Hijacking and Use of Host Kinases by Chlamydiae

Abstract
1. Introduction
2. Host Kinases Important for Chlamydial Entry and Invasion
3. Host Kinases in Nutrient Acquisition by Chlamydia
4. Src Family Kinase Rich Microdomains
5. Phosphorylation of Chlamydia Effectors by Host Kinases
6. Apoptosis Resistance
7. Immunopathology of Chlamydial Infections and Association with Chronic Diseases
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Belland, R.; Ojcius, D.M.; Byrne, G.I. Chlamydia. Nat. Rev. Microbiol. 2004, 2, 530–531. [Google Scholar] [CrossRef] [PubMed]
- Resnikoff, S.; Pascolini, D.; Etya’ale, D.; Kocur, I.; Pararajasegaram, R.; Pokharel, G.P.; Mariotti, S.P. Global data on visual impairment in the year 2002. Bull. World Health Organ. 2004, 82, 844–851. [Google Scholar] [PubMed]
- CDC. Sexually Transmitted Disease Surveillance 2018; Department of Health and Human Services: Atlanta, GA, USA, 2019.
- Zhu, H.; Shen, Z.; Luo, H.; Zhang, W.; Zhu, X. Chlamydia Trachomatis Infection-Associated Risk of Cervical Cancer: A Meta-Analysis. Medicine (Baltimore) 2016, 95, e3077. [Google Scholar] [CrossRef]
- Malhotra, M.; Sood, S.; Mukherjee, A.; Muralidhar, S.; Bala, M. Genital Chlamydia trachomatis: An update. Indian J. Med. Res. 2013, 138, 303–316. [Google Scholar] [PubMed]
- Ngeh, J.; Anand, V.; Gupta, S. Chlamydia pneumoniae and atherosclerosis—What we know and what we don’t. Clin. Microbiol. Infect. 2002, 8, 2–13. [Google Scholar] [CrossRef]
- Campbell, L.A.; Kuo, C.C. Chlamydia pneumoniae—An infectious risk factor for atherosclerosis? Nat. Rev. Microbiol. 2004, 2, 23–32. [Google Scholar] [CrossRef]
- Longbottom, D.; Coulter, L.J. Animal chlamydioses and zoonotic implications. J. Comp. Pathol. 2003, 128, 217–244. [Google Scholar] [CrossRef]
- Barron, A.L.; White, H.J.; Rank, R.G.; Soloff, B.L.; Moses, E.B. A new animal model for the study of Chlamydia trachomatis genital infections: Infection of mice with the agent of mouse pneumonitis. J. Infect. Dis. 1981, 143, 63–66. [Google Scholar] [CrossRef]
- Knittler, M.R.; Sachse, K. Chlamydia psittaci: Update on an underestimated zoonotic agent. Pathog. Dis. 2015, 73, 1–15. [Google Scholar] [CrossRef]
- Ramakers, B.P.; Heijne, M.; Lie, N.; Le, T.N.; van Vliet, M.; Claessen, V.P.J.; Tolsma, P.J.P.; De Rosa, M.; Roest, H.I.J.; Vanrompay, D.; et al. Zoonotic Chlamydia caviae Presenting as Community-Acquired Pneumonia. N. Engl. J. Med. 2017, 377, 992–994. [Google Scholar] [CrossRef]
- van Grootveld, R.; Bilsen, M.P.; Boelsums, T.L.; Heddema, E.R.; Groeneveld, G.H.; Gooskens, J.; de Boer, M.G.J. Chlamydia caviae Causing Community-Acquired Pneumonia: An Emerging Zoonosis. Vector Borne Zoonotic Dis. 2018, 18, 635–637. [Google Scholar] [CrossRef] [PubMed]
- Abdelrahman, Y.M.; Belland, R.J. The chlamydial developmental cycle. FEMS Microbiol. Rev. 2005, 29, 949–959. [Google Scholar] [CrossRef] [PubMed]
- Moulder, J.W. Interaction of chlamydiae and host cells in vitro. Microbiol. Rev. 1991, 55, 143–190. [Google Scholar] [CrossRef] [PubMed]
