Blood Parasites in Endangered Wildlife-Trypanosomes Discovered during a Survey of Haemoprotozoa from the Tasmanian Devil

 ,
, 
 , , , ,
, , , ,  , and
, and 
Abstract
1. Introduction
2. Materials and Methods
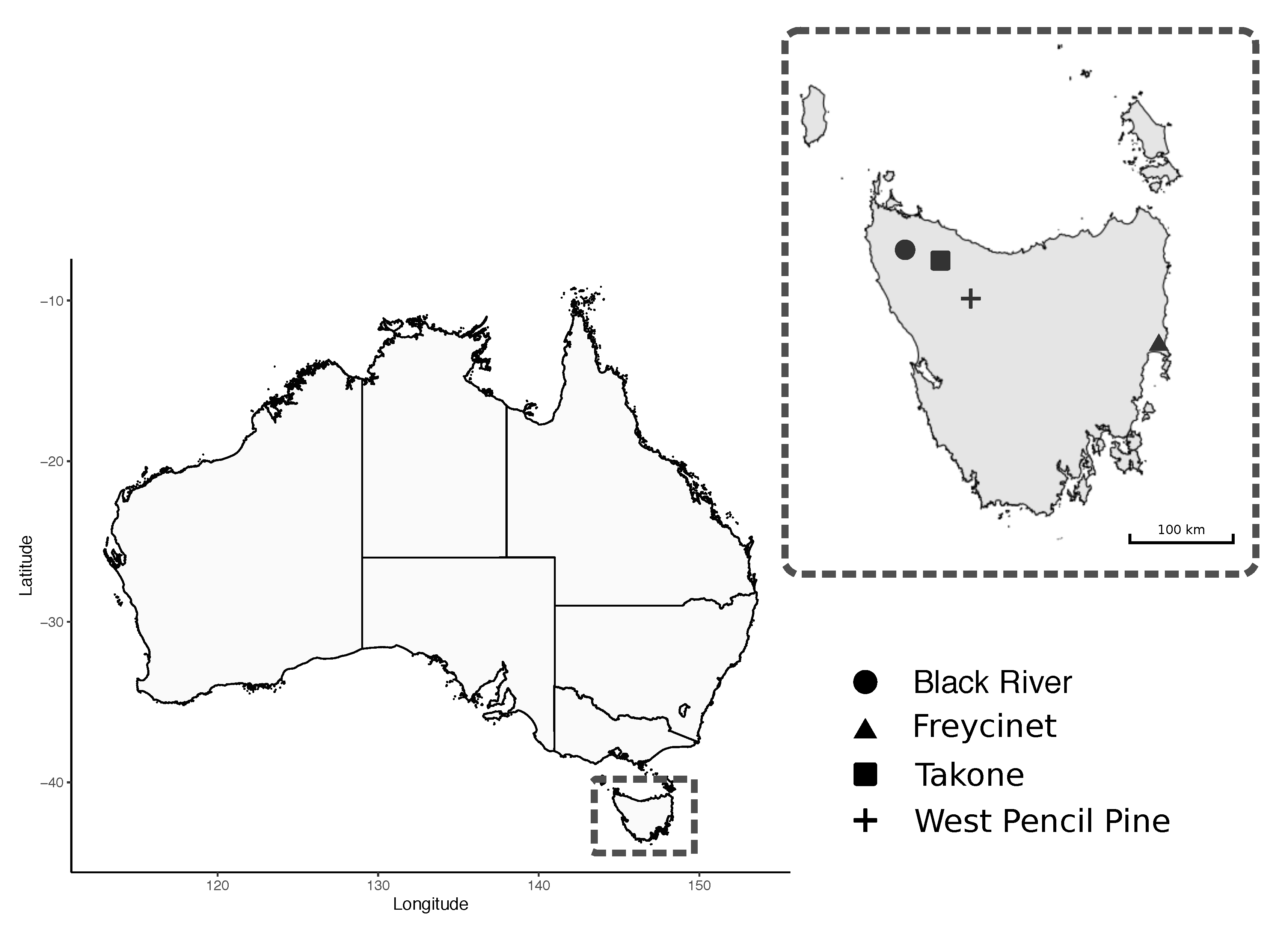
2.1. Study Sites and Sampling
2.2. Molecular Screening
2.2.1. DNA Extraction
2.2.2. PCR Assays
2.2.3. Gel Electrophoresis and Sanger Sequencing
2.2.4. Phylogenetic Analysis
2.3. Statisical Analysis
2.4. Microscopy of Blood Smears
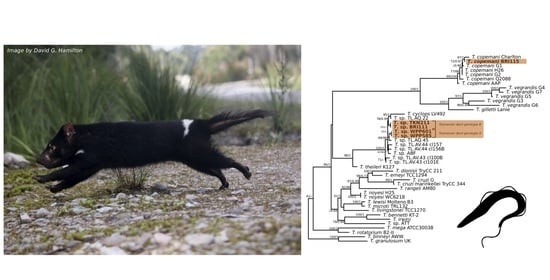
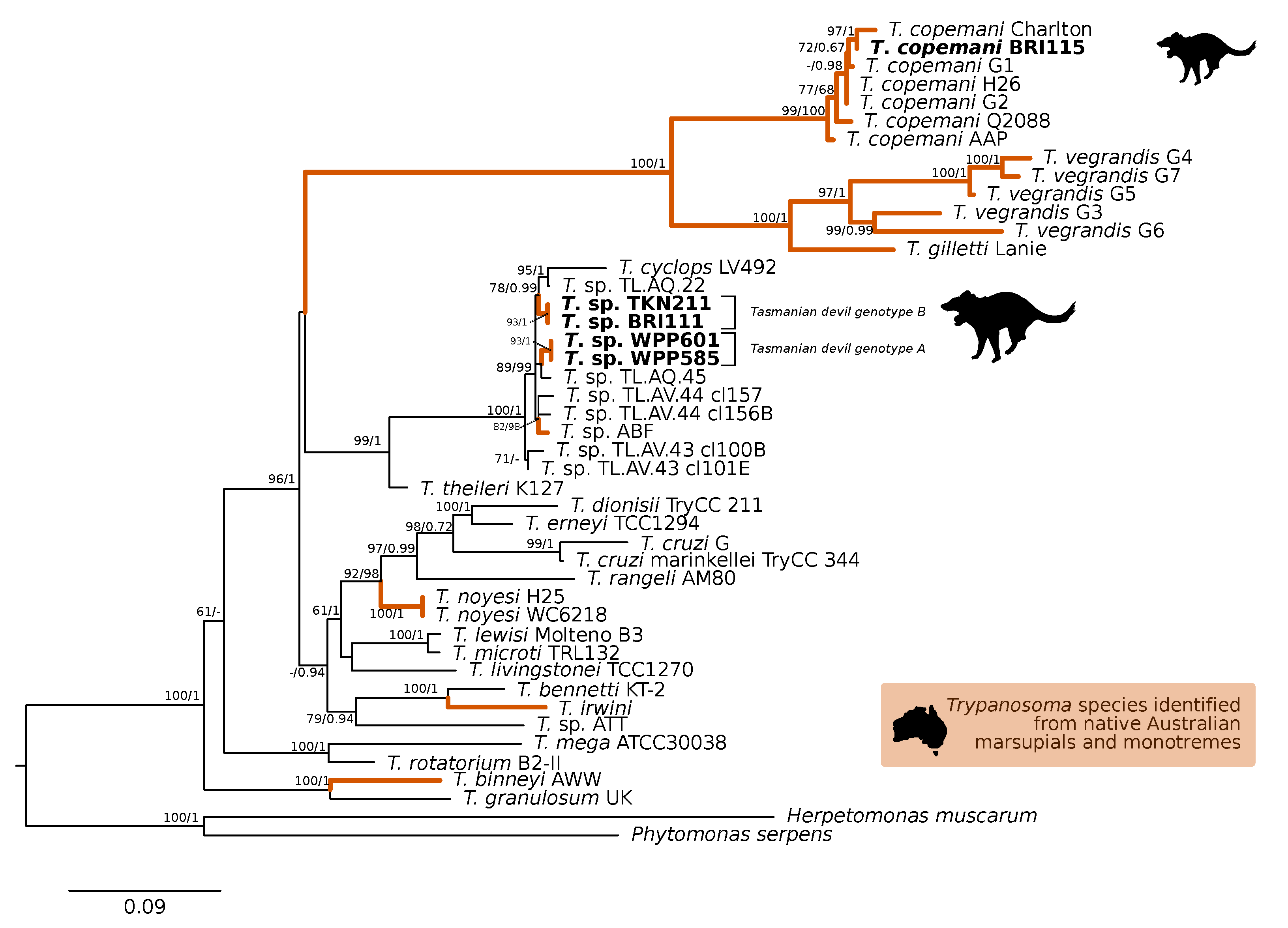
3. Results
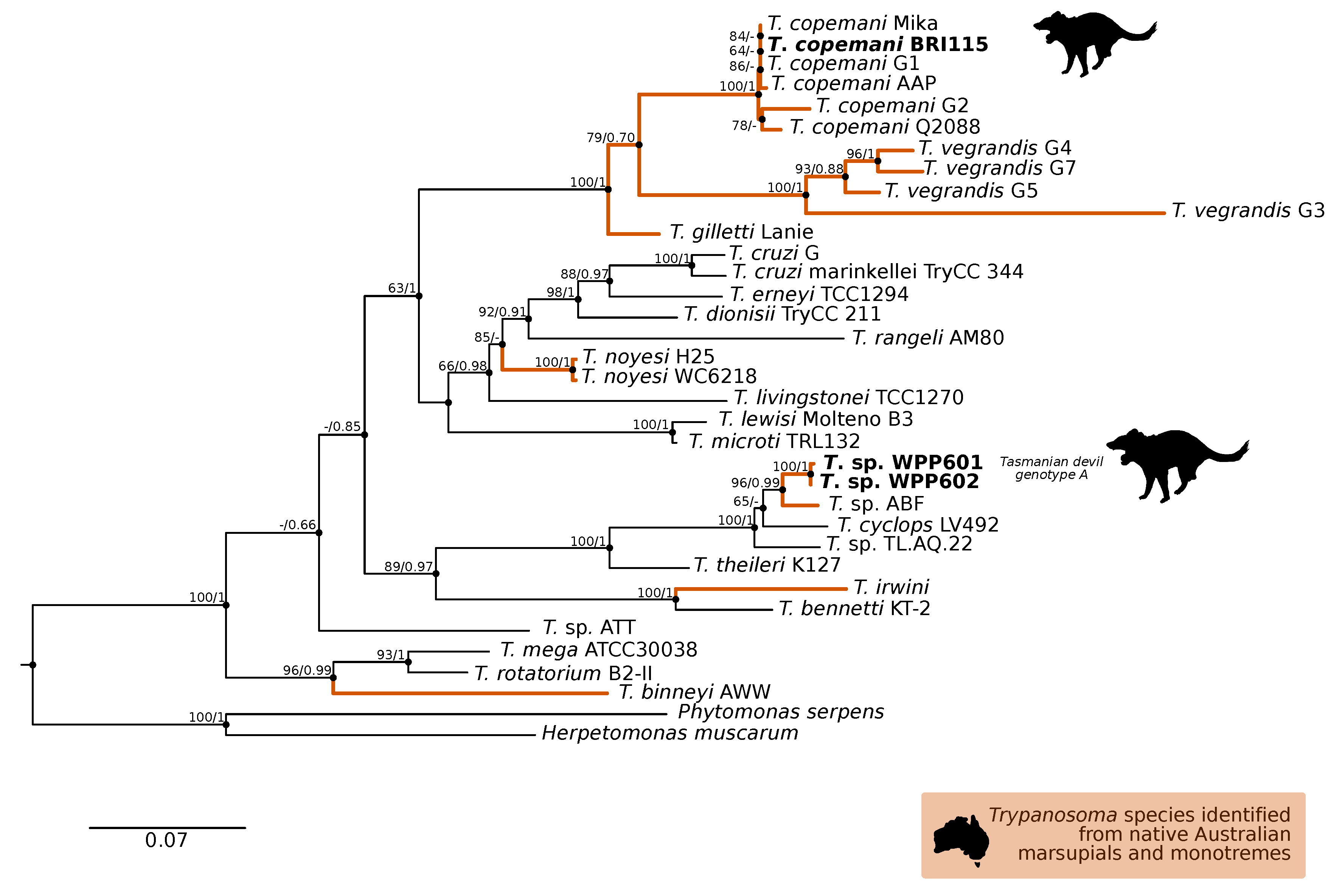
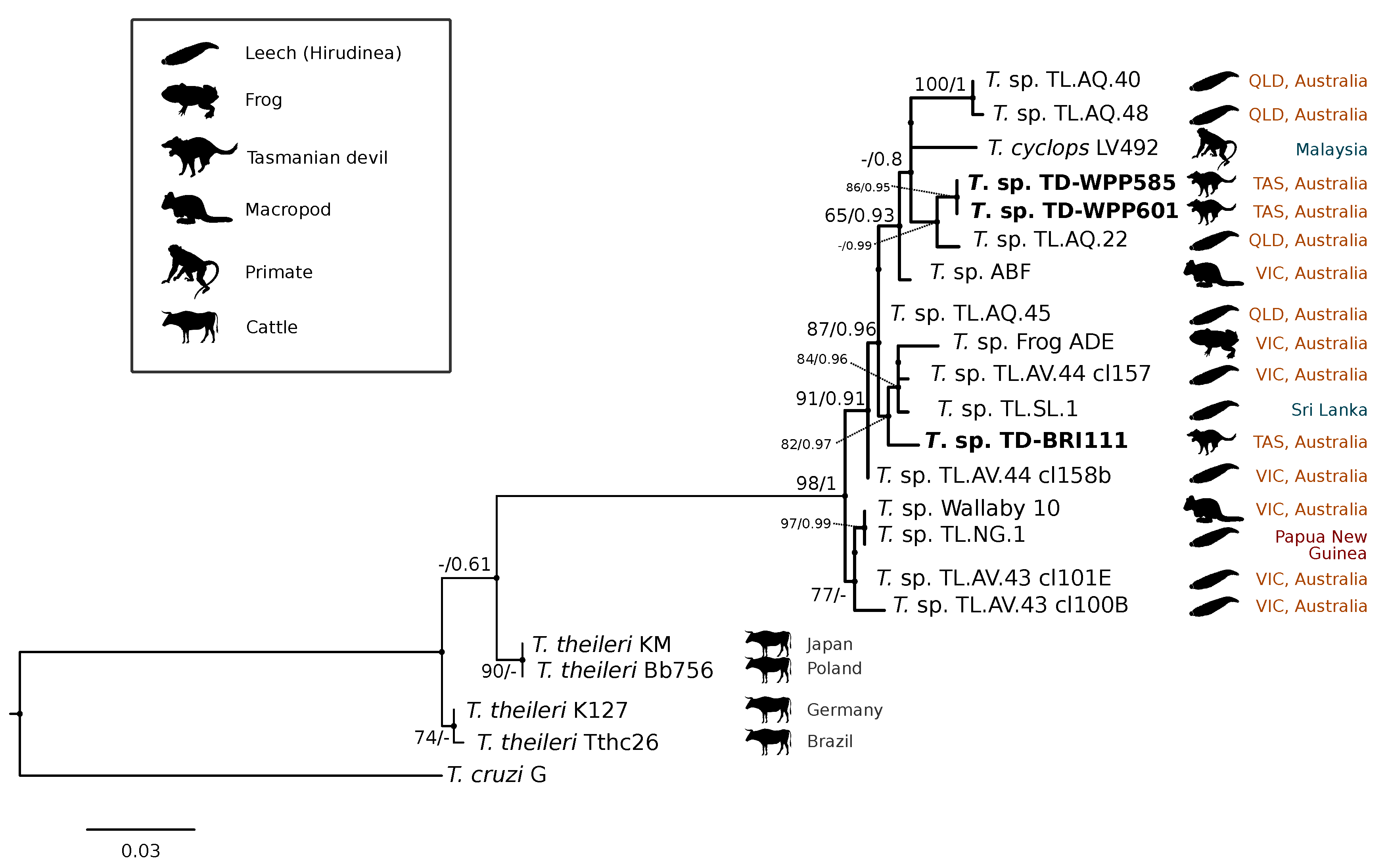
3.1. Molecular Screening
3.2. Microscopy
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| 18S rDNA | 18S ribosomal DNA |
| DFTD | Devil tumour facial disease |
| gDNA | genomic DNA |
| gGAPDH | glycosomal glyceraldehyde phosphate dehydrogenase gene |
| SNP | single nucleotide polymorphism |
References
- O’Donoghue, P. Haemoprotozoa: Making biological sense of molecular phylogenies. Int. J. Parasitol. Parasites Wildl. 2017, 6, 241–256. [Google Scholar] [CrossRef]
- Paparini, A.; Macgregor, J.; Ryan, U.M.; Irwin, P.J. First molecular characterization of Theileria ornithorhynchi Mackerras, 1959: Yet another challenge to the systematics of the piroplasms. Protist 2015, 166, 609–620. [Google Scholar] [CrossRef]
- Clément, L.; Dietrich, M.; Markotter, W.; Fasel, N.; Monadjem, A.; López-Baucells, A.; Scaravelli, D.; Théou, P.; Pigeault, R.; Ruedi, M.; et al. Out of africa: The origins of the protozoan blood parasites of the Trypanosoma cruzi clade found in bats from Africa. Mol. Phylogenet. Evol. 2020, 145, 106705. [Google Scholar] [CrossRef]
