Abstract
Combinatorial antiretroviral therapy (cART) suppresses HIV replication to undetectable levels and has been effective in prolonging the lives of HIV infected individuals. However, cART is not capable of eradicating HIV from infected individuals mainly due to HIV’s persistence in small reservoirs of latently infected resting cells. Latent infection occurs when the HIV-1 provirus becomes transcriptionally inactive and several mechanisms that contribute to the silencing of HIV transcription have been described. Despite these advances, latent infection remains a major hurdle to cure HIV infected individuals. Therefore, there is a need for more understanding of novel mechanisms that are associated with latent infection to purge HIV from infected individuals thoroughly. Caveolin 1(Cav-1) is a multifaceted functional protein expressed in many cell types. The expression of Cav-1 in lymphocytes has been controversial. Recent evidence, however, convincingly established the expression of Cav-1 in lymphocytes. In lieu of this finding, the current review examines the potential role of Cav-1 in HIV latent infection and provides a perspective that helps uncover new insights to understand HIV latent infection.
1. Introduction
The development of HIV vaccines with adequate protection remains elusive. However, there are encouraging studies that will help promote HIV vaccine development. The phase 2b clinical trial in Thailand RV144 showed a reduced acquisition of HIV infection with a 31% efficacy [1]. Although these results are significant and raised hopes, we have not yet reached the goal of attaining an effective HIV vaccine. Combinatorial antiretroviral therapy (cART) suppresses HIV replication to undetectable levels and has been effective in prolonging the lives of HIV infected individuals. The suppression of viral replication achieved by many patients on antiretroviral therapy (ART) initially raised hopes for virus eradication. It is now well documented that HIV persists in a small reservoir of latently infected resting CD4+ cells of patients on ART [2,3,4,5,6,7]. HIV establishes latent infection in the blood and lymphoid tissues as well as multipotent hematopoietic progenitor cells in the bone marrow [8,9]. Besides, monocytes, dendritic cells, and macrophages can contribute to viral persistence as latently infected cells [10,11,12,13,14,15]. The central nervous system (CNS) is another important target for HIV infection [16,17,18]. Many drugs are restricted from crossing the blood–brain and blood–CSF barriers and are therefore suboptimal in penetrating the CNS. Consequently, the CNS serves as a viral reservoir that remains untouched to cART therapy. Residual viral reservoirs in the gastrointestinal (GI) and genitourinary tract, as well as lymphoid tissues in patients under drug treatment, can be an additional obstacle for curing HIV [7,19,20,21,22,23,24]. Despite the advancement of combinatorial drug therapies that have aided in treating HIV infected patients and helped prolong life successfully, current drug therapy is not curative due to HIV’s ability to establish a latent infection that persists in reservoir cells. Consequently, HIV infected individuals will remain on cART treatment throughout their lives. Thus, the use of cART has converted HIV infection into a manageable chronic disease. Although a manageable disease HIV infected individuals under cART are prone to other Non-AIDS diseases [25,26,27]. This enforces the need for a complete understanding of HIV latent infection and subsequently find a strategy for eradication and cure.
Latent infection occurs when the HIV-1 provirus becomes transcriptionally inactive, and several contributing mechanisms already identified as repressive chromatin structure, transcriptional interference from adjacent transcriptional units, increase in histone deacetylase (HDAC) and methyltransferase (HMT) activity and DNA methylation of proviral DNA [28,29,30,31,32]. Reactivation of HIV latent infected cells in the presence of cART would ideally allow for recognition of latent infected cells by the immune system and consequently clearance and cure. Several strategies have been described and examined to reverse latent infection to target and eradicate HIV [28,29,30,31,32,33]. A latent reversing agent (LRA) is one approach as a “shock and kill” strategy in combination with cART to achieve functional cure HIV infection [33,34,35,36,37]. Examples of LRAs tested include protein kinase C (PKC) agonists (e.g., PMA, prostratin, bryostatin, and ingenol molecules) [33,36,38,39,40,41,42,43] and histone deacetylase inhibitors (HDACi; e.g., vorinostat/SAHA, panobinostat, and romidepsin) [33,34,35,44,45,46,47,48,49,50,51], indirect activator of Akt signaling pathways (Disulfiram) [33,52,53,54,55], and CCR5 antagonist (maraviroc) [56]. A recent promising study of reactivation virus in latently CD4+ T cells with the HDACs inhibitor vorinostat shows the cells were susceptible to HIV antigen-specific CD8+ T cells mediated killing in vitro [34]. However, further evidence suggests this process alone may not be sufficient. Global activation of T cells to reactivate virus to target for therapeutic has resulted in a toxic level of immune activation [28,30,32,57,58]. A variety of molecules that induce HIV transcription or inhibit HDACs or HMTs also suffer from lack of specificity, and their safe and efficient in vivo application and clinical trials have yet to be established [28,30,32,34,44,59,60,61,62]. Furthermore, LRAs are inefficient and have limitations in disrupting latency in other cell types such as macrophages and microglial cells. Alternative approaches are emerging to tackle HIV latent infection based on HIV latent promoting agents (LPAs) and proviral DNA scission. The LPA approach is described as “block and lock,” and the principle behind is to effectively silence (block) the promoter region and restrict (lock) viral transcription subsequently impairing viral rebound. For the induction of latency, several methods have been used; however, all these efforts can be categorized in three groups. First approach was to use small inhibitory RNA (siRNA) or natural long non-coding RNA (lncRNA) to prevent either the transcription of HIV-1 RNA or initiate a repressive chromatin modification at the proviral DNA. The most common among these approaches were, siPromA, LTR363, si2A, and S4. All these targeted NF-κB binding region of the proviral DNA to modify the chromatin structure to make them inaccessible for transcription. The second approach was to prevent the Tat, the potent transactivator of HIV-1 transcription. The third approach was used to modulate the signaling by small molecule inhibitors of mTOR pathway. Recently, the use of, the clustered regularly interspaced short palindromic repeats (CRISPR), which rely on CRISPR RNAs (crRNAs) to direct cleavage of complementary sequences via the nuclease activity of CRISPR-associated (Cas9) protein system, has exploded and become a great tool to knock out genes. HIV provirus deletion has been accomplished in vitro using the CRISPR/Cas9 system [63,64,65]. Although targeting proviral genome using CRISPR/Cas9 provided initial success, general variability in viral sequences and escape from the CRISPR by mutation posed hurdles in successful use of the technology [66,67]. Despite these advances, latent infection remains a major hurdle to cure HIV infected individuals. Therefore, to accomplish a complete purge of latent reservoir, further research is critical to understand better the factors that influence and maintain HIV latency. Major critical factors that are missing include complete understanding of the mechanism HIV latency, cell membrane cues that sends the signal for the establishment of latent infection, and markers that help identify latently infected cells.
2. Caveolin-1
One molecule that can be important in HIV latent infection is Caveolin 1 (Cav-1). Contrary to the previous belief, recent findings show that human T cells can express Cav-1 [68,69,70]. Along with this finding, the multiple functions of Cav-1 with relevance to cell regulation can have important implications in HIV latent infection. Caveolin 1 (Cav-1), a 21–24-kDa scaffolding protein, is an important structural component of the caveolae organelle [71]. Cav-1 is also important in establishing specific lipid microdomains (non-caveolar caveolin lipid raft (NCCLR)) and helps compartmentalize signal pathways. Functional studies have shown that Cav-1 is involved in a wide range of cellular processes (Table 1), including cell cycle regulation, signal transduction, endocytosis, cholesterol trafficking, and efflux [72,73,74,75,76,77,78]. Furthermore, Cav-1 engages in crosstalk with the actin cytoskeleton and contributes to mechanosensing and adaptation to various mechanical stimuli and environmental changes that include microbe infection [70,79,80,81,82,83,84,85,86]. Therefore, Cav-1 regulates multiple signaling cascades as well as provides crosstalk with different molecules, and altered expression or/and any perturbation of Cav-1 positioning in the vicinity of the plasma membrane can affect signaling and crosstalk of different biological molecules. Cav-1 is highly expressed in terminally differentiated or quiescent cells, including muscle cells, adipocytes, endothelial cells, monocytes, macrophages, dendritic cells, microglia, and astrocytes [74,87,88,89,90,91,92,93,94,95,96,97]. Initially, it was thought that lymphocytes do not express Cav-1 [74,87,88,89,90,91,92,93,94,95,96,97]. However, several recent reports reveal both T and B primary lymphocytes express Cav-1 at low levels [68,69,70,83,98,99]. The expression of Cav-1 protein at times can be difficult to detect by standard methods. Further studies reveal that Caveolin-1 is important in B cell antigen receptor (BCR) and T cell antigen receptor (TCR) basal membrane organization and also reorganization upon stimulation. Several of the proteins that interact with Cav-1 are suggested to be involved in TCR-regulated membrane dynamics and intracellular signaling [68,69,83,100,101,102]. Cholesterol plays an important role in the resting or activation TCR either by stabilizing the resting TCR conformation or by increasing sensitivity to antigen and cooperation, respectively [83,103,104,105,106,107,108,109,110,111,112]. Since Cav-1 is engaged in cholesterol trafficking and efflux, it can regulate the conformational stage of TCR or other lipid rafts. In addition, Cav-1 is an essential component of the lipid raft platform for the recruitment of signaling proteins to the plasma membrane. Signal transduction most probably happens by linking the plasma membrane and the actin cytoskeleton [83,113]. Therefore, regulation of TCR and other lipid rafts integrates environmental cues such as the concentration of ligand or cholesterol metabolism to modulate TCR responses and T cell resting/activation. NCCR can, thus, regulate membrane dynamics compartmentalization upon receptors activation.

