Gunther, S. Efficacy of favipiravir alone and in combination with ribavirin in a lethal, immunocompetent mouse model of Lassa fever. J. Infect. Dis. 2016, 213, 934–938. [Google Scholar] [CrossRef]
- Delang, L.; Segura Guerrero, N.; Tas, A.; Querat, G.; Pastorino, B.; Froeyen, M.; Dallmeier, K.; Jochmans, D.; Herdewijn, P.; Bello, F.; et al. Mutations in the chikungunya virus non-structural proteins cause resistance to favipiravir (T-705), a broad-spectrum antiviral. J. Antimicrob. Chemother. 2014, 69, 2770–2784. [Google Scholar] [CrossRef]
- Naesens, L.; Guddat, L.W.; Keough, D.T.; van Kuilenburg, A.B.; Meijer, J.; Vande Voorde, J.; Balzarini, J. Role of human hypoxanthine guanine phosphoribosyltransferase in activation of the antiviral agent T-705 (favipiravir). Mol. Pharmacol. 2013, 84, 615–629. [Google Scholar] [CrossRef]
- Sangawa, H.; Komeno, T.; Nishikawa, H.; Yoshida, A.; Takahashi, K.; Nomura, N.; Furuta, Y. Mechanism of action of T-705 ribosyl triphosphate against influenza virus RNA polymerase. Antimicrob. Agents Chemother. 2013, 57, 5202–5208. [Google Scholar] [CrossRef] [PubMed]
- Arias, A.; Thorne, L.; Goodfellow, I. Favipiravir elicits antiviral mutagenesis during virus replication in vivo. eLife 2014, 3, e03679. [Google Scholar] [CrossRef] [PubMed]
- Baranovich, T.; Wong, S.S.; Armstrong, J.; Marjuki, H.; Webby, R.J.; Webster, R.G.; Govorkova, E.A. T-705 (favipiravir) induces lethal mutagenesis in influenza A H1N1 viruses in vitro. J. Virol. 2013, 87, 3741–3751. [Google Scholar] [CrossRef]
- Abdelnabi, R.; Morais, A.T.S.; Leyssen, P.; Imbert, I.; Beaucourt, S.; Blanc, H.; Froeyen, M.; Vignuzzi, M.; Canard, B.; Neyts, J.; et al. Understanding the mechanism of the broad-spectrum antiviral activity of favipiravir (T-705): Key role of the F1 motif of the viral polymerase. J. Virol. 2017, 91, e00487-17. [Google Scholar] [CrossRef]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef]
- Larsson, A. AliView: A fast and lightweight alignment viewer and editor for large datasets. Bioinformatics 2014, 30, 3276–3278. [Google Scholar] [CrossRef]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Neogi, U.; Hill, K.J.; Ambikan, A.T.; Heng, X.; Quinn, T.P.; Byrareddy, S.N.; Sönnerborg, A.; Sarafianos, S.G.; Singh, K. Feasibility of Known RNA Polymerase Inhibitors as Anti-SARS-CoV-2 Drugs. Pathogens 2020, 9, 320. https://doi.org/10.3390/pathogens9050320
Neogi U, Hill KJ, Ambikan AT, Heng X, Quinn TP, Byrareddy SN, Sönnerborg A, Sarafianos SG, Singh K. Feasibility of Known RNA Polymerase Inhibitors as Anti-SARS-CoV-2 Drugs. Pathogens. 2020; 9(5):320. https://doi.org/10.3390/pathogens9050320
Chicago/Turabian StyleNeogi, Ujjwal, Kyle J. Hill, Anoop T Ambikan, Xiao Heng, Thomas P. Quinn, Siddappa N. Byrareddy, Anders Sönnerborg, Stefan G. Sarafianos, and Kamal Singh. 2020. "Feasibility of Known RNA Polymerase Inhibitors as Anti-SARS-CoV-2 Drugs" Pathogens 9, no. 5: 320. https://doi.org/10.3390/pathogens9050320
APA StyleNeogi, U., Hill, K. J., Ambikan, A. T., Heng, X., Quinn, T. P., Byrareddy, S. N., Sönnerborg, A., Sarafianos, S. G., & Singh, K. (2020). Feasibility of Known RNA Polymerase Inhibitors as Anti-SARS-CoV-2 Drugs. Pathogens, 9(5), 320. https://doi.org/10.3390/pathogens9050320