- Brüniche-Olsen, A.; Jones, M.E.; Burridge, C.P.; Murchison, E.P.; Holland, B.R.; Austin, J.J. Ancient DNA tracks the mainland extinction and island survival of the Tasmanian devil. J. Biogeogr. 2018, 45, 963–976. [Google Scholar] [CrossRef]
- Jones, M.E.; Paetkau, D.; Geffen, E.; Moritz, C. Genetic diversity and population structure of Tasmanian devils, the largest marsupial carnivore. Mol. Ecol. 2004, 13, 2197–2209. [Google Scholar] [CrossRef]
- Patton, A.H.; Margres, M.J.; Stahlke, A.R.; Hendricks, S.; Lewallen, K.; Hamede, R.K.; Ruiz-Aravena, M.; Ryder, O.; McCallum, H.I.; Jones, M.E.; et al. Contemporary demographic reconstruction methods are robust to genome assembly quality: A case study in Tasmanian devils. Mol. Biol. Evol. 2019, 36, 2906–2921. [Google Scholar] [CrossRef]
- McCallum, H.; Jones, M.; Hawkins, C.; Hamede, R.; Lachish, S.; Sinn, D.L.; Beeton, N.; Lazenby, B. Transmission dynamics of Tasmanian devil facial tumor disease may lead to disease-induced extinction. Ecology 2009, 90, 3379–3392. [Google Scholar] [CrossRef]
- Lazenby, B.T.; Tobler, M.W.; Brown, W.E.; Hawkins, C.E.; Hocking, G.J.; Hume, F.; Huxtable, S.; Iles, P.; Jones, M.E.; Lawrence, C.; et al. Density trends and demographic signals uncover the long-term impact of transmissible cancer in Tasmanian devils. J. Appl. Ecol. 2018, 55, 1368–1379. [Google Scholar] [CrossRef] [PubMed]
- Pearse, A.M.; Swift, K. Allograft theory: Transmission of devil facial-tumour disease. Nature 2006, 439, 549. [Google Scholar] [CrossRef] [PubMed]