Table 1.
Functions of Cav-1.
3. Potential of Cav-1 in Keeping the Chromatin Open
Cav-1 maintains its presence at the cell surface and segregates different signaling components for the ligand-dependent signaling. In diabetes and ozone injury [116] models, Cav-1 negatively regulates PI3K signaling, a signaling that prevents latency by the activation of NF-κB p65. The expression of Cav-1 itself is prevented by the activation of NF-κB [117]. In macrophages, Cav-1 association with TLR4 prevents pro-inflammatory cytokine production [114] that could add to the viral replication by further activation of NF-κB. Additionally, TLR4 signaling triggers a cascade to degrade Cav-1 by a ring-type E3 ubiquitin ligase, zinc, and ring finger 1 (ZNRF1) [118]. Cav-1 deficient mice maintain a constant inflammatory state; however, T cells deficient in Cav-1 tend to develop into non-inflammatory regulatory T cells. Further research is needed to reconcile these counterintuitive findings. In several human inflammatory diseases, Cav-1 was found lower than the normal levels, suggesting a strong anti-inflammatory role of Cav-1. The cytokine and TLR signaling simultaneously trigger cascade for Mitogen Associated Protein Kinases that control chromatin structure by various mechanisms. Based upon these correlations, we can predict Cav-1 can prevent the transcription of HIV-1 proviral DNA at different levels. Since Cav-1 also ushers signaling components to lipid raft, a careful assessment of Cav-1 in different cells and different condition is necessary (Figure 1).
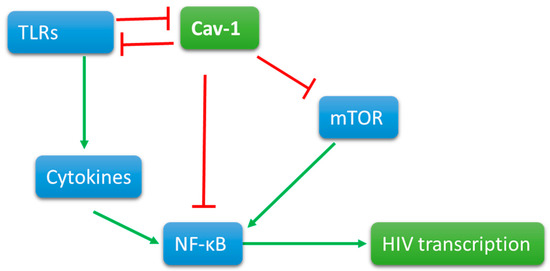
Figure 1.
Cav-1 is present on the cell surface interactions with signaling molecules. When triggered, signaling molecules, such as Toll-like receptors, activate directly and indirectly by cytokine production NFkB. Viral promoter at the 5’ end of the proviral DNA contains the promoter that has NFKB binding site for viral transcription. Cav-1 directly or indirectly interacts with TLR signaling and mTOR pathways to reduce the activation of NFkB and thus the transcription of proviral DNA.
4. Caveolin-1 and the Potential Link to HIV Latent Infection
The participation of Cav-1 in environmental cues makes this molecule an important regulator of cell physiology. Interestingly, activation or antigenic stimulation of the TCR results in enhanced expression of Cav-1 in both CD4+ and CD8+ T cells [68,69,70,83,98,99]. In complete lack of Cav-1 genes in CD8+, T cells cripple their function by LFA-1 redistribution and polarity [69,70]. Based on these findings, higher Cav-1 would benefit the host by more active T cell function. However, hyperactivation of CD8+ T cells has not been reported in HIV-1 infected patients. The major caveat of such studies is complete lack of Cav-1. The function of most of the signaling molecules depends upon their relative concentration. A complete lack of a molecule suggests a prominent role in the system; however, it cannot provide a clear picture for its position. Future experiments should be carried out with a varied expression of Cav-1 to know its real function. Several molecules modulate the expression of Cav-1 by different mechanisms [86]. The expression of Cav-1 is activated or enhanced by microbes and microbial byproduct lipopolysaccharide (LPS) or oxidative stress [85,119,120,121], strengthening the notion that Cav-1 role in sensing environmental cues. Oxidized low-density lipoprotein (oxLDL) or simvastatin also upregulates the expression Cav-1 [75,122]. Furthermore, cholesterol is an important modulator of Cav-1 expression. In addition, cellular growth factors can modulate Cav-1 expression through transcriptional mechanisms [123,124,125,126]. Multiple studies suggest that both HIV infection and antiretroviral drugs induce reactive oxygen species (ROS) production in HIV infected individuals [121,127], which subsequently can result in induction of Cav-1 expression. Such regulation of Cav-1 expression implies that Cav-1 is a critical molecule for cell survival and adjustment to stress.
Oxidative Stress: Oxidative damage has been linked to cellular senescence in aging animals as well as stress induced premature senescence. Cellular senescence is a mechanism of irreversible growth arrest to protect against proliferating aging cells and/or damaged cells to halt transmission of damage to daughter cells [128]. The HIV proteins Tat, Nef, Vpr, and gp120 have been shown to independently increase ROS while decreasing antioxidants establishing HIV-mediated oxidative stress [Reviewed in [127]]. Extensive studies also suggest that cART is a major contributor of ROS increase in HIV infected patients [127,129,130,131,132,133,134]. ROS is involved in a variety of cellular processes including proliferation, differentiation, host-defense, and wound healing ((Reviewed in [135]). The levels of increased ROS production is regulated by antioxidants such as superoxide dismutase glutathione peroxidase, catalase and vitamins E and C, and glutathione [135]. Uncontrolled increased levels of ROS lead to damage of macromolecules and causes cellular apoptosis and senescence [136,137,138,139,140,141,142,143,144]. ROS, therefore, promotes oxidative stress, which then results in cellular dysfunction and tissue destruction. Enhanced ROS production by HIV, thus, will advance the breakdown of cellular tight junctions of the epithelium at the mucosal surface as well as the blood-brain barrier vasculature, amplifying HIV infection. Furthermore, increased levels of ROS production due to HIV infection and/or cART treatment contribute to HIV associated vascular disease. Therefore, oxidative stress is a central contributing priming factor to many parameters leading to pathogenesis in HIV infected individuals irrespective of ART treatment. Oxidative damage has been linked to cellular senescence in aging animals as well as stress induced premature senescence.
Cav-1 and HIV inhibition: Caveolin 1 (Cav-1) plays a major role in controlling cellular senescence [119]. Several studies have shown that oxidative stress upregulates Cav-1 and enhanced expression of Cav-1 plays a central role in stress-induced cellular senescence [119]. Therefore, in response to environmental cues, Cav-1 can be induced to promote growth arrest of cells. Relevant to such notion, low expression of Cav-1 expression correlates with transformed cells, and inverse relationship between Cav-1 expression and cell transformation is established [83,145,146,147]. Furthermore, Cav-1 modulates cell cycle progression [72,83,145,148,149,150]. Cell cycle analyses show that Cav-1 maintains cell cycle at the quiescent stage, whereas the deletion of Cav-1 results cell cycle to progress from G0 to G1 and G2/M phases [72,83,148]. For example, Caveolin 1 is important in the regulation of cyclin D [149,151]. Previous studies of gene expression patterns in combination with epigenetic information revealed that the cell cycle regulator cyclin D2 might have an important role in maintaining the HIV latency [152], implicating a role for Cav-1 plays in HIV latency. Similar to other activators, we and others have demonstrated that HIV infection induces Cav-1 expression in macrophages [153]. Since the recent finding that Cav-1 expresses in human lymphocytes, we have also established HIV infection enhances the expression of Cav-1 in T cells (Unpublished results). Furthermore, we have previously demonstrated inhibition of HIV replication in Cav-1 over-expressing primary CD4+T cells and monocyte-derived macrophages [115,153,154]. The mechanism of induction of Cav-1 during HIV infection is not completely understood. Lin et al. [153] established that Tat is important in Cav-1′s upregulation at the transcriptional level, and this upregulation involves p53 protein. The study further shows that the p53 expression level is not affected by HIV infection. However, the phosphorylation of p53 at Ser15 and Ser46 is enhanced significantly. This suggests that the level of p53 activity plays an important role enhancement of Tat-mediated Cav-1 expression. Inhibitor of the p38 mitogen-activated protein kinase (MAPK) blocked the phosphorylation of the p53, subsequently reducing the induction Cav-1 significantly. These results suggest that in HIV-infected cells, Tat mediates activation of p38 MAPK, promoting the phosphorylation of p53, subsequently upregulating Cav-1 expression. However, further studies are needed to determine the factors and pathways involved in the mechanisms of Cav-1 up-regulation by HIV. These include epigenetic elements such as upstream factors as well as transcription factors and cis-acting elements. For example, the promoter region of Cav-1 NF-KB cis-element important for Cav-1 expression and regulation has not been examined in the context of the induction of Cav-1 expression mediated by HIV infection.