- Fields, K.A.; Hackstadt, T. The chlamydial inclusion: Escape from the endocytic pathway. Annu. Rev. Cell Dev. Biol. 2002, 18, 221–245. [Google Scholar] [CrossRef] [PubMed]
- Hybiske, K.; Stephens, R.S. Mechanisms of host cell exit by the intracellular bacterium Chlamydia. Proc. Natl. Acad. Sci. USA 2007, 104, 11430–11435. [Google Scholar] [CrossRef] [PubMed]
- Clifton, D.R.; Fields, K.A.; Grieshaber, S.S.; Dooley, C.A.; Fischer, E.R.; Mead, D.J.; Carabeo, R.A.; Hackstadt, T. A chlamydial type III translocated protein is tyrosine-phosphorylated at the site of entry and associated with recruitment of actin. Proc. Natl. Acad. Sci. USA 2004, 101, 10166–10171. [Google Scholar] [CrossRef]
- Elwell, C.A.; Ceesay, A.; Kim, J.H.; Kalman, D.; Engel, J.N. RNA interference screen identifies Abl kinase and PDGFR signaling in Chlamydia trachomatis entry. PLoS Pathog. 2008, 4, e1000021. [Google Scholar] [CrossRef]
- Mehlitz, A.; Banhart, S.; Hess, S.; Selbach, M.; Meyer, T.F. Complex kinase requirements for Chlamydia trachomatis Tarp phosphorylation. FEMS Microbiol. Lett. 2008, 289, 233–240. [Google Scholar] [CrossRef]
- Jewett, T.J.; Dooley, C.A.; Mead, D.J.; Hackstadt, T. Chlamydia trachomatis tarp is phosphorylated by src family tyrosine kinases. Biochem. Biophys. Res. Commun. 2008, 371, 339–344. [Google Scholar] [CrossRef]
- Carpenter, V.; Chen, Y.S.; Dolat, L.; Valdivia, R.H. The Effector TepP Mediates Recruitment and Activation of Phosphoinositide 3-Kinase on Early Chlamydia trachomatis Vacuoles. mSphere 2017, 2. [Google Scholar] [CrossRef]
- Chen, Y.S.; Bastidas, R.J.; Saka, H.A.; Carpenter, V.K.; Richards, K.L.; Plano, G.V.; Valdivia, R.H. The Chlamydia trachomatis type III secretion chaperone Slc1 engages multiple early effectors, including TepP, a tyrosine-phosphorylated protein required for the recruitment of CrkI-II to nascent inclusions and innate immune signaling. PLoS Pathog. 2014, 10, e1003954. [Google Scholar] [CrossRef] [PubMed]
- Coombes, B.K.; Mahony, J.B. Identification of MEK- and phosphoinositide 3-kinase-dependent signalling as essential events during Chlamydia pneumoniae invasion of HEp2 cells. Cell. Microbiol. 2002, 4, 447–460. [Google Scholar] [CrossRef] [PubMed]
- Thwaites, T.; Nogueira, A.T.; Campeotto, I.; Silva, A.P.; Grieshaber, S.S.; Carabeo, R.A. The Chlamydia effector TarP mimics the mammalian leucine-aspartic acid motif of paxillin to subvert the focal adhesion kinase during invasion. J. Biol. Chem. 2014, 289, 30426–30442. [Google Scholar] [CrossRef] [PubMed]
- Mital, J.; Hackstadt, T. Diverse requirements for SRC-family tyrosine kinases distinguish chlamydial species. mBio 2011, 2. [Google Scholar] [CrossRef]
- Mital, J.; Hackstadt, T. Role for the SRC family kinase Fyn in sphingolipid acquisition by chlamydiae. Infect. Immun. 2011, 79, 4559–4568. [Google Scholar] [CrossRef] [PubMed]
- Mital, J.; Miller, N.J.; Fischer, E.R.; Hackstadt, T. Specific chlamydial inclusion membrane proteins associate with active Src family kinases in microdomains that interact with the host microtubule network. Cell. Microbiol. 2010, 12, 1235–1249. [Google Scholar] [CrossRef] [PubMed]