- Hamede, R.K.; McCallum, H.; Jones, M. Biting injuries and transmission of tasmanian devil facial tumour disease. J. Anim. Ecol. 2013, 82, 182–190. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, D.G.; Jones, M.E.; Cameron, E.Z.; McCallum, H.; Storfer, A.; Hohenlohe, P.A.; Hamede, R.K. Rate of intersexual interactions affects injury likelihood in Tasmanian devil contact networks. Behav. Ecol. 2019, 30, 1087–1095. [Google Scholar] [CrossRef]
- Pye, R.J.; Woods, G.M.; Kreiss, A. Devil facial tumor disease. Vet. Pathol. 2016, 53, 726–736. [Google Scholar] [CrossRef] [PubMed]
- Wait, L.F.; Peck, S.; Fox, S.; Power, M.L. A review of parasites in the Tasmanian devil (Sarcophilus harrisii). Biodivers. Conserv. 2017, 26, 509–526. [Google Scholar] [CrossRef]
- Munday, B.L.; Mason, R.W.; Hartley, W.J.; Presidente, P.J.A.; Obendorf, D. Sarcocystis and related organisms in Australian wildife: I. Survey findings in mammals. J. Wildl. Dis. 1978, 14, 417–433. [Google Scholar] [CrossRef] [PubMed]
- Hollings, T.; Jones, M.; Mooney, N.; McCallum, H. Wildlife disease ecology in changing landscapes: Mesopredator release and toxoplasmosis. Int. J. Parasitol. Parasites Wildl. 2013, 2. [Google Scholar] [CrossRef] [PubMed]
- Wait, L.F.; Fox, S.; Peck, S.; Power, M.L. Molecular characterization of (Cryptosporidum) and (Giardia) from the Tasmanian Devil (Sarcophilus harrisii). PLoS ONE 2017, 12, e0174994. [Google Scholar] [CrossRef] [PubMed]
- Austen, J.M.; Reid, S.A.; Robinson, D.R.; Friend, J.A.; Ditcham, W.G.; Irwin, P.J.; Ryan, U. Investigation of the morphological diversity of the potentially zoonotic (Trypanosoma copemani) in quokkas and Gilbert’s potoroos. Parasitology 2015, 142, 1443–1452. [Google Scholar] [CrossRef] [PubMed]