Caveolin-1 present on the cells surface and acts as an entry point for various infectious agents, including HIV-1. HIV-1 infects immune cells, mainly dendritic cells, macrophages, and T cells. The virus spread either by actively transferred to T cells from dendritic cells [155], direct interaction of virion with their cognate receptor, or through the virological synapses (VS) [156]. Dendritic captured virion presented on the dendrites, which are formed with actin filaments, and inhibition of Caveolin-interacting tetraspanin and dynamin prevented the process [155]. When a free virion interacts with a CD4/CXCR4 on T cells, they form a cluster; a requisite event for the viral attachment is controlled by Filamin-A, a Caveolin-interacting molecule [157,158]. These interactions among viruses and other cytoskeletal molecules are critical for transporting the cytoplasm for replication and finally reaching the nucleus. There is a defined role for the T cell synapse to occur for the caveolin-1 [70], which is needed for T cell activation. It is still unclear whether virological synapse on T cells for viral spread requires Caveloin-1; however, for efficient VS to form, it requires various actin-associated proteins that may be interacting with the Cav-1. Like the entry, viral budding also needs actin cytoskeletal rearrangements attached to Caveolin-1 at the surface [159]. Similarly, there is limited information on the mechanism of inhibition of HIV replication by Cav-1. Since Cav-1 participates in many cellular functions, the inhibition of HIV can involve several mechanisms (Figure 2). Two independent studies have demonstrated that Cav-1 inhibits HIV replication by transcriptional repression mediated through NF-κB [115,160]. However, the upstream factors involved in transcriptional repression mediated through NF-κB by Cav-1 is not known. Wang et al. [161] has proposed a potential mechanism for Cav-1’s ability to inhibit HIV replication that involves the association of Cav-1 and the HIV envelope. Cav-1 significantly suppressed Env-induced membrane hemifusion by possible interaction with the gp41. The env glycoprotein stimulates viral transcription and increases infection by modulating cellular machinery [162]. The increase in Cav-1 expression during HIV infection along its possible interaction and sequestration of Env can result in inhibition of Env mediated manipulation of cellular machinery to stimulate viral transcription and infectivity of virus progeny, subsequently contributing to HIV latent infection. Since surface lipid composition is essential in cell fusion of the block Env mediated fusion with target cells during an increased expression of Cav-1 may have to do with Cav-1’s role in cholesterol metabolism. Cav-1 is an essential regulator of cholesterol metabolism. Furthermore, Cav-1 is vital in cholesterol transport from the ER to the cell membrane. An increase in Cav-1 expression leads to the restoration of cholesterol efflux impaired by HIV via Nef and negatively affects virus replication [154]. Therefore, the specific interaction of Env with Cav-1 blocking fusion with target cells and the role of Cav-1 in cholesterol trafficking have important implication the establishment of HIV persistent in the patient under cART. Since cholesterol plays an important role in the resting or activation TCR, the HIV Env binding and/or fusion can lead to similar signal transduction affecting cell physiology. Furthermore, since Cav-1 is engaged in cholesterol efflux, an increase in its expression can modulate the confirmation stage of the lipid rafts and essential component of the lipid raft platform for the recruitment of signaling proteins to the plasma membrane. These changes would have an impact on several cellular functions, including the resting and cycling status of cells and including the establishment of HIV latent infection (Figure 2).
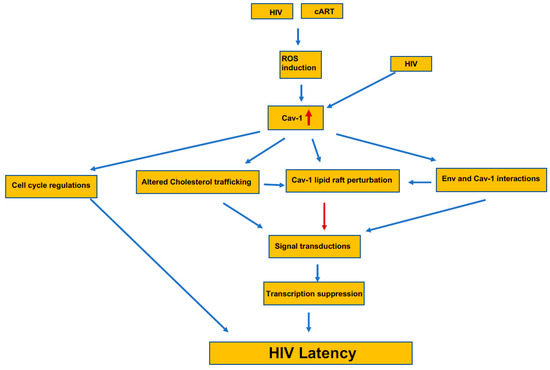
Figure 2.
The induction and the pathway for the potential contribution of Cav-1 to the establishment of HIV latency. The presence of Caveolin-1 increases in HIV-1 infected and in latently infected cells by the generation of ROS. This increase in Cav-1 interferes with the cell cycle regulation that adds to the HIV latency. Simultaneously, increased Cav-1 alters cholesterol trafficking and thus the lipid raft composition, which directly and indirectly interferes with the interaction with Cav-1 and the virus’ envelop. Changes at the lipid raft that recruits signaling components alters the overall cellular signaling and initiates transcriptional suppression that further adds to the latency process.
5. Conclusions
Cav-1/NCCLR membrane compartmentalization, along with its interaction with the cytoskeleton, can be an important environmental cue sensing mechanism, including ligand binding and HIV infection. The system regulates the steady and activation states of a cell equilibrium. The induction of Cav-1 expression can be a cell surviving mechanism to push the equilibrium from activated to a steady state by promoting cells to move into the resting stage. During HIV infection, most cells are overwhelmed by virus replication showing cytopathology, whereas a few cells become quiescent with latent infection. Since Cav-1 regulates cell cycle, signaling involving NCCLR can be important and play an essential role in promoting infected cells to become quiescent, subsequently resulting in latent infection as a cell survival mechanism (Figure 2). The upregulation of Cav-1 during viral infection can alter cholesterol composition modulate NCCLR, serving as initial danger signal and consequently leading to cell arrest and repression of viral transcription contributing to latent infection.
Author Contributions
B.S. and A.M. equally contributed to the writing of the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
The work is supported from indirect cost returns from previous grant from the National Institutes of Health (AI39126) to Ayalew Mergia.
Conflicts of Interest
The authors declare no competing or financial interests.
References
- Rerks-Ngarm, S.; Pitisuttithum, P.; Nitayaphan, S.; Kaewkungwal, J.; Chiu, J.; Paris, R.; Premsri, N.; Namwat, C.; De Souza, M.; Adams, E.; et al. Vaccination with ALVAC and AIDSVAX to Prevent HIV-1 Infection in Thailand. N. Engl. J. Med. 2009, 361, 2209–2220. [Google Scholar] [CrossRef] [PubMed]
- Blankson, J.N.; Persaud, D.; Siliciano, R.F. The Challenge of Viral Reservoirs in HIV-1 Infection. Annu. Rev. Med. 2002, 53, 557–593. [Google Scholar] [CrossRef] [PubMed]
- Finzi, D.; Blankson, J.N.; Siliciano, J.D.; Margolick, J.B.; Chadwick, K.; Pierson, T.C.; A Smith, K.; Lisziewicz, J.; Lori, F.; Flexner, C.; et al. Latent infection of CD4+ T cells provides a mechanism for lifelong persistence of HIV-1, even in patients on effective combination therapy. Nat. Med. 1999, 5, 512–517. [Google Scholar] [CrossRef] [PubMed]
- Palmer, S.; Josefsson, L.; Coffin, J.M. HIV reservoirs and the possibility of a cure for HIV infection. J. Intern. Med. 2011, 270, 550–560. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.Z.; Wightman, F.; Lewin, S.R. HIV Reservoirs and Strategies for Eradication. Curr. HIV/AIDS Rep. 2012, 9, 5–15. [Google Scholar] [CrossRef] [PubMed]
- Chomont, N.; El-Far, M.; Ancuta, P.; Trautmann, L.; A Procopio, F.; Yassine-Diab, B.; Boucher, G.; Boulassel, M.-R.; Ghattas, G.; Brenchley, J.M.; et al. HIV reservoir size and persistence are driven by T cell survival and homeostatic proliferation. Nat. Med. 2009, 15, 893–900. [Google Scholar] [CrossRef] [PubMed]
- Chun, T.-W.; Carruth, L.; Finzi, D.; Shen, X.; DiGiuseppe, J.A.; Taylor, H.; Hermankova, M.; Chadwick, K.; Margolick, J.; Quinn, T.C.; et al. Quantification of latent tissue reservoirs and total body viral load in HIV-1 infection. Nat. Cell Biol. 1997, 387, 183–188. [Google Scholar] [CrossRef]
- Carter, C.C.; Onafuwa-Nuga, A.; McNamara, L.A.; Riddell, J.; Bixby, D.; Savona, M.R.; Collins, K.L. HIV-1 infects multipotent progenitor cells causing cell death and establishing latent cellular reservoirs. Nat. Med. 2010, 16, 446–451. [Google Scholar] [CrossRef]
- Carter, C.C.; McNamara, L.A.; Onafuwa-Nuga, A.; Shackleton, M.; Riddell, J.; Bixby, D.; Savona, M.R.; Morrison, S.J.; Collins, K.L. HIV-1 Utilizes the CXCR4 Chemokine Receptor to Infect Multipotent Hematopoietic Stem and Progenitor Cells. Cell Host Microbe 2011, 9, 223–234. [Google Scholar] [CrossRef]
- Bailey, J.R.; Sedaghat, A.R.; Kieffer, T.; Brennan, T.; Lee, P.K.; Wind-Rotolo, M.; Haggerty, C.M.; Kamireddi, A.R.; Liu, Y.; Lee, J.; et al. Residual Human Immunodeficiency Virus Type 1 Viremia in Some Patients on Antiretroviral Therapy Is Dominated by a Small Number of Invariant Clones Rarely Found in Circulating CD4+ T Cells. J. Virol. 2006, 80, 6441–6457. [Google Scholar] [CrossRef]
- Keele, B.F.; Tazi, L.; Gartner, S.; Liu, Y.; Burgon, T.B.; Estes, J.D.; Thacker, T.C.; Crandall, K.A.; McArthur, J.C.; Burton, G.F. Characterization of the Follicular Dendritic Cell Reservoir of Human Immunodeficiency Virus Type 1. J. Virol. 2008, 82, 5548–5561. [Google Scholar] [CrossRef] [PubMed]
- Zhu, T.; Muthui, D.; Holte, S.; Nickle, D.; Feng, F.; Brodie, S.; Hwangbo, Y.; Mullins, J.I.; Corey, L. Evidence for Human Immunodeficiency Virus Type 1 Replication In Vivo in CD14+ Monocytes and Its Potential Role as a Source of Virus in Patients on Highly Active Antiretroviral Therapy. J. Virol. 2002, 76, 707–716. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Wigdahl, B. Cellular Reservoirs of HIV-1 and their Role in Viral Persistence. Curr. HIV Res. 2008, 6, 388–400. [Google Scholar] [CrossRef]
- Alexaki, A.; Wigdahl, B. HIV-1 Infection of Bone Marrow Hematopoietic Progenitor Cells and Their Role in Trafficking and Viral Dissemination. PLoS Pathog. 2008, 4, e1000215. [Google Scholar] [CrossRef]
- Coleman, C.M.; Wu, L. HIV interactions with monocytes and dendritic cells: Viral latency and reservoirs. Retrovirology 2009, 6, 51. [Google Scholar] [CrossRef]
- Churchill, M.J.; Wesselingh, S.L.; Cowley, D.; Pardo, C.A.; McArthur, J.C.; Brew, B.J.; Gorry, P.R. Extensive astrocyte infection is prominent in human immunodeficiency virus-associated dementia. Ann. Neurol. 2009, 66, 253–258. [Google Scholar] [CrossRef] [PubMed]
- Fischer-Smith, T.; Croul, S.; Sverstiuk, A.E.; Capini, C.; L’Heureux, D.; Regulier, E.G.; Richardson, M.W.; Amini, S.; Morgello, S.; Khalili, K.; et al. CNS invasion by CD14+/CD16+ peripheral blood-derived monocytes in HIV dementia: Perivascular accumulation and reservoir of HIV infection. J. Neurovirol. 2001, 77, 528–541. [Google Scholar] [CrossRef]
- Edén, A.; Fuchs, D.; Hagberg, L.; Nilsson, S.; Spudich, S.; Svennerholm, B.; Price, R.W.; Gisslén, M. HIV-1 Viral Escape in Cerebrospinal Fluid of Subjects on Suppressive Antiretroviral Treatment. J. Infect. Dis. 2010, 202, 1819–1825. [Google Scholar] [CrossRef]
- Chun, T.; Nickle, D.C.; Justement, J.S.; Meyers, J.H.; Roby, G.; Hallahan, C.W.; Kottilil, S.; Moir, S.; Mican, J.M.; Mullins, J.I.; et al. Persistence of HIV in Gut-Associated Lymphoid Tissue despite Long-Term Antiretroviral Therapy. J. Infect. Dis. 2008, 197, 714–720. [Google Scholar] [CrossRef]
- Cu-Uvin, S.; Delong, A.K.; Venkatesh, K.K.; Hogan, J.W.; Ingersoll, J.; Kurpewski, J.; De Pasquale, M.P.; D’aquila, R.; Caliendo, A.M. Genital tract HIV-1 RNA shedding among women with below detectable plasma viral load. AIDS 2010, 24, 2489–2497. [Google Scholar] [CrossRef]
- Thacker, T.C.; Zhou, X.; Estes, J.D.; Jiang, Y.; Keele, B.F.; Elton, T.S.; Burton, G.F. Follicular Dendritic Cells and Human Immunodeficiency Virus Type 1 Transcription in CD4+ T Cells. J. Virol. 2008, 83, 150–158. [Google Scholar] [CrossRef] [PubMed]
- Van Leeuwen, E.; Ter Heine, R.; Van Der Veen, F.; Repping, S.; Beijnen, J.H.; Prins, J.M. Penetration of Atazanavir in Seminal Plasma of Men Infected with Human Immunodeficiency Virus Type 1. Antimicrob. Agents Chemother. 2006, 51, 335–337. [Google Scholar] [CrossRef] [PubMed]
- Yukl, S.A.; Gianella, S.; Sinclair, E.; Epling, L.; Li, Q.; Duan, L.; Choi, A.L.M.; Girling, V.; Ho, T.; Li, P.; et al. Differences in HIV Burden and Immune Activation within the Gut of HIV-Positive Patients Receiving Suppressive Antiretroviral Therapy. J. Infect. Dis. 2010, 202, 1553–1561. [Google Scholar] [CrossRef] [PubMed]
- Yukl, S.A.; Shergill, A.K.; McQuaid, K.; Gianella, S.; Lampiris, H.; Hare, C.B.; Pandori, M.; Sinclair, E.; Günthard, H.F.; Fischer, M.; et al. Effect of raltegravir-containing intensification on HIV burden and T-cell activation in multiple gut sites of HIV-positive adults on suppressive antiretroviral therapy. AIDS 2010, 24, 2451–2460. [Google Scholar] [CrossRef]
- Luetkemeyer, A.F.; Havlir, D.V.; Currier, J.S. Complications of HIV disease and antiretroviral treatment. Top. HIV Med. Publ. Int. AIDS Soc. USA 2010, 18, 57–65. [Google Scholar]
- Gomo, Z.A.; Hakim, J.; Walker, A.S.; Tinago, W.; Mandozana, G.; Kityo, C.M.; Munderi, P.; Katabira, E.; Reid, A.; Gibb, D.M.; et al. Impact of second-line antiretroviral regimens on lipid profiles in an African setting: The DART trial sub-study. AIDS Res. Ther. 2014, 11, 32. [Google Scholar] [CrossRef][Green Version]
- Matoga, M.; Hosseinipour, M.C.; Aga, E.; Ribaudo, H.J.; Kumarasamy, N.; Bartlett, J.; Hughes, M.D.; the ACTG A5230 Study Team. Hyperlipidaemia in HIV-infected patients on lopinavir/ritonavir monotherapy in resource-limited settings. Antivir. Ther. 2016, 22, 205–213. [Google Scholar] [CrossRef]
- Coiras, M.; López-Huertas, M.R.; Pérez-Olmeda, M.; Alcamí, J. Understanding HIV-1 latency provides clues for the eradication of long-term reservoirs. Nat. Rev. Genet. 2009, 7, 798–812. [Google Scholar] [CrossRef]
- Hakre, S.; Chavez, L.; Shirakawa, K.; Verdin, E. Epigenetic regulation of HIV latency. Curr. Opin. HIV AIDS 2011, 6, 19–24. [Google Scholar] [CrossRef]
- Archin, N.M.; Sung, J.M.; Garrido, C.; Soriano-Sarabia, N.; Margolis, D.M. Eradicating HIV-1 infection: Seeking to clear a persistent pathogen. Nat. Rev. Genet. 2014, 12, 750–764. [Google Scholar] [CrossRef]
- Taube, R.; Peterlin, B.M. Lost in Transcription: Molecular Mechanisms that Control HIV Latency. Viruses 2013, 5, 902–927. [Google Scholar] [CrossRef]
- Mbonye, U.; Karn, J. Transcriptional control of HIV latency: Cellular signaling pathways, epigenetics, happenstance and the hope for a cure. Virology 2014, 455, 328–339. [Google Scholar] [CrossRef] [PubMed]
- Castro, S.; Colomer-Lluch, M.; Serra-Moreno, R. Barriers for HIV Cure: The Latent Reservoir. AIDS Res. Hum. Retroviruses 2018, 34. [Google Scholar] [CrossRef]
- Archin, N.M.; Liberty, A.L.; Kashuba, A.D.; Choudhary, S.K.; Kuruc, J.D.; Crooks, A.M.; Parker, D.C.; Anderson, E.M.; Kearney, M.F.; Strain, M.C.; et al. Administration of vorinostat disrupts HIV-1 latency in patients on antiretroviral therapy. Nature 2012, 487, 482–485. [Google Scholar] [CrossRef] [PubMed]
- Shan, L.; Deng, K.; Shroff, N.S.; Durand, C.M.; Rabi, S.A.; Yang, H.-C.; Zhang, H.; Margolick, J.B.; Blankson, J.N.; Siliciano, R.F. Stimulation of HIV-1-Specific Cytolytic T Lymphocytes Facilitates Elimination of Latent Viral Reservoir after Virus Reactivation. Immunity 2012, 36, 491–501. [Google Scholar] [CrossRef] [PubMed]
- Spivak, A.M.; Planelles, V. HIV-1 Eradication: Early Trials (and Tribulations). Trends Mol. Med. 2016, 22, 10–27. [Google Scholar] [CrossRef] [PubMed]
- Spivak, A.M.; Planelles, V. Novel Latency Reversal Agents for HIV-1 Cure. Annu. Rev. Med. 2018, 69, 421–436. [Google Scholar] [CrossRef] [PubMed]
- Jiang, G.; Dandekar, S. Targeting NF-kappaB signaling with protein kinase C agonists as an emerging strategy for combating HIV latency. Aids Res. Hum. Retrovir. 2015, 31, 4–12. [Google Scholar] [CrossRef]
- Spina, C.A.; Anderson, J.; Archin, N.M.; Bosque, A.; Chan, J.; Famiglietti, M.; Greene, W.C.; Kashuba, A.; Lewin, S.R.; Margolis, D.M.; et al. An In-Depth Comparison of Latent HIV-1 Reactivation in Multiple Cell Model Systems and Resting CD4+ T Cells from Aviremic Patients. PLoS Pathog. 2013, 9, e1003834. [Google Scholar] [CrossRef]
- Bullen, C.K.; Laird, G.M.; Durand, C.M.; Siliciano, J.D.; Siliciano, R.F. New ex vivo approaches distinguish effective and ineffective single agents for reversing HIV-1 latency in vivo. Nat. Med. 2014, 20, 425–429. [Google Scholar] [CrossRef]
- Laird, G.M.; Bullen, C.K.; Rosenbloom, D.I.; Martin, A.R.; Hill, A.L.; Durand, C.M.; Siliciano, J.D.; Siliciano, R.F. Ex vivo analysis identifies effective HIV-1 latency–reversing drug combinations. J. Clin. Investig. 2015, 125, 1901–1912. [Google Scholar] [CrossRef]
- Cary, D.C.; Fujinaga, K.; Peterlin, B.M. Euphorbia Kansui Reactivates Latent HIV. PLoS ONE 2016, 11, e0168027. [Google Scholar] [CrossRef]
- Martin, A.R.; Pollack, R.A.; Capoferri, A.; Ambinder, R.F.; Durand, C.M.; Siliciano, R.F. Rapamycin-mediated mTOR inhibition uncouples HIV-1 latency reversal from cytokine-associated toxicity. J. Clin. Investig. 2017, 127, 651–656. [Google Scholar] [CrossRef]
- Van Lint, C.; Emiliani, S.; Ott, M.; Verdin, E. Transcriptional activation and chromatin remodeling of the HIV-1 promoter in response to histone acetylation. EMBO J. 1996, 15, 1112–1120. [Google Scholar] [CrossRef] [PubMed]
- Archin, N.M.; Espeseth, A.; Parker, D.; Cheema, M.; Hazuda, D.; Margolis, D.M. Expression of Latent HIV Induced by the Potent HDAC Inhibitor Suberoylanilide Hydroxamic Acid. AIDS Res. Hum. Retroviruses 2009, 25, 207–212. [Google Scholar] [CrossRef] [PubMed]
- Contreras, X.; Schweneker, M.; Chen, C.-S.; McCune, J.M.; Deeks, S.G.; Martin, J.; Peterlin, B.M. Suberoylanilide Hydroxamic Acid Reactivates HIV from Latently Infected Cells. J. Biol. Chem. 2009, 284, 6782–6789. [Google Scholar] [CrossRef] [PubMed]
- Ke, R.; Lewin, S.R.; Elliott, J.H.; Perelson, A.S. Modeling the Effects of Vorinostat In Vivo Reveals both Transient and Delayed HIV Transcriptional Activation and Minimal Killing of Latently Infected Cells. PLoS Pathog. 2015, 11, e1005237. [Google Scholar] [CrossRef] [PubMed]
- Olesen, R.; Viganò, S.; Rasmussen, T.A.; Søgaard, O.S.; Ouyang, Z.; Buzon, M.J.; Bashirova, A.; Carrington, M.; Palmer, S.; Brinkmann, C.R.; et al. Innate Immune Activity Correlates with CD4 T Cell-Associated HIV-1 DNA Decline during Latency-Reversing Treatment with Panobinostat. J. Virol. 2015, 89, 10176–10189. [Google Scholar] [CrossRef] [PubMed]
- Elliott, J.H.; Wightman, F.; Solomon, A.; Ghneim, K.; Ahlers, J.; Cameron, M.J.; Smith, M.Z.; Spelman, T.; McMahon, J.; Velayudham, P.; et al. Activation of HIV Transcription with Short-Course Vorinostat in HIV-Infected Patients on Suppressive Antiretroviral Therapy. PLoS Pathog. 2014, 10, e1004473. [Google Scholar] [CrossRef]
- Rasmussen, T.A.; Tolstrup, M.; Brinkmann, C.R.; Olesen, R.; Erikstrup, C.; Solomon, A.; Winckelmann, A.; Palmer, S.; Dinarello, C.; Buzon, M.J.; et al. Panobinostat, a histone deacetylase inhibitor, for latent-virus reactivation in HIV-infected patients on suppressive antiretroviral therapy: A phase 1/2, single group, clinical trial. Lancet HIV 2014, 1, e13–e21. [Google Scholar] [CrossRef]
- Søgaard, O.S.; Graversen, M.E.; Leth, S.; Olesen, R.; Brinkmann, C.R.; Nissen, S.K.; Kjaer, A.S.; Schleimann, M.H.; Denton, P.W.; Hey-Cunningham, W.J.; et al. The Depsipeptide Romidepsin Reverses HIV-1 Latency In Vivo. PLoS Pathog. 2015, 11, e1005142. [Google Scholar] [CrossRef]
- Xing, S.; Bullen, C.K.; Shroff, N.S.; Shan, L.; Yang, H.-C.; Manucci, J.L.; Bhat, S.; Zhang, H.; Margolick, J.B.; Quinn, T.C.; et al. Disulfiram Reactivates Latent HIV-1 in a Bcl-2-Transduced Primary CD4+ T Cell Model without Inducing Global T Cell Activation. J. Virol. 2011, 85, 6060–6064. [Google Scholar] [CrossRef] [PubMed]
- Doyon, G.; Zerbato, J.; Mellors, J.W.; Sluis-Cremer, N. Disulfiram reactivates latent HIV-1 expression through depletion of the phosphatase and tensin homolog. AIDS 2013, 27, F7–F11. [Google Scholar] [CrossRef]
- Elliott, J.H.; McMahon, J.H.; Chang, C.C.; Lee, S.A.; Hartogensis, W.; Bumpus, N.N.; Savic, R.; Roney, J.; Hoh, R.; Solomon, A.; et al. Short-term administration of disulfiram for reversal of latent HIV infection: A phase 2 dose-escalation study. Lancet HIV 2015, 2, e520–e529. [Google Scholar] [CrossRef]
- Spivak, A.M.; Andrade, A.; Eisele, E.; Hoh, R.; Bacchetti, P.; Bumpus, N.N.; Emad, F.; Buckheit, R., III; McCance-Katz, E.F.; Lai, J.; et al. A pilot study assessing the safety and latency-reversing activity of disulfiram in HIV-1-infected adults on antiretroviral therapy. Clin. Infect. Dis. 2014, 58, 883–890. [Google Scholar] [CrossRef] [PubMed]
- Madrid-Elena, N.; Garcia-Bermejo, M.L.; Serrano-Villar, S.; Diaz-de Santiago, A.; Sastre, B.; Gutierrez, C.; Dronda, C.; Díaz, M.C.; Domínguez, E.; López-Huertas, M.R.; et al. Maraviroc is associated with latent HIV-1 reactivation through NF-kappaB activation in resting CD4(+) T cells from HIV-Infected Individuals on Suppressive Antiretroviral Therapy. J. Virol. 2018, 92, e01931-17. [Google Scholar] [CrossRef]
- Beans, E.J.; Fournogerakis, D.; Gauntlett, C.; Heumann, L.V.; Kramer, R.; Marsden, M.D.; Murray, D.; Chun, T.-W.; Zack, J.A.; Wender, P.A. Highly potent, synthetically accessible prostratin analogs induce latent HIV expression in vitro and ex vivo. Proc. Natl. Acad. Sci. USA 2013, 110, 11698–11703. [Google Scholar] [CrossRef] [PubMed]
- Muñoz, E.; Vo, M.Q.; Perez, M.; Calzado, M.A.; Moreno, S.; Appendino, G.; Munoz, E. Activation of Latent HIV-1 Expression by Protein Kinase C Agonists. A Novel Therapeutic Approach to Eradicate HIV-1 Reservoirs. Curr. Drug Targets 2011, 12, 348–356. [Google Scholar] [CrossRef]
- Friedman, J.; Cho, W.-K.; Chu, C.K.; Keedy, K.S.; Archin, N.M.; Margolis, D.M.; Karn, J. Epigenetic Silencing of HIV-1 by the Histone H3 Lysine 27 Methyltransferase Enhancer of Zeste 2. J. Virol. 2011, 85, 9078–9089. [Google Scholar] [CrossRef]
- Imai, K.; Togami, H.; Okamoto, T. Involvement of Histone H3 Lysine 9 (H3K9) Methyltransferase G9a in the Maintenance of HIV-1 Latency and Its Reactivation by BIX01294. J. Biol. Chem. 2010, 285, 16538–16545. [Google Scholar] [CrossRef]
- Bouchat, S.; Gatot, J.-S.; Kabeya, K.; Cardona, C.; Colin, L.; Herbein, G.; De Wit, S.; Clumeck, N.; Lambotte, O.; Rouzioux, C.; et al. Histone methyltransferase inhibitors induce HIV-1 recovery in resting CD4+ T cells from HIV-1-infected HAART-treated patients. AIDS 2012, 26, 1473–1482. [Google Scholar] [CrossRef]
- Bernhard, W.; Barreto, K.; Saunders, A.; Dahabieh, M.S.; Johnson, P.; Sadowski, I. The Suv39H1 methyltransferase inhibitor chaetocin causes induction of integrated HIV-1 without producing a T cell response. FEBS Lett. 2011, 585, 3549–3554. [Google Scholar] [CrossRef] [PubMed]
- Ebina, H.; Misawa, N.; Kanemura, Y.; Koyanagi, Y. Harnessing the CRISPR/Cas9 system to disrupt latent HIV-1 provirus. Sci. Rep. 2013, 3, srep02510. [Google Scholar] [CrossRef]
- Hu, W.; Kaminski, R.; Yang, F.; Zhang, Y.; Cosentino, L.; Li, F.; Luo, B.; Alvarez-Carbonell, D.; Garcia-Mesa, Y.; Karn, J.; et al. RNA-directed gene editing specifically eradicates latent and prevents new HIV-1 infection. Proc. Natl. Acad. Sci. USA 2014, 111, 11461–11466. [Google Scholar] [CrossRef]
- Manjunath, M.N.; Yi, G.; Dang, Y.; Shankar, P. Newer Gene Editing Technologies toward HIV Gene Therapy. Viruses 2013, 5, 2748–2766. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Pan, Q.; Gendron, P.; Zhu, W.; Guo, F.; Cen, S.; Wainberg, M.A.; Liang, C. CRISPR/Cas9-Derived Mutations Both Inhibit HIV-1 Replication and Accelerate Viral Escape. Cell Rep. 2016, 15, 481–489. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Wang, W.; Cui, Y.C.; Pan, Q.; Zhu, W.; Gendron, P.; Guo, F.; Cen, S.; Witcher, M.; Liang, C. HIV-1 Employs Multiple Mechanisms to Resist Cas9/Single Guide RNA Targeting the Viral Primer Binding Site. J. Virol. 2018, 92, 01135-18. [Google Scholar] [CrossRef]
- Schönle, A.; Hartl, F.A.; Mentzel, J.; Nöltner, T.; Rauch, K.S.; Prestipino, A.; Wohlfeil, S.; Apostolova, P.; Hechinger, A.-K.; Melchinger, W.; et al. Caveolin-1 regulates TCR signal strength and regulatory T-cell differentiation into alloreactive T cells. Blood 2016, 127, 1930–1939. [Google Scholar] [CrossRef]
- Borger, J.G.; Morrison, V.L.; Filby, A.; Garcia, C.; Uotila, L.M.; Simbari, F.; Fagerholm, S.C.; Zamoyska, R. Caveolin-1 Influences LFA-1 Redistribution upon TCR Stimulation in CD8 T Cells. J. Immunol. 2017, 199, 874–884. [Google Scholar] [CrossRef]
- Tomassian, T.; Humphries, L.A.; Liu, S.D.; Silva, O.; Brooks, D.G.; Miceli, M.C. Caveolin-1 orchestrates TCR synaptic polarity, signal specificity, and function in CD8 T cells. J. Immunol. 2011, 187, 2993–3002. [Google Scholar] [CrossRef]
- Rothberg, K.G.; Heuser, J.E.; Donzell, W.C.; Ying, Y.-S.; Glenney, J.R.; Anderson, R.G. Caveolin, a protein component of caveolae membrane coats. Cell 1992, 68, 673–682. [Google Scholar] [CrossRef]
- Galbiati, F.; Volonte, D.; Liu, J.; Capozza, F.; Frank, P.G.; Zhu, L.; Pestell, R.G.; Lisanti, M.P. Caveolin-1 expression negatively regulates cell cycle progression by inducing G(0)/G(1) arrest via a p53/p21(WAF1/Cip1)-dependent mechanism. Mol. Biol. Cell 2001, 12, 2229–2244. [Google Scholar] [CrossRef] [PubMed]
- Fielding, P.E.; Russel, J.S.; Spencer, T.A.; Hakamata, H.; Nagao, K.; Fielding, C.J. Sterol Efflux to Apolipoprotein A-I Originates from Caveolin-Rich Microdomains and Potentiates PDGF-Dependent Protein Kinase Activity†. Biochemistry 2002, 41, 4929–4937. [Google Scholar] [CrossRef] [PubMed]
- Gargalovic, P.; Dory, L. Caveolins and macrophage lipid metabolism. J. Lipid Res. 2002, 44, 11–21. [Google Scholar] [CrossRef]
- Gargalovic, P.; Dory, L. Cellular apoptosis is associated with increased caveolin-1 expression in macrophages. J. Lipid Res. 2003, 44, 1622–1632. [Google Scholar] [CrossRef]
- Le Feuvre, R.; Guay, G.; Altschuler, Y.; Nabi, I.R. Caveolin-1 Is a Negative Regulator of Caveolae-mediated Endocytosis to the Endoplasmic Reticulum. J. Biol. Chem. 2001, 277, 3371–3379. [Google Scholar] [CrossRef]
- Chao, W.T.; Fan, S.S.; Chen, J.K.; Yang, V.C. Visualizing caveolin-1 and HDL in cholesterol-loaded aortic endothelial cells. J. Lipid Res. 2003, 44, 1094–1099. [Google Scholar] [CrossRef]
- Fu, Y.; Hoang, A.; Escher, G.; Parton, R.G.; Krozowski, Z.; Sviridov, D.; Ge, K.; Roeder, R.G.; Hankinson, O.; Wang, S. Expression of Caveolin-1 Enhances Cholesterol Efflux in Hepatic Cells. J. Biol. Chem. 2004, 279, 14140–14146. [Google Scholar] [CrossRef]
- Boyd, N.L.; Park, H.; Yi, H.; Boo, Y.C.; Sorescu, G.P.; Sykes, M.; Jo, H. Chronic shear induces caveolae formation and alters ERK and Akt responses in endothelial cells. Am. J. Physiol. Circ. Physiol. 2003, 285, H1113–H1122. [Google Scholar] [CrossRef] [PubMed]
- Muriel, O.; Echarri, A.; Hellriegel, C.; Pavon, D.M.; Beccari, L.; Del Pozo, M.A. Phosphorylated filamin A regulates actin-linked caveolae dynamics. J. Cell Sci. 2011, 124, 2763–2776. [Google Scholar] [CrossRef] [PubMed]
- Sinha, B.; Koester, D.; Ruez, R.; Gonnord, P.; Bastiani, M.; Abankawa, D.; Stan, R.V.; Butler-Browne, G.; Vedie, B.; Johannes, L.; et al. Cells Respond to Mechanical Stress by Rapid Disassembly of Caveolae. Cell 2011, 144, 402–413. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Vicente, R.; Pavón, D.M.; Martín-Padura, I.; Català-Montoro, M.; Díez-Sánchez, A.; Quílez-Álvarez, A.; López, J.A.; Sánchez-Álvarez, M.; Vázquez, J.; Strippoli, R.; et al. Caveolin-1 Modulates Mechanotransduction Responses to Substrate Stiffness through Actin-Dependent Control of YAP. Cell Rep. 2018, 25, 1622–1635. [Google Scholar] [CrossRef] [PubMed]
- Fiala, G.J.; Minguet, S. Caveolin-1: The Unnoticed Player in TCR and BCR Signaling. Adv. Immunol. 2018, 137, 83–133. [Google Scholar] [CrossRef]
- Lei, M.G.; Morrison, D.C. Differential Expression of Caveolin-1 in Lipopolysaccharide-Activated Murine Macrophages. Infect. Immun. 2000, 68, 5084–5089. [Google Scholar] [CrossRef]
- Lei, M.G.; Tan, X.; Qureshi, N.; Morrison, D.C. Regulation of Cellular Caveolin-1 Protein Expression in Murine Macrophages by Microbial Products. Infect. Immun. 2005, 73, 8136–8143. [Google Scholar] [CrossRef] [PubMed]
- Mergia, A. The Role of Caveolin 1 in HIV Infection and Pathogenesis. Viruses 2017, 9, 129. [Google Scholar] [CrossRef] [PubMed]
- Palade GEFsobc. Fine structure of blood capillaries. J. Appl. Phys. 1953, 24, 1424. [Google Scholar]
- Yamada, E. The fine structure of the gall bladder epithelium of the mouse. J. Cell Biol. 1955, 1, 445–458. [Google Scholar] [CrossRef]
- Harris, J.; Werling, D.; Koss, M.; Monaghan, P.; Taylor, G.; Howard, C.J. Expression of caveolin by bovine lymphocytes and antigen-presenting cells. Immunology 2002, 105, 190–195. [Google Scholar] [CrossRef]
- Quest, A.F.G.; Leyton, L.; Párraga, M. Caveolins, caveolae, and lipid rafts in cellular transport, signaling, and disease. Biochem. Cell Biol. 2004, 82, 129–144. [Google Scholar] [CrossRef]
- Stan, R.V.; Tkachenko, E.; Niesman, I.R. PV1 Is a Key Structural Component for the Formation of the Stomatal and Fenestral Diaphragms. Mol. Biol. Cell 2004, 15, 3615–3630. [Google Scholar] [CrossRef] [PubMed]
- Stan, R.V. Structure of caveolae. Biochim. Biophys. Acta (BBA) Bioenerg. 2005, 1746, 334–348. [Google Scholar] [CrossRef] [PubMed]
- Parton, R.G.; Simons, K. The multiple faces of caveolae. Nat. Rev. Mol. Cell Biol. 2007, 8, 185–194. [Google Scholar] [CrossRef] [PubMed]
- Mercier, I.; Jasmin, J.-F.; Pavlides, S.; Minetti, C.; Flomenberg, N.; Pestell, R.G.; Frank, P.G.; Sotgia, F.; Lisanti, M.P. Clinical and translational implications of the caveolin gene family: Lessons from mouse models and human genetic disorders. Lab. Investig. 2009, 89, 614–623. [Google Scholar] [CrossRef]
- Fra, A.M.; Williamson, E.; Simons, K.; Parton, R.G. De novo formation of caveolae in lymphocytes by expression of VIP21-caveolin. Proc. Natl. Acad. Sci. USA 1995, 92, 8655–8659. [Google Scholar] [CrossRef]
- Hatanaka, M.; Maedab, T.; Ikemotoc, T.; Morib, H.; Seyad, T.; Shimizuac, A. Expression of Caveolin-1 in Human T Cell Leukemia Cell Lines. Biochem. Biophys. Res. Commun. 1998, 253, 382–387. [Google Scholar] [CrossRef]
- Vallejo, J.; Hardin, C.D. Expression of caveolin-1 in lymphocytes induces caveolae formation and recruitment of phosphofructokinase to the plasma membrane. FASEB J. 2005, 19, 1–19. [Google Scholar] [CrossRef]
- Medina, F.A.; Williams, T.M.; Sotgia, F.; Tanowitz, H.B.; Lisanti, M.P. A Novel Role for Caveolin-1 in B Lymphocyte Function and the Development of Thymus-Independent Immune Responses. Cell Cycle 2006, 5, 1865–1871. [Google Scholar] [CrossRef]
- Minguet, S.; Kläsener, K.; Schaffer, A.-M.; Fiala, G.J.; Osteso-Ibánez, T.; Raute, K.; Navarro-Lérida, I.; Hartl, F.A.; Seidl, M.; Reth, M.; et al. Caveolin-1-dependent nanoscale organization of the BCR regulates B cell tolerance. Nat. Immunol. 2017, 18, 1150–1159. [Google Scholar] [CrossRef]
- Stahlhut, M.; Van Deurs, B. Identification of Filamin as a Novel Ligand for Caveolin-1: Evidence for the Organization of Caveolin-1–associated Membrane Domains by the Actin Cytoskeleton. Mol. Biol. Cell 2000, 11, 325–337. [Google Scholar] [CrossRef]
- Cao, H.; Courchesne, W.E.; Mastick, C.C. A Phosphotyrosine-dependent Protein Interaction Screen Reveals a Role for Phosphorylation of Caveolin-1 on Tyrosine 14. J. Biol. Chem. 2002, 277, 8771–8774. [Google Scholar] [CrossRef] [PubMed]
- Yao, Q.; Chen, J.; Cao, H.; Orth, J.D.; McCaffery, J.M.; Stan, R.V.; McNiven, M.A. Caveolin-1 Interacts Directly with Dynamin-2. J. Mol. Biol. 2005, 348, 491–501. [Google Scholar] [CrossRef] [PubMed]
- Schamel, W.W.; Arechaga, I.; RisueñoR, M.; Van Santen, H.M.; Cabezas, P.; Risco, C.; Valpuesta, J.M.; Alarcón, B. Coexistence of multivalent and monovalent TCRs explains high sensitivity and wide range of response. J. Exp. Med. 2005, 202, 493–503. [Google Scholar] [CrossRef] [PubMed]
- Alarcón, B.; Swamy, M.; Van Santen, H.M.; Schamel, W.W. T-cell antigen-receptor stoichiometry: Pre-clustering for sensitivity. EMBO Rep. 2006, 7, 490–495. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Martin, N.; Risueno, R.M.; Morreale, A.; Zaldivar, I.; Fernandez-Arenas, E.; Herranz, F.; Ortiz, A.R.; Alarcon, B. Cooperativity between T cell receptor complexes revealed by conformational mutants of CD3epsilon. Sci. Signal. 2009, 2, ra43. [Google Scholar] [CrossRef]
- Molnar, E.; Deswal, S.; Schamel, W.W. Pre-clustered TCR complexes. FEBS Lett. 2010, 584, 4832–4837. [Google Scholar] [CrossRef]
- Kumar, R.; Ferez, M.; Swamy, M.; Arechaga, I.; Rejas, M.T.; Valpuesta, J.; Schamel, W.W.; Alarcón, B.; Van Santen, H.M. Increased Sensitivity of Antigen-Experienced T Cells through the Enrichment of Oligomeric T Cell Receptor Complexes. Immunity 2011, 35, 375–387. [Google Scholar] [CrossRef]
- Schamel, W.W.; Alarcón, B. Organization of the resting TCR in nanoscale oligomers. Immunol. Rev. 2012, 251, 13–20. [Google Scholar] [CrossRef]
- Swamy, M.; Beck-Garcia, K.; Beck-Garcia, E.; Hartl, F.A.; Morath, A.; Yousefi, O.S.; Dopfer, E.P.; Molnar, E.; Schulze, A.K.; Blanco, R.; et al. A Cholesterol-Based Allostery Model of T Cell Receptor Phosphorylation. Immunity 2016, 44, 1091–1101. [Google Scholar] [CrossRef]
- Wang, F.; Beck-García, K.; Zorzin, C.; Schamel, W.W.A.; Davis, M.M. Inhibition of T cell receptor signaling by cholesterol sulfate, a naturally occurring derivative of membrane cholesterol. Nat. Immunol. 2016, 17, 844–850. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Bai, Y.; Xiong, Y.; Zhang, J.; Chen, S.; Zheng, X.; Meng, X.; Li, L.; Wang, J.; Xu, C.; et al. Potentiating the antitumour response of CD8+ T cells by modulating cholesterol metabolism. Nat. Cell Biol. 2016, 531, 651–655. [Google Scholar] [CrossRef]
- Schamel, W.W.; Alarcon, B.; Höfer, T.; Minguet, S. The Allostery Model of TCR Regulation. J. Immunol. 2016, 198, 47–52. [Google Scholar] [CrossRef]
- Head, B.P.; Patel, H.H.; Insel, P.A. Interaction of membrane/lipid rafts with the cytoskeleton: Impact on signaling and function. Biochim. Biophys. Acta (BBA) Biomembr. 2014, 1838, 532–545. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.M.; Kim, H.P.; Song, R.; Choi, A.M. Caveolin-1 confers antiinflammatory effects in murine macrophages via the MKK3/p38 MAPK pathway. Am. J. Respir. Cell Mol. Biol. 2006, 34, 434–442. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.M.; Nadeau, P.E.; Lin, S.; Abbott, J.E.; Mergia, A. Caveolin 1 inhibits HIV replication by transcriptional repression mediated through NF-κB Virol. J. Virol. 2011, 85, 5483–5493. [Google Scholar] [CrossRef]
- Fakhrzadeh, L.; Laskin, J.D.; Laskin, D.L. Regulation of caveolin-1 expression, nitric oxide production and tissue injury by tumor necrosis factor-α following ozone inhalation. Toxicol. Appl. Pharmacol. 2008, 227, 380–389. [Google Scholar] [CrossRef][Green Version]
- Thangavel, C.; Gomes, C.M.; Zderic, S.A.; Javed, E.; Addya, S.; Singh, J.; Das, S.; Birbe, R.; Den, R.B.; Rattan, S.; et al. NF-κB and GATA-Binding Factor 6 Repress Transcription of Caveolins in Bladder Smooth Muscle Hypertrophy. Am. J. Pathol. 2019, 189, 847–867. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.-Y.; Lai, T.-Y.; Tsai, M.-K.; Chang, Y.-C.; Ho, Y.-H.; Yu, I.-S.; Yeh, T.-W.; Chou, C.-C.; Lin, Y.-S.; Lawrence, T.; et al. The ubiquitin ligase ZNRF1 promotes caveolin-1 ubiquitination and degradation to modulate inflammation. Nat. Commun. 2017, 8, 15502. [Google Scholar] [CrossRef] [PubMed]
- Zou, H.; Stoppani, E.; Volonte, D.; Galbiati, F. Caveolin-1, cellular senescence and age-related diseases. Mech. Ageing Dev. 2011, 132, 533–542. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.M.; Kim, H.P.; Nakahira, K.; Ryter, S.W.; Choi, A.M. The Heme Oxygenase-1/Carbon Monoxide Pathway Suppresses TLR4 Signaling by Regulating the Interaction of TLR4 with Caveolin-1. J. Immunol. 2009, 182, 3809–3818. [Google Scholar] [CrossRef]
- Marincowitz, C.; Genis, A.; Goswami, N.; De Boever, P.; Nawrot, T.S.; Strijdom, H. Vascular endothelial dysfunction in the wake of HIV and ART. FEBS J. 2018, 286, 1256–1270. [Google Scholar] [CrossRef]
- Chau-Chung, W.; Wang, S.-H.; Kuan, I.-I.; Tseng, W.-K.; Chen, M.-F.; Wu, J.-C.; Chen, Y.-L. OxLDL upregulates caveolin-1 expression in macrophages: Role for caveolin-1 in the adhesion of oxLDL-treated macrophages to endothelium. J. Cell. Biochem. 2009, 107, 460–472. [Google Scholar] [CrossRef]
- Engelman, J.A.; Lee, R.J.; Karnezis, A.; Bearss, D.; Webster, M.; Siegel, P.; Muller, W.J.; Windle, J.J.; Pestell, R.G.; Lisanti, M.P. Reciprocal Regulation of Neu Tyrosine Kinase Activity and Caveolin-1 Protein Expressionin Vitroandin Vivo. J. Biol. Chem. 1998, 273, 20448–20455. [Google Scholar] [CrossRef]
- Koleske, A.J.; Baltimore, D.; Lisanti, M.P. Reduction of caveolin and caveolae in oncogenically transformed cells. Proc. Natl. Acad. Sci. USA 1995, 92, 1381–1385. [Google Scholar] [CrossRef]
- Park, D.S.; Razani, B.; Lasorella, A.; Schreiber-Agus, N.; Pestell, R.G.; Iavarone, A.; Lisanti, M.P. Evidence that Myc isoforms transcriptionally repress caveolin-1 gene expression via an INR-dependent mechanism. Biochemistry 2001, 40, 3354–3362. [Google Scholar] [CrossRef]
- Timme, T.L.; Goltsov, A.; Tahir, S.; Li, L.; Wang, J.; Ren, C.; Johnston, R.N.; Thompson, T.C. Caveolin-1 is regulated by c-myc and suppresses c-myc-induced apoptosis. Oncogene 2000, 19, 3256–3265. [Google Scholar] [CrossRef]
- Porter, K.M.; Sutliff, R.L. HIV-1, reactive oxygen species, and vascular complications. Free. Radic. Biol. Med. 2012, 53, 143–159. [Google Scholar] [CrossRef]
- Sabin, R.J.; Anderson, R.M. Cellular Senescence - its role in cancer and the response to ionizing radiation. Genome Integr. 2011, 2, 7. [Google Scholar] [CrossRef]
- Sutliff, R.L.; Dikalov, S.; Weiss, D.; Parker, J.; Raidel, S.; Racine, A.K.; Russ, R.; Haase, C.P.; Taylor, W.R.; Lewis, W. Nucleoside reverse transcriptase inhibitors impair endothelium-dependent relaxation by increasing superoxide. Am. J. Physiol. Circ. Physiol. 2002, 283, H2363–H2370. [Google Scholar] [CrossRef]
- Kline, E.R.; Bassit, L.; Hernandez-Santiago, B.I.; Detorio, M.A.; Liang, B.; Kleinhenz, D.J.; Walp, E.R.; Dikalov, S.; Jones, D.P.; Schinazi, R.F.; et al. Long-Term Exposure to AZT, but not d4T, Increases Endothelial Cell Oxidative Stress and Mitochondrial Dysfunction. Cardiovasc. Toxicol. 2008, 9, 1–12. [Google Scholar] [CrossRef]
- Pacher, P.; Gao, R.Y.; Mukhopadhyay, P.; Mohanraj, R.; Wang, H.; Horvath, B.; Yin, S. Resveratrol attenuates azidothymidine-induced cardiotoxicity by decreasing mitochondrial reactive oxygen species generation in human cardiomyocytes. Mol. Med. Rep. 2010, 4, 151–155. [Google Scholar] [CrossRef]
- Mak, I.T.; Nedelec, L.F.; Weglicki, W.B. Pro-oxidant properties and cytotoxicity of AZT-monophosphate and AZT. Cardiovasc. Toxicol. 2004, 4, 109–116. [Google Scholar] [CrossRef]
- Ben-Romano, R.; Rudich, A.; Etzion, S.; Potashnik, R.; Kagan, E.; Greenbaum, U.; Bashan, N. Nelfinavir induces adipocyte insulin resistance through the induction of oxidative stress: Differential protective effect of antioxidant agents. Antivir. Ther. 2006, 11, 1051. [Google Scholar]
- Mondal, D.; Pradhan, L.; Ali, M.; Agrawal, K.C. HAART Drugs Induce Oxidative Stress in Human Endothelial Cells and Increase Endothelial Recruitment of Mononuclear Cells: Exacerbation by Inflammatory Cytokines and Amelioration by Antioxidants. Cardiovasc. Toxicol. 2004, 4, 287–302. [Google Scholar] [CrossRef]
- Edeas, M. Strategies to Target Mitochondria and Oxidative Stress by Antioxidants: Key Points and Perspectives. Pharm. Res. 2011, 28, 2771–2779. [Google Scholar] [CrossRef]
- Chen, Q.; Ames, B.N. Senescence-like growth arrest induced by hydrogen peroxide in human diploid fibroblast F65 cells. Proc. Natl. Acad. Sci. USA 1994, 91, 4130–4134. [Google Scholar] [CrossRef]
- Chen, Q.M.; Bartholomew, J.C.; Campisi, J.; Acosta, M.; Reagan, J.D.; Ames, B.N. Molecular analysis of H2O2-induced senescent-like growth arrest in normal human fibroblasts: p53 and Rb control G1 arrest but not cell replication. Biochem. J. 1998, 332, 43–50. [Google Scholar] [CrossRef]
- Marnett, L.J. Oxyradicals and DNA damage. Carcinogenesis 2000, 21, 361–370. [Google Scholar] [CrossRef]
- Starke-Reed, P.; Yu, B. Protein Oxidation. In Methods in Aging Research; Informa UK Limited: London, UK, 1998; Volume 899, pp. 637–655. [Google Scholar]
- Yla-Herttuala, S. Oxidized LDL and atherogenesis. Ann. N. Y. Acad. Sci. 1999, 874, 134–137. [Google Scholar] [CrossRef]
- Finkel, T. Oxidant signals and oxidative stress. Curr. Opin. Cell Biol. 2003, 15, 247–254. [Google Scholar] [CrossRef]
- Frippiat, C.; Chen, Q.M.; Zdanov, S.; Magalhaes, J.-P.; Remacle, J.; Toussaint, O. Subcytotoxic H2O2Stress Triggers a Release of Transforming Growth Factor-β1, Which Induces Biomarkers of Cellular Senescence of Human Diploid Fibroblasts. J. Biol. Chem. 2000, 276, 2531–2537. [Google Scholar] [CrossRef]
- Frippiat, C.; Dewelle, J.; Remacle, J.; Toussaint, O. Signal transduction in H2O2-induced senescence-like phenotype in human diploid fibroblasts. Free Radic. Biol. Med. 2002, 33, 1334–1346. [Google Scholar] [CrossRef]
- Martindale, J.L.; Holbrook, N.J. Cellular response to oxidative stress: Signaling for suicide and survival. J. Cell. Physiol. 2002, 192, 1–15. [Google Scholar] [CrossRef]
- Williams, T.M.; Lisanti, M.P. Caveolin-1 in oncogenic transformation, cancer, and metastasis. Am. J. Physiol. Physiol. 2005, 288, C494–C506. [Google Scholar] [CrossRef]
- Lajoie, P.; Partridge, E.A.; Guay, G.; Goetz, J.G.; Pawling, J.; Lagana, A.; Joshi, B.; Dennis, J.W.; Nabi, I.R. Plasma membrane domain organization regulates EGFR signaling in tumor cells. J. Cell Biol. 2007, 179, 341–356. [Google Scholar] [CrossRef]
- Lajoie, P.; Nabi, I. Regulation of raft-dependent endocytosis. J. Cell. Mol. Med. 2007, 11, 644–653. [Google Scholar] [CrossRef]
- Bai, L.; Shi, G.; Zhang, L.; Guan, F.; Ma, Y.; Li, Q.; Cong, Y.-S. Cav-1 deletion impaired hematopoietic stem cell function. Cell Death Dis. 2014, 5, e1140. [Google Scholar] [CrossRef]
- Hulit, J.; Bash, T.; Fu, M.; Galbiati, F.; Albanese, C.; Sage, D.R.; Schlegel, A.; Zhurinsky, J.; Shtutman, M.S.; Ben-Ze’Ev, A.; et al. The Cyclin D1 Gene Is Transcriptionally Repressed by Caveolin-1. J. Biol. Chem. 2000, 275, 21203–21209. [Google Scholar] [CrossRef]
- Lee, S.W.; Reimer, C.L.; Oh, P.; Campbell, D.B.; E Schnitzer, J. Tumor cell growth inhibition by caveolin re-expression in human breast cancer cells. Oncogene 1998, 16, 1391–1397. [Google Scholar] [CrossRef]
- Zeng, W.; Tang, J.; Li, H.; Xu, H.; Lu, H.; Peng, H.; Lin, C.; Gao, R.; Lin, S.; Lin, K.; et al. Caveolin-1 deficiency protects pancreatic beta cells against palmitate-induced dysfunction and apoptosis. Cell Signal. 2018, 47, 65–78. [Google Scholar] [CrossRef]
- Park, J.; Lim, C.H.; Ham, S.; Kim, S.S.; Choi, B.-S.; Roh, T.-Y. Genome-wide analysis of histone modifications in latently HIV-1 infected T cells. AIDS 2014, 28, 1719–1728. [Google Scholar] [CrossRef]
- Lin, S.; Wang, X.M.; Nadeau, P.E.; Mergia, A. HIV Infection Upregulates Caveolin 1 Expression to Restrict Virus Production. J. Virol. 2010, 84, 9487–9496. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.; Nadeau, P.E.; Wang, X.; Mergia, A. Caveolin-1 reduces HIV-1 infectivity by restoration of HIV Nef mediated impairment of cholesterol efflux by apoA-I. Retrovirology 2012, 9, 85. [Google Scholar] [CrossRef] [PubMed]
- Ménager, M.M.; Littman, D.R. Actin Dynamics Regulates Dendritic Cell-Mediated Transfer of HIV-1 to T Cells. Cell 2016, 164, 695–709. [Google Scholar] [CrossRef]
- Piguet, V.; Sattentau, Q. Dangerous liaisons at the virological synapse. J. Clin. Investig. 2004, 114, 605–610. [Google Scholar] [CrossRef]
- Jiménez-Baranda, S.; Gómez-Moutón, C.; Rojas, A.M.; Martínez-Prats, L.; Mira, E.; LaCalle, R.A.; Valencia, A.; Dimitrov, D.S.; Viola, A.; Delgado, R.; et al. Filamin-A regulates actin-dependent clustering of HIV receptors. Nat. Cell Biol. 2007, 9, 838–846. [Google Scholar] [CrossRef] [PubMed]
- Sverdlov, M.; Shinin, V.; Place, A.T.; Castellon, M.; Minshall, R.D. Filamin A Regulates Caveolae Internalization and Trafficking in Endothelial Cells. Mol. Biol. Cell 2009, 20, 4531–4540. [Google Scholar] [CrossRef]
- Hovanessian, A.G.; Briand, J.-P.; Said, E.A.; Svab, J.; Ferris, S.; Dali, H.; Muller, S.; Desgranges, C.; Krust, B. The Caveolin-1 Binding Domain of HIV-1 Glycoprotein gp41 Is an Efficient B Cell Epitope Vaccine Candidate against Virus Infection. Immunity 2004, 21, 617–627. [Google Scholar] [CrossRef]
- Simmons, G.E.; Jr Taylor, H.E.; Hildreth, J.E. Caveolin-1 suppresses human immunodeficiency virus-1 replication by inhibiting acetylation of NF-kappaB. Virology 2012, 432, 110–119. [Google Scholar] [CrossRef]
- Wang, X.M.; Nadeau, P.E.; Lo, Y.-T.; Mergia, A. Caveolin-1 Modulates HIV-1 Envelope-Induced Bystander Apoptosis through gp41. J. Virol. 2010, 84, 6515–6526. [Google Scholar] [CrossRef]
- Ran, X.; Xiaozhuo, R.; Trajtman, A.; Xu, W.; Kobinger, G.; Keynan, Y.; Yao, X. HIV-1 envelope glycoprotein stimulates viral transcription and increases the infectivity of the progeny virus through the manipulation of cellular machinery. Sci. Rep. 2017, 7, 1–16. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).