- Lutter, E.I.; Barger, A.C.; Nair, V.; Hackstadt, T. Chlamydia trachomatis inclusion membrane protein CT228 recruits elements of the myosin phosphatase pathway to regulate release mechanisms. Cell Rep. 2013, 3, 1921–1931. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, P.H.; Lutter, E.I.; Hackstadt, T. Chlamydia trachomatis inclusion membrane protein MrcA interacts with the inositol 1,4,5-trisphosphate receptor type 3 (ITPR3) to regulate extrusion formation. PLoS Pathog. 2018, 14, e1006911. [Google Scholar] [CrossRef]
- Shaw, J.H.; Key, C.E.; Snider, T.A.; Sah, P.; Shaw, E.I.; Fisher, D.J.; Lutter, E.I. Genetic Inactivation of Chlamydia trachomatis Inclusion Membrane Protein CT228 Alters MYPT1 Recruitment, Extrusion Production, and Longevity of Infection. Front. Cell. Infect. Microbiol. 2018, 8, 415. [Google Scholar] [CrossRef]
- Kim, J.H.; Jiang, S.; Elwell, C.A.; Engel, J.N. Chlamydia trachomatis co-opts the FGF2 signaling pathway to enhance infection. PLoS Pathog. 2011, 7, e1002285. [Google Scholar] [CrossRef]
- Molleken, K.; Becker, E.; Hegemann, J.H. The Chlamydia pneumoniae invasin protein Pmp21 recruits the EGF receptor for host cell entry. PLoS Pathog. 2013, 9, e1003325. [Google Scholar] [CrossRef] [PubMed]
- Patel, A.L.; Chen, X.; Wood, S.T.; Stuart, E.S.; Arcaro, K.F.; Molina, D.P.; Petrovic, S.; Furdui, C.M.; Tsang, A.W. Activation of epidermal growth factor receptor is required for Chlamydia trachomatis development. BMC Microbiol. 2014, 14, 277. [Google Scholar] [CrossRef] [PubMed]
- Subbarayal, P.; Karunakaran, K.; Winkler, A.C.; Rother, M.; Gonzalez, E.; Meyer, T.F.; Rudel, T. EphrinA2 receptor (EphA2) is an invasion and intracellular signaling receptor for Chlamydia trachomatis. PLoS Pathog. 2015, 11, e1004846. [Google Scholar] [CrossRef] [PubMed]
- Moorhead, A.M.; Jung, J.Y.; Smirnov, A.; Kaufer, S.; Scidmore, M.A. Multiple host proteins that function in phosphatidylinositol-4-phosphate metabolism are recruited to the chlamydial inclusion. Infect. Immun. 2010, 78, 1990–2007. [Google Scholar] [CrossRef] [PubMed]
- Rajalingam, K.; Sharma, M.; Lohmann, C.; Oswald, M.; Thieck, O.; Froelich, C.J.; Rudel, T. Mcl-1 is a key regulator of apoptosis resistance in Chlamydia trachomatis-infected cells. PLoS ONE 2008, 3, e3102. [Google Scholar] [CrossRef] [PubMed]
- Siegl, C.; Prusty, B.K.; Karunakaran, K.; Wischhusen, J.; Rudel, T. Tumor suppressor p53 alters host cell metabolism to limit Chlamydia trachomatis infection. Cell Rep. 2014, 9, 918–929. [Google Scholar] [CrossRef]
- Verbeke, P.; Welter-Stahl, L.; Ying, S.; Hansen, J.; Hacker, G.; Darville, T.; Ojcius, D.M. Recruitment of BAD by the Chlamydia trachomatis vacuole correlates with host-cell survival. PLoS Pathog. 2006, 2, e45. [Google Scholar] [CrossRef]
- Lane, B.J.; Mutchler, C.; Al Khodor, S.; Grieshaber, S.S.; Carabeo, R.A. Chlamydial entry involves TARP binding of guanine nucleotide exchange factors. PLoS Pathog. 2008, 4, e1000014. [Google Scholar] [CrossRef]
- Gurumurthy, R.K.; Maurer, A.P.; Machuy, N.; Hess, S.; Pleissner, K.P.; Schuchhardt, J.; Rudel, T.; Meyer, T.F. A loss-of-function screen reveals Ras- and Raf-independent MEK-ERK signaling during Chlamydia trachomatis infection. Sci. Signal. 2010, 3, ra21. [Google Scholar] [CrossRef]
- Capmany, A.; Gambarte Tudela, J.; Alonso Bivou, M.; Damiani, M.T. Akt/AS160 Signaling Pathway Inhibition Impairs Infection by Decreasing Rab14-Controlled Sphingolipids Delivery to Chlamydial Inclusions. Front. Microbiol. 2019, 10, 666. [Google Scholar] [CrossRef]
- Al-Zeer, M.A.; Xavier, A.; Abu Lubad, M.; Sigulla, J.; Kessler, M.; Hurwitz, R.; Meyer, T.F. Chlamydia trachomatis Prevents Apoptosis Via Activation of PDPK1-MYC and Enhanced Mitochondrial Binding of Hexokinase II. EBioMedicine 2017, 23, 100–110. [Google Scholar] [CrossRef] [PubMed]
- Flores, R.; Zhong, G. The Chlamydia pneumoniae Inclusion Membrane Protein Cpn1027 Interacts with Host Cell Wnt Signaling Pathway Regulator Cytoplasmic Activation/Proliferation-Associated Protein 2 (Caprin2). PLoS ONE 2015, 10, e0127909. [Google Scholar] [CrossRef] [PubMed]
- Sah, P.; Nelson, N.H.; Shaw, J.H.; Lutter, E.I. Chlamydia trachomatis recruits protein kinase C during infection. Pathog. Dis. 2019, 77, ftz061. [Google Scholar] [CrossRef]
- Tse, S.M.; Mason, D.; Botelho, R.J.; Chiu, B.; Reyland, M.; Hanada, K.; Inman, R.D.; Grinstein, S. Accumulation of diacylglycerol in the Chlamydia inclusion vacuole: Possible role in the inhibition of host cell apoptosis. J. Biol. Chem. 2005, 280, 25210–25215. [Google Scholar] [CrossRef] [PubMed]
- Shivshankar, P.; Lei, L.; Wang, J.; Zhong, G. Rottlerin inhibits chlamydial intracellular growth and blocks chlamydial acquisition of sphingolipids from host cells. Appl. Environ. Microbiol. 2008, 74, 1243–1249. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Elwell, C.A.; Engel, J.N. Lipid acquisition by intracellular Chlamydiae. Cell. Microbiol. 2012, 14, 1010–1018. [Google Scholar] [CrossRef]
- Su, H.; McClarty, G.; Dong, F.; Hatch, G.M.; Pan, Z.K.; Zhong, G. Activation of Raf/MEK/ERK/cPLA2 signaling pathway is essential for chlamydial acquisition of host glycerophospholipids. J. Biol. Chem. 2004, 279, 9409–9416. [Google Scholar] [CrossRef]
- Buchholz, K.R.; Stephens, R.S. The extracellular signal-regulated kinase/mitogen-activated protein kinase pathway induces the inflammatory factor interleukin-8 following Chlamydia trachomatis infection. Infect. Immun. 2007, 75, 5924–5929. [Google Scholar] [CrossRef]
- Chen, F.; Cheng, W.; Zhang, S.; Zhong, G.; Yu, P. Induction of IL-8 by Chlamydia trachomatis through MAPK pathway rather than NF-kappaB pathway. Zhong Nan Da Xue Xue Bao Yi Xue Ban 2010, 35, 307–313. [Google Scholar]
- Chumduri, C.; Gurumurthy, R.K.; Zadora, P.K.; Mi, Y.; Meyer, T.F. Chlamydia infection promotes host DNA damage and proliferation but impairs the DNA damage response. Cell Host Microbe 2013, 13, 746–758. [Google Scholar] [CrossRef]
- Du, K.; Zhou, M.; Li, Q.; Liu, X.Z. Chlamydia trachomatis inhibits the production of pro-inflammatory cytokines in human PBMCs through induction of IL-10. J. Med. Microbiol. 2018, 67, 240–248. [Google Scholar] [CrossRef] [PubMed]
- Kun, D.; Xiang-Lin, C.; Ming, Z.; Qi, L. Chlamydia inhibit host cell apoptosis by inducing Bag-1 via the MAPK/ERK survival pathway. Apoptosis 2013, 18, 1083–1092. [Google Scholar] [CrossRef] [PubMed]
- Zadora, P.K.; Chumduri, C.; Imami, K.; Berger, H.; Mi, Y.; Selbach, M.; Meyer, T.F.; Gurumurthy, R.K. Integrated Phosphoproteome and Transcriptome Analysis Reveals Chlamydia-Induced Epithelial-to-Mesenchymal Transition in Host Cells. Cell Rep. 2019, 26, 1286–1302.e8. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.; Wang, F.; Lu, Z.; Ying, H.; Zhang, H.; Ding, W.; Wang, C.; Shi, L. MAPK kinase 3 potentiates Chlamydia HSP60-induced inflammatory response through distinct activation of NF-kappaB. J. Immunol. 2013, 191, 386–394. [Google Scholar] [CrossRef]
- Rupp, J.; Hellwig-Burgel, T.; Wobbe, V.; Seitzer, U.; Brandt, E.; Maass, M. Chlamydia pneumoniae infection promotes a proliferative phenotype in the vasculature through Egr-1 activation in vitro and in vivo. Proc. Natl. Acad. Sci. USA 2005, 102, 3447–3452. [Google Scholar] [CrossRef] [PubMed]
- Sasu, S.; LaVerda, D.; Qureshi, N.; Golenbock, D.T.; Beasley, D. Chlamydia pneumoniae and chlamydial heat shock protein 60 stimulate proliferation of human vascular smooth muscle cells via toll-like receptor 4 and p44/p42 mitogen-activated protein kinase activation. Circ. Res. 2001, 89, 244–250. [Google Scholar] [CrossRef]
- Du, K.; Zheng, Q.; Zhou, M.; Zhu, L.; Ai, B.; Zhou, L. Chlamydial antiapoptotic activity involves activation of the Raf/MEK/ERK survival pathway. Curr. Microbiol. 2011, 63, 341–346. [Google Scholar] [CrossRef]
- Yang, S.; Traore, Y.; Jimenez, C.; Ho, E.A. Autophagy induction and PDGFR-beta knockdown by siRNA-encapsulated nanoparticles reduce chlamydia trachomatis infection. Sci. Rep. 2019, 9, 1306. [Google Scholar] [CrossRef]
- Carabeo, R.A.; Dooley, C.A.; Grieshaber, S.S.; Hackstadt, T. Rac interacts with Abi-1 and WAVE2 to promote an Arp2/3-dependent actin recruitment during chlamydial invasion. Cell. Microbiol. 2007, 9, 2278–2288. [Google Scholar] [CrossRef]
- Carabeo, R.A.; Grieshaber, S.S.; Hasenkrug, A.; Dooley, C.; Hackstadt, T. Requirement for the Rac GTPase in Chlamydia trachomatis invasion of non-phagocytic cells. Traffic 2004, 5, 418–425. [Google Scholar] [CrossRef]
- Subtil, A.; Wyplosz, B.; Balana, M.E.; Dautry-Varsat, A. Analysis of Chlamydia caviae entry sites and involvement of Cdc42 and Rac activity. J. Cell Sci. 2004, 17 Pt 117, 3923–3933. [Google Scholar] [CrossRef][Green Version]
- Thwaites, T.R.; Pedrosa, A.T.; Peacock, T.P.; Carabeo, R.A. Vinculin Interacts with the Chlamydia Effector TarP Via a Tripartite Vinculin Binding Domain to Mediate Actin Recruitment and Assembly at the Plasma Membrane. Front. Cell. Infect. Microbiol. 2015, 5, 88. [Google Scholar] [CrossRef] [PubMed]
- Clifton, D.R.; Dooley, C.A.; Grieshaber, S.S.; Carabeo, R.A.; Fields, K.A.; Hackstadt, T. Tyrosine phosphorylation of the chlamydial effector protein Tarp is species specific and not required for recruitment of actin. Infect. Immun. 2005, 73, 3860–3868. [Google Scholar] [CrossRef] [PubMed]
- Stephens, R.S.; Kalman, S.; Lammel, C.; Fan, J.; Marathe, R.; Aravind, L.; Mitchell, W.; Olinger, L.; Tatusov, R.L.; Zhao, Q.; et al. Genome sequence of an obligate intracellular pathogen of humans: Chlamydia trachomatis. Science 1998, 282, 754–759. [Google Scholar] [CrossRef] [PubMed]
- Robertson, D.K.; Gu, L.; Rowe, R.K.; Beatty, W.L. Inclusion biogenesis and reactivation of persistent Chlamydia trachomatis requires host cell sphingolipid biosynthesis. PLoS Pathog. 2009, 5, e1000664. [Google Scholar] [CrossRef] [PubMed]
- Van Ooij, C.; Kalman, L.; Van Ijzendoorn, S.; Nishijima, M.; Hanada, K.; Mostov, K.; Engel, J.N. Host cell-derived sphingolipids are required for the intracellular growth of Chlamydia trachomatis. Cell. Microbiol. 2000, 2, 627–637. [Google Scholar] [CrossRef] [PubMed]
- Wylie, J.L.; Hatch, G.M.; McClarty, G. Host cell phospholipids are trafficked to and then modified by Chlamydia trachomatis. J. Bacteriol. 1997, 179, 7233–7242. [Google Scholar] [CrossRef]
- Wu-Zhang, A.X.; Newton, A.C. Protein kinase C pharmacology: Refining the toolbox. Biochem. J. 2013, 452, 195–209. [Google Scholar] [CrossRef]
- Derre, I.; Swiss, R.; Agaisse, H. The lipid transfer protein CERT interacts with the Chlamydia inclusion protein IncD and participates to ER-Chlamydia inclusion membrane contact sites. PLoS Pathog. 2011, 7, e1002092. [Google Scholar] [CrossRef]
- Elwell, C.A.; Jiang, S.; Kim, J.H.; Lee, A.; Wittmann, T.; Hanada, K.; Melancon, P.; Engel, J.N. Chlamydia trachomatis co-opts GBF1 and CERT to acquire host sphingomyelin for distinct roles during intracellular development. PLoS Pathog. 2011, 7, e1002198. [Google Scholar] [CrossRef]
- Weber, M.M.; Bauler, L.D.; Lam, J.; Hackstadt, T. Expression and localization of predicted inclusion membrane proteins in Chlamydia trachomatis. Infect. Immun. 2015, 83, 4710–4718. [Google Scholar] [CrossRef] [PubMed]
- Betts-Hampikian, H.J.; Fields, K.A. The Chlamydial Type III Secretion Mechanism: Revealing Cracks in a Tough Nut. Front. Microbiol. 2010, 1, 114. [Google Scholar] [CrossRef] [PubMed]
- Caven, L.; Carabeo, R.A. Pathogenic Puppetry: Manipulation of the Host Actin Cytoskeleton by Chlamydia trachomatis. Int. J. Mol. Sci. 2019, 21, 90. [Google Scholar] [CrossRef] [PubMed]
- Lutter, E.I.; Bonner, C.; Holland, M.J.; Suchland, R.J.; Stamm, W.E.; Jewett, T.J.; McClarty, G.; Hackstadt, T. Phylogenetic analysis of Chlamydia trachomatis Tarp and correlation with clinical phenotype. Infect. Immun. 2010, 78, 3678–3688. [Google Scholar] [CrossRef] [PubMed]
- Dehoux, P.; Flores, R.; Dauga, C.; Zhong, G.; Subtil, A. Multi-genome identification and characterization of chlamydiae-specific type III secretion substrates: The Inc proteins. BMC Genom. 2011, 12, 109. [Google Scholar] [CrossRef]
- Bannantine, J.P.; Griffiths, R.S.; Viratyosin, W.; Brown, W.J.; Rockey, D.D. A secondary structure motif predictive of protein localization to the chlamydial inclusion membrane. Cell. Microbiol. 2000, 2, 35–47. [Google Scholar] [CrossRef]
- Rockey, D.D.; Scidmore, M.A.; Bannantine, J.P.; Brown, W.J. Proteins in the chlamydial inclusion membrane. Microbes Infect. 2002, 4, 333–340. [Google Scholar] [CrossRef]
- Lutter, E.I.; Martens, C.; Hackstadt, T. Evolution and conservation of predicted inclusion membrane proteins in chlamydiae. Comp. Funct. Genom. 2012, 2012, 362104. [Google Scholar] [CrossRef]
- Rockey, D.D.; Grosenbach, D.; Hruby, D.E.; Peacock, M.G.; Heinzen, R.A.; Hackstadt, T. Chlamydia psittaci IncA is phosphorylated by the host cell and is exposed on the cytoplasmic face of the developing inclusion. Mol. Microbiol. 1997, 24, 217–228. [Google Scholar] [CrossRef]
- Rockey, D.D.; Heinzen, R.A.; Hackstadt, T. Cloning and characterization of a Chlamydia psittaci gene coding for a protein localized in the inclusion membrane of infected cells. Mol. Microbiol. 1995, 15, 617–626. [Google Scholar] [CrossRef]
- Scidmore, M.A.; Hackstadt, T. Mammalian 14-3-3beta associates with the Chlamydia trachomatis inclusion membrane via its interaction with IncG. Mol. Microbiol. 2001, 39, 1638–1650. [Google Scholar] [CrossRef] [PubMed]
- Aeberhard, L.; Banhart, S.; Fischer, M.; Jehmlich, N.; Rose, L.; Koch, S.; Laue, M.; Renard, B.Y.; Schmidt, F.; Heuer, D. The Proteome of the Isolated Chlamydia trachomatis Containing Vacuole Reveals a Complex Trafficking Platform Enriched for Retromer Components. PLoS Pathog. 2015, 11, e1004883. [Google Scholar] [CrossRef] [PubMed]
- Mirrashidi, K.M.; Elwell, C.A.; Verschueren, E.; Johnson, J.R.; Frando, A.; Von Dollen, J.; Rosenberg, O.; Gulbahce, N.; Jang, G.; Johnson, T.; et al. Global Mapping of the Inc-Human Interactome Reveals that Retromer Restricts Chlamydia Infection. Cell Host Microbe 2015, 18, 109–121. [Google Scholar] [CrossRef] [PubMed]
- Bastidas, R.J.; Elwell, C.A.; Engel, J.N.; Valdivia, R.H. Chlamydial intracellular survival strategies. Cold Spring Harb. Perspect. Med. 2013, 3, a010256. [Google Scholar] [CrossRef] [PubMed]
- Elwell, C.; Mirrashidi, K.; Engel, J. Chlamydia cell biology and pathogenesis. Nat. Rev. Microbiol. 2016, 14, 385–400. [Google Scholar] [CrossRef]
- Mehlitz, A.; Banhart, S.; Maurer, A.P.; Kaushansky, A.; Gordus, A.G.; Zielecki, J.; Macbeath, G.; Meyer, T.F. Tarp regulates early Chlamydia-induced host cell survival through interactions with the human adaptor protein SHC1. J. Cell Biol. 2010, 190, 143–157. [Google Scholar] [CrossRef]
- Kintner, J.; Moore, C.G.; Whittimore, J.D.; Butler, M.; Hall, J.V. Inhibition of Wnt Signaling Pathways Impairs Chlamydia trachomatis Infection in Endometrial Epithelial Cells. Front. Cell. Infect. Microbiol. 2017, 7, 501. [Google Scholar] [CrossRef]
- Stephens, R.S. The cellular paradigm of chlamydial pathogenesis. Trends Microbiol. 2003, 11, 44–51. [Google Scholar] [CrossRef]
- Igietseme, J.U.; Omosun, Y.; Stuchlik, O.; Reed, M.S.; Partin, J.; He, Q.; Joseph, K.; Ellerson, D.; Bollweg, B.; George, Z.; et al. Role of Epithelial-Mesenchyme Transition in Chlamydia Pathogenesis. PLoS ONE 2015, 10, e0145198. [Google Scholar] [CrossRef]
- Krull, M.; Klucken, A.C.; Wuppermann, F.N.; Fuhrmann, O.; Magerl, C.; Seybold, J.; Hippenstiel, S.; Hegemann, J.H.; Jantos, C.A.; Suttorp, N. Signal transduction pathways activated in endothelial cells following infection with Chlamydia pneumoniae. J. Immunol. 1999, 162, 4834–4841. [Google Scholar]
- Kol, A.; Bourcier, T.; Lichtman, A.H.; Libby, P. Chlamydial and human heat shock protein 60s activate human vascular endothelium, smooth muscle cells, and macrophages. J. Clin. Investig. 1999, 103, 571–577. [Google Scholar] [CrossRef] [PubMed]
- Bulut, Y.; Faure, E.; Thomas, L.; Karahashi, H.; Michelsen, K.S.; Equils, O.; Morrison, S.G.; Morrison, R.P.; Arditi, M. Chlamydial heat shock protein 60 activates macrophages and endothelial cells through Toll-like receptor 4 and MD2 in a MyD88-dependent pathway. J. Immunol. 2002, 168, 1435–1440. [Google Scholar] [CrossRef] [PubMed]



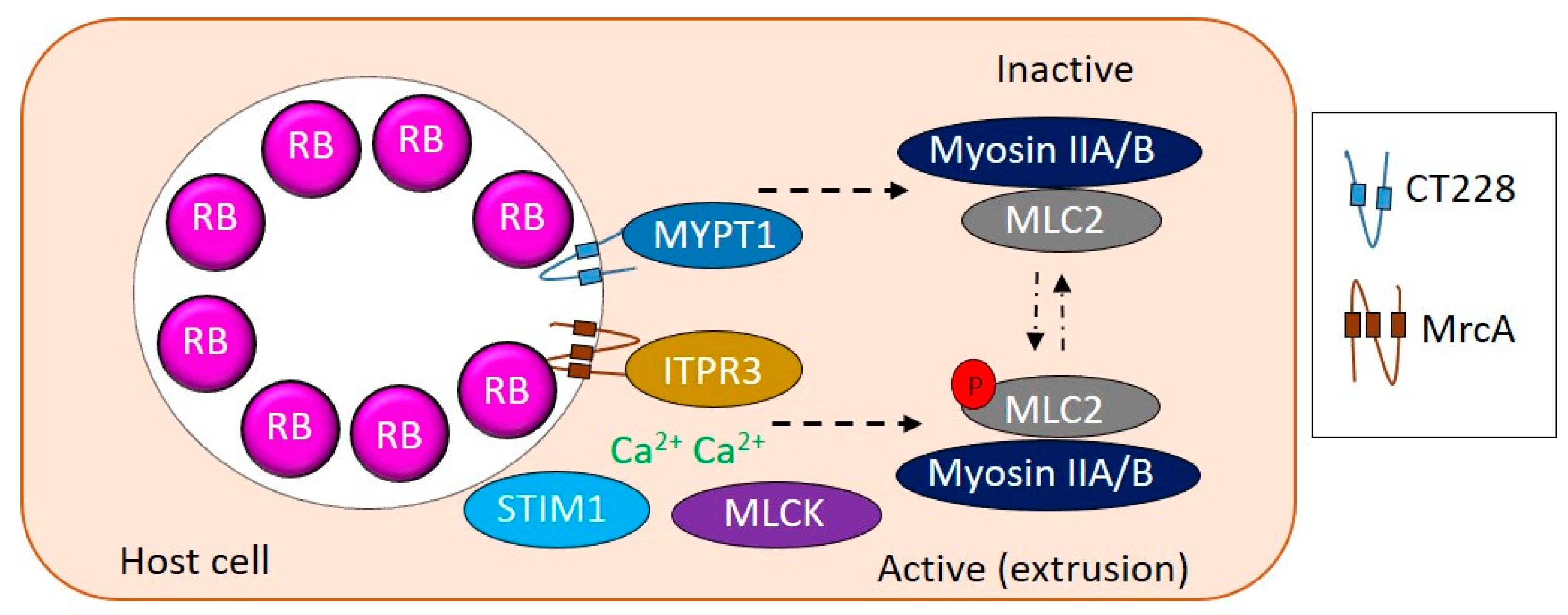{kind=link}
{kind=link}
| Host Kinase | Implicated Roles in Chlamydial Infection | References |
|---|---|---|
| Src, Abl, Syk | Phosphorylation T3SS effectors TarP and TepP. Important for entry, inclusion development and regulation of innate immune response | [17,18,19,20,21,22] |
| FAK | Recruited by TarP Actin remodeling during entry | [23,24] |
| SFKs (Src, Fyn) | Microdomain localization Microtubule dependent trafficking of inclusion to MTOC Intracellular development Lipid acquisition | [25,26,27] |
| MLCK | Microdomain localization with other elements of Myosin pathway Regulation of extrusion | [28,29,30] |
| RTKs (PDGFR, FGFR, EGFR, EPHA2) | Chlamydial entry and invasion F-actin assembly around inclusion Host cell survival | [18,31,32,33,34] |
| PI4KIIα | Generation of a pool of PI4P around inclusion Important for infectious progeny and inclusion formation | [35] |
| PI3K/AKT | Invasion Apoptosis resistance Chlamydial replication Dampening of innate immune response Sphingolipid acquisition Reduction of tumor suppressor protein p53 | [21,34,36,37,38,39,40,41] |
| PDPK1 | Apoptosis resistance | [42] |
| GSK3β | Interacts with Cpn1027 Recruitment by TepP | [21,43] |
| PKC | Resistance to apoptosis Microdomain localization Potential role in lipid acquisition | [44,45,46,47] |
| MEK/ERK | Lipid acquisition Bacterial replication Apoptosis resistance cytokine IL-8, IL-10 induction Chlamydia induced epithelial-to-mesenchymal transition Proliferation of vasculature smooth muscle cells | [18,23,31,32,36,40,48,49,50,51,52,53,54,55,56,57,58] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sah, P.; Lutter, E.I. Hijacking and Use of Host Kinases by Chlamydiae. Pathogens 2020, 9, 1034. https://doi.org/10.3390/pathogens9121034
Sah P, Lutter EI. Hijacking and Use of Host Kinases by Chlamydiae. Pathogens. 2020; 9(12):1034. https://doi.org/10.3390/pathogens9121034
Chicago/Turabian StyleSah, Prakash, and Erika I. Lutter. 2020. "Hijacking and Use of Host Kinases by Chlamydiae" Pathogens 9, no. 12: 1034. https://doi.org/10.3390/pathogens9121034
APA StyleSah, P., & Lutter, E. I. (2020). Hijacking and Use of Host Kinases by Chlamydiae. Pathogens, 9(12), 1034. https://doi.org/10.3390/pathogens9121034