- Barbosa, A.D.; Austen, J.; Portas, T.J.; Friend, J.A.; Ahlstrom, L.A.; Oskam, C.L.; Ryan, U.M.; Irwin, P.J. Sequence analyses at mitochondrial and nuclear loci reveal a novel Theileria sp. and aid in the phylogenetic resolution of piroplasms from Australian marsupials and ticks. PLoS ONE 2019, 14, e0225822. [Google Scholar] [CrossRef]
- Northover, A.S.; Godfrey, S.S.; Keatley, S.; Lymbery, A.J.; Wayne, A.F.; Cooper, C.; Pallant, L.; Morris, K.; Thompson, R.C.A. Increased Trypanosoma spp. richness and prevalence of haemoparasite co-infection following translocation. Parasites Vectors 2019, 12. [Google Scholar] [CrossRef]
- Thompson, C.K.; Thompson, R.A. Trypanosomes of Australian mammals: Knowledge gaps regarding transmission and biosecurity. Trends Parasitol. 2015, 31, 553–562. [Google Scholar] [CrossRef]
- Munson, L.; Terio, K.A.; Kock, R.; Mlengeya, T.; Roelke, M.E.; Dubovi, E.; Summers, B.; Sinclair, A.R.E.; Packer, C. Climate extremes promote fatal co-infections during canine distemper epidemics in African lions. PLoS ONE 2008, 3, e2545. [Google Scholar] [CrossRef] [PubMed]
- Botero, A.; Thompson, C.K.; Peacock, C.S.; Clode, P.L.; Nicholls, P.K.; Wayne, A.F.; Lymbery, A.J.; Thompson, R.C. Trypanosomes genetic diversity, polyparasitism and the population decline of the critically endangered Australian marsupial, the brush tailed bettong or woylie (Bettongia penicillata). Int. J. Parasitol. Parasites Wildl. 2013, 2, 77–89. [Google Scholar] [CrossRef] [PubMed]
- Thompson, C.K.; Wayne, A.F.; Godfrey, S.S.; Thompson, R.C.A. Temporal and spatial dynamics of trypanosomes infecting the brush-tailed bettong (Bettongia penicillata): A cautionary note of disease-induced population decline. Parasites Vectors 2014, 7, 169. [Google Scholar] [CrossRef] [PubMed]
- McInnes, L.M.; Gillett, A.; Hanger, J.; Reid, S.A.; Ryan, U.M. The potential impact of native Australian trypanosome infections on the health of koalas (Phascolarctos cinereus). Parasitology 2011, 138, 873–883. [Google Scholar] [CrossRef]
- Maslov, D.A.; Opperdoes, F.R.; Kostygov, A.Y.; Hashimi, H.; Lukeš, J.; Yurchenko, V. Recent advances in trypanosomatid research: Genome organization, expression, metabolism, taxonomy and evolution. Parasitology 2019, 146, 1–27. [Google Scholar] [CrossRef]
- d’Avila-Levy, C.M.; Boucinha, C.; Kostygov, A.; Santos, H.L.C.; Morelli, K.A.; Grybchuk-Ieremenko, A.; Duval, L.; Votýpka, J.; Yurchenko, V.; Grellier, P.; et al. Exploring the environmental diversity of kinetoplastid flagellates in the high-throughput DNA sequencing era. Mem. Inst. Oswaldo Cruz 2015, 110, 956–965. [Google Scholar] [CrossRef]
- Lukeš, J.; Butenko, A.; Hashimi, H.; Maslov, D.A.; Votýpka, J.; Yurchenko, V. Trypanosomatids are much more than just trypanosomes: Clues from the expanded family tree. Trend Parasitol. 2018, 34, 466–480. [Google Scholar] [CrossRef]
- Jackson, A.P. Genome evolution in trypanosomatid parasites. Parasitology 2015, 142, S40–S56. [Google Scholar] [CrossRef]
- Mackerras, M.J. The haematozoa of Australian mammals. Aus. J. Zool. 1959, 7, 105–135. [Google Scholar] [CrossRef]
- Austen, J.M.; Ryan, U.M.; Friend, J.A.; Ditchman, W.G.F.; Reid, S.A. Vector of Trypanosoma copemani identified as Ixodes sp. Parasitology 2011, 138, 866–872. [Google Scholar] [CrossRef]
- Botero, A.; Cooper, C.; Thompson, C.K.; Clode, P.L.; Rose, K.; Thompson, R.A. Morphological and phylogenetic description of Trypanosoma noyesi sp. nov.: An Australian wildlife trypanosome within the T. cruzi clade. Protist 2016, 167, 425–439. [Google Scholar] [CrossRef] [PubMed]
- McInnes, L.M.; Gillett, A.; Ryan, U.M.; Austen, J.; Campbell, R.S.; Hanger, J.; Reid, S.A. Trypanosoma irwini n. sp (Sarcomastigophora: Trypanosomatidae) from the koala (Phascolarctos cinereus). Parasitology 2009, 136, 875–885. [Google Scholar] [CrossRef] [PubMed]
- Maslov, D.A.; Lukeš, J.; Jirku, M.; Simpson, L. Phylogeny of trypanosomes as inferred from the small and large Subunit rRNAs: Implications for the evolution of parasitism in the trypanosomatid protozoa. Mol. Biochem. Parasitol. 1996, 75, 197–205. [Google Scholar] [CrossRef]
- Hamilton, P.B.; Stevens, J.R.; Gaunt, M.W.; Gidley, J.; Gibson, W.C. Trypanosomes are monophyletic: Evidence from genes for glyceraldehyde phosphate dehydrogenase and small subunit ribosomal RNA. Int. J. Parasitol. 2004, 34, 1393–1404. [Google Scholar] [CrossRef]
- Jefferies, R.; Ryan, U.M.; Irwin, P.J. PCR-RFLP for the detection and differentiation of the canine piroplasm species and its use with filter paper-based technologies. Vet. Parasitol. 2007, 144, 20–27. [Google Scholar] [CrossRef]
- Ujvari, B.; Madsen, T.; Olsson, M. High prevalence of Hepatozoon spp. (Apicomplexa, Hepatozoidae) infection in water pythons (Liasis fuscus) from tropical australia. J. Parasitol. 2004, 90, 670–672. [Google Scholar] [CrossRef]
- Waldenström, J.; Bensch, S.; Hasselquist, D.; Östman, Ö. A new nested polymerase chain reaction method very efficient in detecting Plasmodium and Haemoproteus infections from avian blood. J. Parasitol. 2004, 90, 191–194. [Google Scholar] [CrossRef]
- Bensch, S.; Stjernman, M.; Hasselquist, D.; Örjan, Ö.; Hannson, B.; Westerdahl, H.; Pinheiro, R.T. Host specificity in avian blood parasites: A study of Plasmodium and Haemoproteus mitochondrial DNA amplified from birds. Proc. R. Soc. B 2000, 267, 1583–1589. [Google Scholar] [CrossRef]
- Yang, R.; Murphy, C.; Song, Y.; Ng-Hublin, J.; Estcourt, A.; Hijjawi, N.; Chalmers, R.; Hadfield, S.; Bath, A.; Gordon, C.; et al. Specific and quantitative detection and identification of Cryptosporidium hominis and C. parvum Parvum in clinical and environmental samples. Exp. Parasitol. 2013, 135, 142–147. [Google Scholar] [CrossRef]
- Zhang, Z.; Schwartz, S.; Wagner, L.; Miller, W. A greedy algorithm for aligning DNA sequences. J. Comput. Biol. 2000, 7, 203–214. [Google Scholar] [CrossRef] [PubMed]
- Morgulis, A.; Coulouris, G.; Raytselis, Y.; Madden, T.L.; Agarwala, R.; Schäffer, A.A. Database indexing for production MegaBLAST searches. Bioinformatics 2008, 24, 1757–1764. [Google Scholar] [CrossRef]
- Benson, D.A.; Cavanaugh, M.; Clark, K.; Karsch-Mizrachi, I.; Lipman, D.J.; Ostell, J.; Sayers, E.W. GenBank. Nucleic Acids Res. 2017, 45, D37–D42. [Google Scholar] [CrossRef]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef]
- Nguyen, L.T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Hoang, D.T.; Chernomor, O.; von Haeseler, A.; Minh, B.Q.; Vinh, L.S. UFBoot2: Improving the ultrafast bootstrap approximation. Mol. Biol. Evol. 2018, 35, 518–522. [Google Scholar] [CrossRef]
- Posada, D. Using MODELTEST and PAUP* to select a model of nucleotide substitution. In Current Protocols in Bioinformatics, 1st ed.; Davison, D., Page, R., Petsko, G., Stein, L., Stormo, G., Eds.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2003; pp. 6.5.1–6.5.14. [Google Scholar]
- Gu, X.; Fu, X.Y.; Li, W.H. Maximum likelihood estimation of the heterogeneity of substitution rate among nucleotide sites. Mol. Biol. Evol. 1995, 12, 546–557. [Google Scholar] [CrossRef]
- Hamilton, P.B.; Stevens, J.R. Resolving relationships between Australian trypanosomes using DNA barcoding data. Trend Parasit. 2011, 27, 99. [Google Scholar] [CrossRef]
- Kimura, M. A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J. Mol. Evol. 1980, 16, 111–120. [Google Scholar] [CrossRef]
- Tamura, K.; Nei, M. Estimation of the Number of Nucleotide Substitutions in the Control Region of Mitochondrial DNA in Humans and Chimpanzees. Mol. Biol. Evol. 1993, 10, 512–526. [Google Scholar] [CrossRef]
- Weinman, D. Trypanosoma cyclops n. sp.: A pigmented trypanosome from the Malaysian primates Macaca nemestrina and M. ira. Trans. R. Soc. Trop. Med. Hyg. 1972, 66, 628–636. [Google Scholar] [CrossRef]
- Hamilton, P.; Stevens, J.; Gidley, J.; Holz, P.; Gibson, W. A New Lineage of trypanosomes from Australian vertebrates and terrestrial bloodsucking leeches (Haemadipsidae). Int. J. Parasitol. 2005, 35, 431–443. [Google Scholar] [CrossRef] [PubMed]
- Werszko, J.; Szewczyk, T.; Steiner-Bogdaszewska, Ż.; Wróblewski, P.; Karbowiak, G.; Laskowski, Z. Molecular detection of Megatrypanum trypanosomes in tabanid flies. Med. Vet. