Molecular Changes in Dengue Envelope Protein Domain III upon Interaction with Glycosaminoglycans

 ,
,  , , and
, , and
Abstract

1. Introduction
2. Materials and Methods
2.1. Protein Expression from E. coli
2.2. Protein Expression in E. coli Using Matchout Deuterated Minimal Medium
2.3. Protein Refolding and Purification
2.4. Glycosaminoglycan’s (GAGs)
2.5. Circular Dichroism (CD)
2.6. Heparin Affinity Column
2.7. Differential Scanning Fluorimetry (DSF)
2.8. Small Angle X-ray Scattering (SAXS)
2.9. Small Angle Neutron Scattering (SANS)
2.10. Ab Initio Shape Determination
3. Results and Discussion
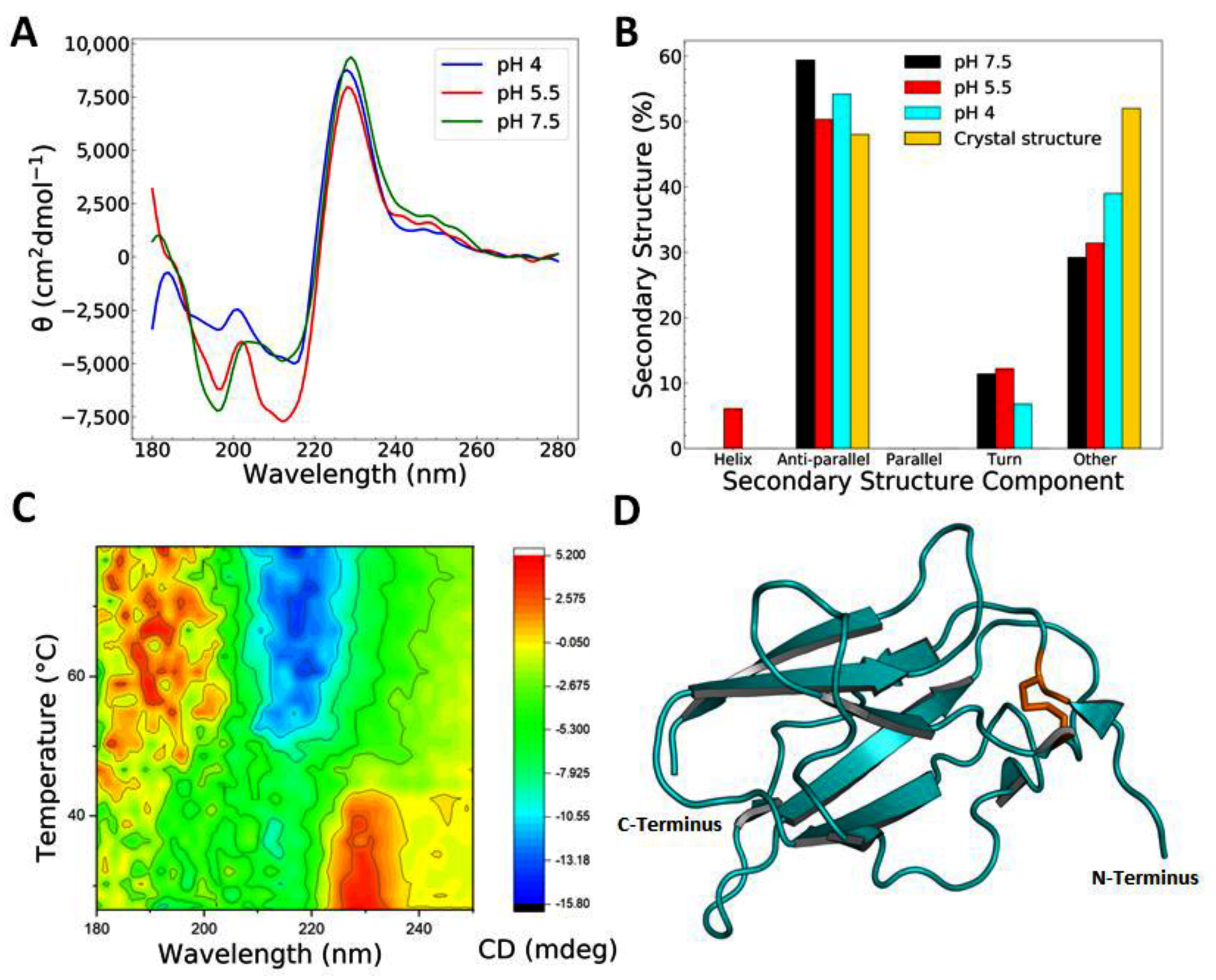
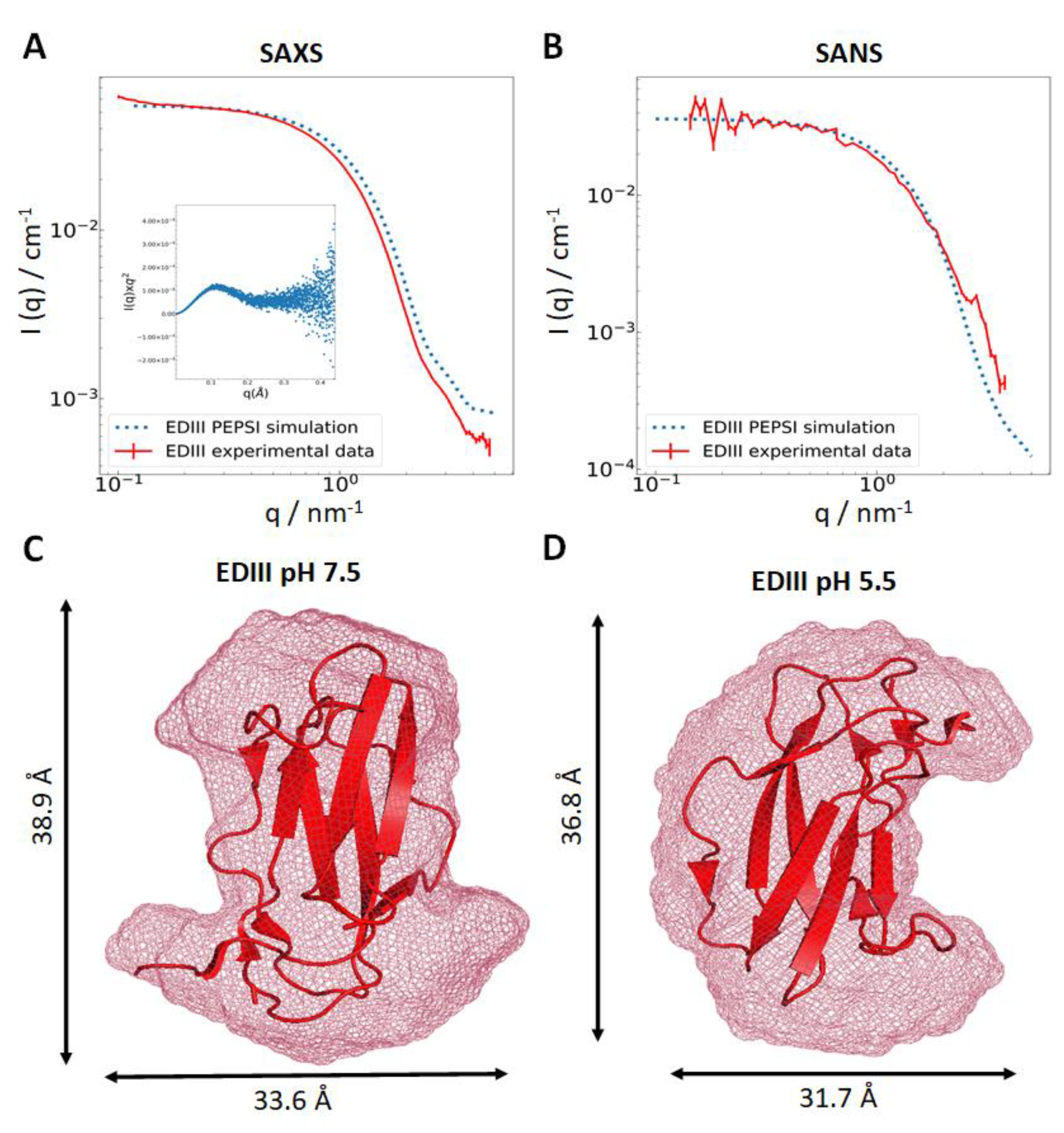
3.1. Recombinant EDIII Is a Monomeric, Globular, and Well-Folded Protein
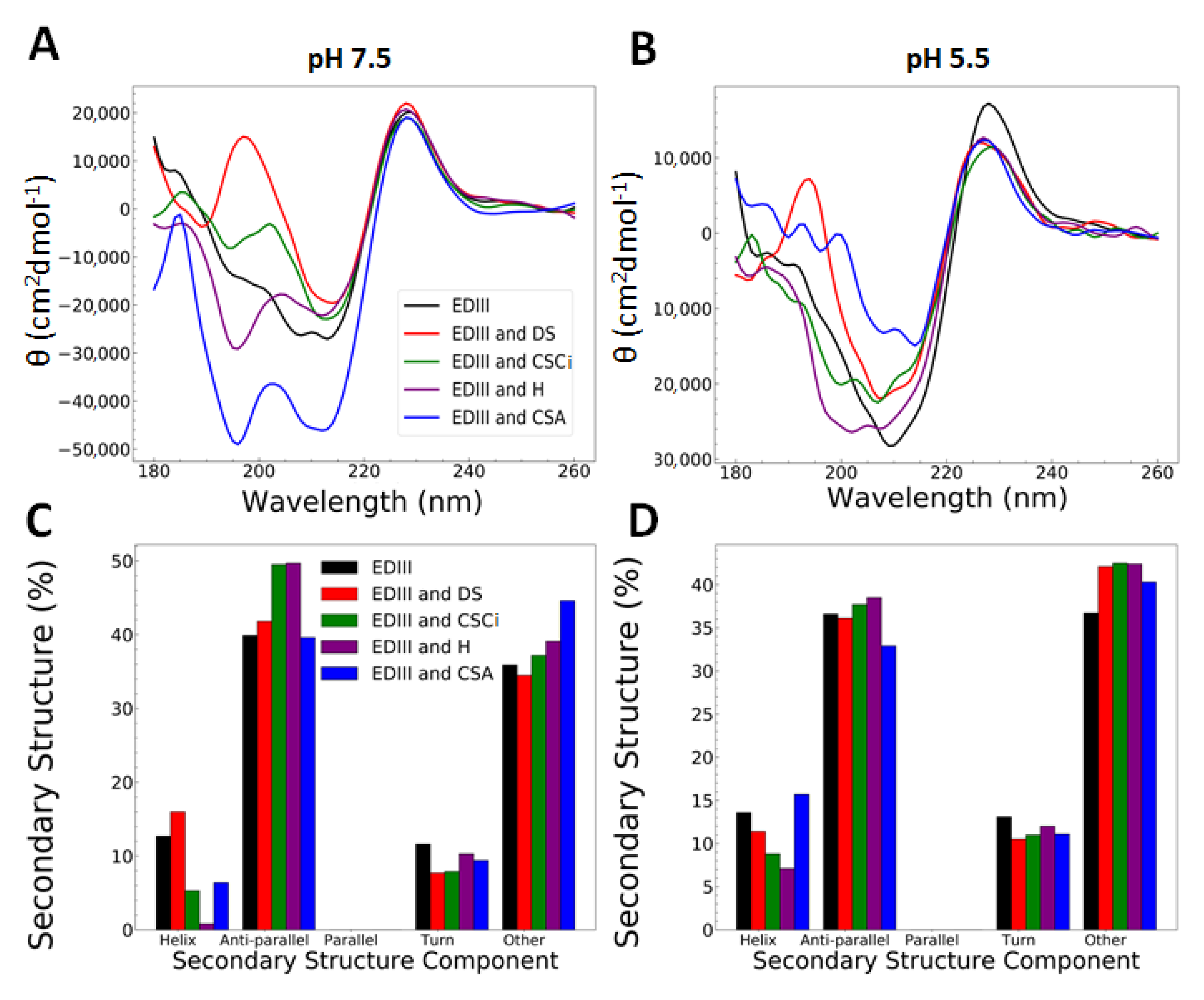
3.2. Interactions with Heparin and Chondroitin Sulphate C Are pH-Dependent and Cause Changes at a Molecular Level
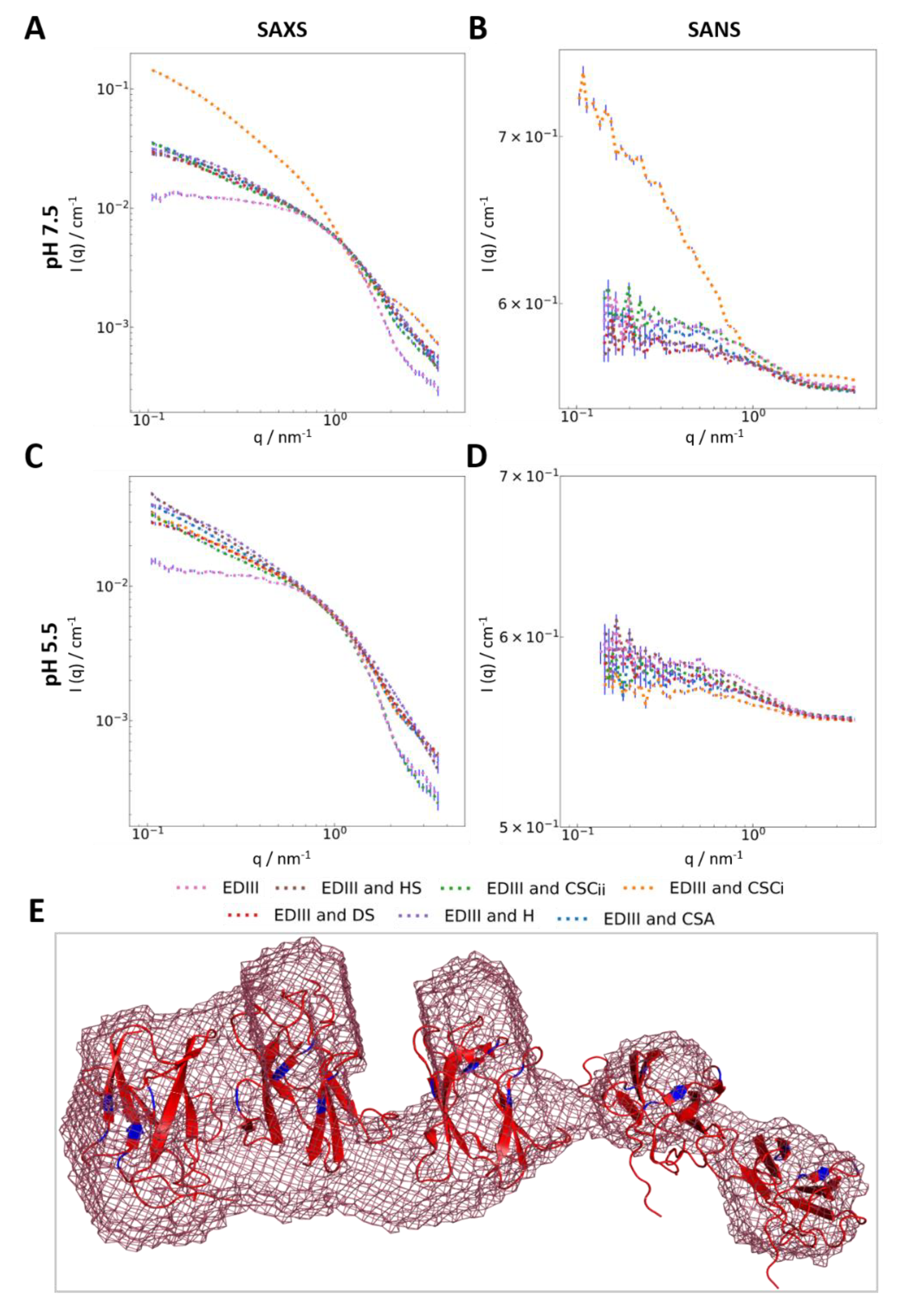
3.3. Interaction with Chondroitin Sulphate C Indicates Higher-Order Molecular Organisation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Brady, O.J.; Gething, P.W.; Bhatt, S.; Messina, J.P.; Brownstein, J.S.; Hoen, A.G.; Moyes, C.L.; Farlow, A.W.; Scott, T.W.; Hay, S.I. Refining the Global Spatial Limits of Dengue Virus Transmission by Evidence-Based Consensus. PLoS Negl. Trop. Dis. 2012, 6, e1760. [Google Scholar] [CrossRef]
- Bhatt, S.; Gething, P.W.; Brady, O.J.; Messina, J.P.; Farlow, A.W.; Moyes, C.L.; Drake, J.M.; Brownstein, J.S.; Hoen, A.G.; Sankoh, O.; et al. The global distribution and burden of dengue. Nature 2013, 496, 504–507. [Google Scholar] [CrossRef]
- Dengue and Severe Dengue. Available online: https://www.who.int/en/news-room/fact-sheets/detail/dengue-and-severe-dengue (accessed on 28 November 2019).
- Germi, R.; Crance, J.M.; Garin, D.; Guimet, J.; Lortat-Jacob, H.; Ruigrok, R.W.H.; Zarski, J.P.; Drouet, E. Heparan sulfate-mediated binding of infectious dengue virus type 2 and yellow fever virus. Virology 2002, 292, 162–168. [Google Scholar] [CrossRef]
- Kozlovskaya, L.I.; Osolodkin, D.I.; Shevtsova, A.S.; Romanova, L.I.; Rogova, Y.V.; Dzhivanian, T.I.; Lyapustin, V.N.; Pivanova, G.P.; Gmyl, A.P.; Palyulin, V.A.; et al. GAG-binding variants of tick-borne encephalitis virus. Virology 2010, 398, 262–272. [Google Scholar] [CrossRef]
- Kim, S.Y.; Zhao, J.; Liu, X.; Fraser, K.; Lin, L.; Zhang, X.; Zhang, F.; Dordick, J.S.; Linhardt, R.J. Interaction of Zika Virus Envelope Protein with Glycosaminoglycans. Biochemistry 2017, 56, 1151–1162. [Google Scholar] [CrossRef] [PubMed]
- Cruz-Oliveira, C.; Freire, J.M.; Conceicao, T.M.; Higa, L.M.; Castanho, M.A.R.B.; Da Poian, A.T. Receptors and routes of dengue virus entry into the host cells. FEMS Microbiol. Rev. 2015, 39, 155–170. [Google Scholar] [CrossRef] [PubMed]
- Modis, Y.; Ogata, S.; Clements, D.; Harrison, S.C. Structure of the dengue virus envelope protein after membrane fusion. Nature 2004, 427, 313–319. [Google Scholar] [CrossRef]
- Rudd, T.R.; Skidmore, M.A.; Guerrini, M.; Hricovini, M.; Powell, A.K.; Siligardi, G.; Yates, E.A. The conformation and structure of GAGs: Recent progress and perspectives. Curr. Opin. Struct. Biol. 2010, 20, 567–574. [Google Scholar] [CrossRef] [PubMed]
- Gandhi, N.S.; Mancera, R.L. The structure of glycosaminoglycans and their interactions with proteins. Chem. Biol. Drug Des. 2008, 72, 455–482. [Google Scholar] [CrossRef] [PubMed]
- Toida, T.; Yoshida, H.; Toyoda, H.; Koshiishi, I.; Imanari, T.; Hileman, R.E.; Fromm, J.R.; Linhardt, R.J. Structural differences and the presence of unsubstituted amino groups in heparan sulphates from different tissues and species. Biochem. J. 1997, 322, 499–506. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Maguire, T.; Hileman, R.E.; Fromm, J.R.; Esko, J.D.; Linhardt, R.J.; Marks, R.M. Dengue virus infectivity depends on envelope protein binding to target cell heparan sulfate. Nat. Med. 1997, 3, 866–871. [Google Scholar] [CrossRef] [PubMed]
- Hilgard, P.; Stockert, R. Heparan sulfate proteoglycans initiate dengue virus infection of hepatocytes. Hepatology 2000, 32, 1069–1077. [Google Scholar] [CrossRef] [PubMed]
- Dalrymple, N.; Mackow, E.R. Productive Dengue Virus Infection of Human Endothelial Cells Is Directed by Heparan Sulfate-Containing Proteoglycan Receptors. J. Virol. 2011, 85, 9478–9485. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.Y.; Li, B.; Linhardt, R.J. Pathogenesis and inhibition of flaviviruses from a carbohydrate perspective. Pharmaceuticals 2017, 10, 44. [Google Scholar] [CrossRef]
- Thaisomboonsuk, B.K.; Clayson, E.T.; Pantuwatana, S.; Vaughn, D.W.; Endy, T.P. Characterization of dengue-2 virus binding to surfaces of mammalian and insect cells. Am. J. Trop. Med. Hyg. 2005, 72, 375–383. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, W.; Ogata, S.; Clements, D.; Strauss, J.H.; Baker, T.S.; Kuhn, R.J.; Rossmann, M.G. Conformational changes of the flavivirus E glycoprotein. Structure 2004, 12, 1607–1618. [Google Scholar] [CrossRef]
- Modis, Y. Relating structure to evolution in class II viral membrane fusion proteins. Curr. Opin. Virol. 2014, 5, 34–41. [Google Scholar] [CrossRef]
- Rey, F.A.; Heinz, F.X.; Mandl, C.; Kunz, C.; Harrison, S.C. The envelope glycoprotein from tick-borne encephalitis virus at 2 Å resolution. Nature 1995, 375, 291–298. [Google Scholar] [CrossRef]
- Nybakken, G.E.; Nelson, C.A.; Chen, B.R.; Diamond, M.S.; Fremont, D.H. Crystal Structure of the West Nile Virus Envelope Glycoprotein. J. Virol. 2006. [Google Scholar] [CrossRef]
- Luca, V.C.; AbiMansour, J.; Nelson, C.A.; Fremont, D.H. Crystal Structure of the Japanese Encephalitis Virus Envelope Protein. J. Virol. 2012. [Google Scholar] [CrossRef]
- Sirohi, D.; Chen, Z.; Sun, L.; Klose, T.; Pierson, T.C.; Rossmann, M.G.; Kuhn, R.J. The 3.8 Å resolution cryo-EM structure of Zika virus. Science 2016, 352, 467–470. [Google Scholar] [CrossRef] [PubMed]
- Ji, G.H.; Deng, Y.Q.; Yu, X.J.; Jiang, T.; Wang, H.J.; Shi, X.; Zhang, D.P.; Li, X.F.; Zhu, S.Y.; Zhao, H.; et al. Characterization of a novel dengue serotype 4 virus-specific neutralizing epitope on the envelope protein domain III. PLoS ONE 2015, 10, e0139741. [Google Scholar] [CrossRef] [PubMed]
- Crill, W.D.; Roehrig, J.T. Monoclonal Antibodies That Bind to Domain III of Dengue Virus E Glycoprotein Are the Most Efficient Blockers of Virus Adsorption to Vero Cells. J. Virol. 2001, 75, 7769–7773. [Google Scholar] [CrossRef] [PubMed]
- Erb, S.M.; Butrapet, S.; Moss, K.J.; Luy, B.E.; Childers, T.; Calvert, A.E.; Silengo, S.J.; Roehrig, J.T.; Huang, C.Y.H.; Blair, C.D. Domain-III FG loop of the dengue virus type 2 envelope protein is important for infection of mammalian cells and Aedes aegypti mosquitoes. Virology 2010, 406, 328–335. [Google Scholar] [CrossRef] [PubMed]
- Haertlein, M.; Moulin, M.; Devos, J.M.; Laux, V.; Dunne, O.; Trevor Forsyth, V. Biomolecular Deuteration for Neutron Structural Biology and Dynamics. In Methods in Enzymology; Academic Press: Cambridge, MA, USA, 2016. [Google Scholar]
- Micsonai, A.; Wien, F.; Bulyaki, E.; Kun, J.; Moussong, E.; Lee, Y.-H.; Goto, Y.; Refregiers, M.; Kardos, J. BeStSel: A web server for accurate protein secondary structure prediction and fold recognition from the circular dichroism spectra. Nucleic Acids Res. 2018, 46, W315–W322. [Google Scholar] [CrossRef] [PubMed]
- Hunter, J.D. Matplotlib: A 2D graphics environment. Comput. Sci. Eng. 2007. [Google Scholar] [CrossRef]
- Grudinin, S.; Garkavenko, M.; Kazennov, A. Pepsi-SAXS: An adaptive method for rapid and accurate computation of small-angle X-ray scattering profiles. Acta Crystallogr. Sect. D Struct. Biol. 2017, D73, 449–464. [Google Scholar] [CrossRef]
- Breßler, I.; Kohlbrecher, J.; Thünemann, A.F. SASfit: A tool for small-angle scattering data analysis using a library of analytical expressions. J. Appl. Crystallogr. 2015, 48, 1587–1598. [Google Scholar] [CrossRef] [PubMed]
- Classen, S.; Hura, G.L.; Holton, J.M.; Rambo, R.P.; Rodic, I.; McGuire, P.J.; Dyer, K.; Hammel, M.; Meigs, G.; Frankel, K.A.; et al. Implementation and performance of SIBYLS: A dual endstation small-angle X-ray scattering and macromolecular crystallography beamline at the Advanced Light Source. J. Appl. Crystallogr. 2013, 46, 1–13. [Google Scholar] [CrossRef]
- Franke, D.; Svergun, D.I. DAMMIF, a program for rapid ab-initio shape determination in small-angle scattering. J. Appl. Crystallogr. 2009, 42, 342–346. [Google Scholar] [CrossRef]
- Svergun, D.I.; Petoukhov, M.V.; Koch, M.H.J. Determination of domain structure of proteins from x-ray solution scattering. Biophys. J. 2001, 80, 2946–2953. [Google Scholar] [CrossRef]
- Grant, T.D. Ab initio electron density determination directly from solution scattering data. Nat. Methods 2018, 15, 191–193. [Google Scholar] [CrossRef] [PubMed]
- Modis, Y.; Ogata, S.; Clements, D.; Harrison, S.C. A ligand-binding pocket in the dengue virus envelope glycoprotein. Proc. Natl. Acad. Sci. USA 2003, 100, 6986–6991. [Google Scholar] [CrossRef] [PubMed]
- Maillard, R.A.; Jordan, M.; Beasley, D.W.C.; Barrett, A.D.T.; Lee, J.C. Long range communication in the envelope protein domain III and its effect on the resistance of West Nile virus to antibody-mediated neutralization. J. Biol. Chem. 2008, 283, 613–622. [Google Scholar] [CrossRef] [PubMed]
- Volk, D.E.; Beasley, D.W.C.; Kallick, D.A.; Holbrook, M.R.; Barrett, A.D.T.; Gorenstein, D.G. Solution structure and antibody binding studies of the envelope protein domain III from the New York strain of West Nile virus. J. Biol. Chem. 2004, 279, 38755–38761. [Google Scholar] [CrossRef] [PubMed]
- Volk, D.E.; Lee, Y.C.; Li, X.; Thiviyanathan, V.; Gromowski, G.D.; Li, L.; Lamb, A.R.; Beasley, D.W.C.; Barrett, A.D.T.; Gorenstein, D.G. Solution structure of the envelope protein domain III of dengue-4 virus. Virology 2007, 364, 147–154. [Google Scholar] [CrossRef]
- Yu, S.; Wuu, A.; Basu, R.; Holbrook, M.R.; Barrett, A.D.T.; Lee, J.C. Solution structure and structural dynamics of envelope protein domain III of mosquito- and tick-borne flaviviruses. Biochemistry 2004, 43, 9168–9176. [Google Scholar] [CrossRef]
- Li, Z.; Hirst, J.D. Quantitative first principles calculations of protein circular dichroism in the near-ultraviolet. Chem. Sci. 2017, 8, 4318–4333. [Google Scholar] [CrossRef]
- Andersson, D.; Carlsson, U.; Freskgard, P.O. Contribution of tryptophan residues to the CD spectrum of the extracellular domain of human tissue factor: Application in folding studies and prediction of secondary structure. Eur. J. Biochem. 2001, 268, 1118–1128. [Google Scholar] [CrossRef]
- Yu, I.M.; Zhang, W.; Holdaway, H.A.; Li, L.; Kostyuchenko, V.A.; Chipman, P.R.; Kuhn, R.J.; Rossmann, M.G.; Chen, J. Structure of the immature dengue virus at low pH primes proteolytic maturation. Science 2008, 319, 1834–1837. [Google Scholar] [CrossRef] [PubMed]
- Fuzo, C.A.; Degrève, L. The pH dependence of flavivirus envelope protein structure: Insights from molecular dynamics simulations. J. Biomol. Struct. Dyn. 2014, 32, 1563–1574. [Google Scholar] [CrossRef] [PubMed]
- Su, C.M.; Liao, C.L.; Lee, Y.L.; Lin, Y.L. Highly sulfated forms of heparin sulfate are involved in Japanese encephalitis virus infection. Virology 2001, 286, 206–215. [Google Scholar] [CrossRef] [PubMed]
- Dunne, O.; Weidenhaupt, M.; Callow, P.; Martel, A.; Moulin, M.; Perkins, S.J.; Haertlein, M.; Forsyth, V.T. Matchout deuterium labelling of proteins for small-angle neutron scattering studies using prokaryotic and eukaryotic expression systems and high cell-density cultures. Eur. Biophys. J. 2017, 46, 425–432. [Google Scholar] [CrossRef] [PubMed]
- Hammouda, B. Small-angle scattering from branched polymers. Macromol. Theory Simul. 2012, 21. [Google Scholar] [CrossRef]
- Zheng, A.; Yuan, F.; Kleinfelter, L.M.; Kielian, M. A toggle switch controls the low pH-triggered rearrangement and maturation of the dengue virus envelope proteins. Nat. Commun. 2014, 5, 3877. [Google Scholar] [CrossRef] [PubMed]
- Kempson, G.E.; Muir, H.; Swanson, S.A.V.; Freeman, M.A.R. Correlations between stiffness and the chemical constituents of cartilage on the human femoral head. BBA Gen. Subj. 1970, 215, 70–77. [Google Scholar] [CrossRef]
- Volpi, N. Analytical aspects of pharmaceutical grade chondroitin sulfates. J. Pharm. Sci. 2007, 96, 3168–3180. [Google Scholar] [CrossRef]
- Tanaka, A.; Tumkosit, U.; Nakamura, S.; Motooka, D.; Kishishita, N.; Priengprom, T.; Sa-ngasang, A.; Kinoshita, T.; Takeda, N.; Maeda, Y. Genome-Wide Screening Uncovers the Significance of N-Sulfation of Heparan Sulfate as a Host Cell Factor for Chikungunya Virus Infection. J. Virol. 2017, 91, e00432-17. [Google Scholar] [CrossRef]
- Leonova, G.N.; Belikov, S.I. Effect of Glycosaminoglycans on Pathogenic Properties Far-Eastern Tick-Borne Encephalitis Virus. Bull. Exp. Biol. Med. 2019, 167, 482–485. [Google Scholar] [CrossRef]
- Nurunnabi, M.; Revuri, V.; Huh, K.M.; Lee, Y. Chapter 14—Polysaccharide based nano/microformulation: An effective and versatile oral drug delivery system. In Micro and Nano Technologies; Andronescu, E., Grumezescu, A.M.B.T.-N., Eds.; Elsevier: Amsterdam, The Netherlands, 2017; pp. 409–433. ISBN 978-0-323-47720-8. [Google Scholar]
- Thelin, M.A.; Svensson, K.J.; Shi, X.; Bagher, M.; Axelsson, J.; Isinger-Ekstrand, A.; Van Kuppevelt, T.H.; Johansson, J.; Nilbert, M.; Zaia, J.; et al. Dermatan sulfate is involved in the tumorigenic properties of esophagus squamous cell carcinoma. Cancer Res. 2012, 72, 1943–1952. [Google Scholar] [CrossRef]
- Jinno, A.; Park, P.W. Role of glycosaminoglycans in infectious disease. Methods Mol. Biol. 2015, 1229, 567–585. [Google Scholar] [CrossRef] [PubMed]
- Jinno-Oue, A.; Tanaka, A.; Shimizu, N.; Mori, T.; Sugiura, N.; Kimata, K.; Isomura, H.; Hoshino, H. Inhibitory effect of chondroitin sulfate type E on the binding step of human T-cell leukemia virus type 1. AIDS Res. Hum. Retrovir. 2013, 29, 621–629. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | pH 7.5 | pH 5.5 |
|---|---|---|
| EDIII | 52.38 | 50.9 |
| EDIII & H | n/a | 50.9 |
| EDIII & CSCi | 53.12 | 49.5 |
| EDIII & CSCii | 55 | 59.8 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hyatt, J.G.; Prévost, S.; Devos, J.M.; Mycroft-West, C.J.; Skidmore, M.A.; Winter, A. Molecular Changes in Dengue Envelope Protein Domain III upon Interaction with Glycosaminoglycans. Pathogens 2020, 9, 935. https://doi.org/10.3390/pathogens9110935
Hyatt JG, Prévost S, Devos JM, Mycroft-West CJ, Skidmore MA, Winter A. Molecular Changes in Dengue Envelope Protein Domain III upon Interaction with Glycosaminoglycans. Pathogens. 2020; 9(11):935. https://doi.org/10.3390/pathogens9110935
Chicago/Turabian StyleHyatt, James G., Sylvain Prévost, Juliette M. Devos, Courtney J. Mycroft-West, Mark A. Skidmore, and Anja Winter. 2020. "Molecular Changes in Dengue Envelope Protein Domain III upon Interaction with Glycosaminoglycans" Pathogens 9, no. 11: 935. https://doi.org/10.3390/pathogens9110935
APA StyleHyatt, J. G., Prévost, S., Devos, J. M., Mycroft-West, C. J., Skidmore, M. A., & Winter, A. (2020). Molecular Changes in Dengue Envelope Protein Domain III upon Interaction with Glycosaminoglycans. Pathogens, 9(11), 935. https://doi.org/10.3390/pathogens9110935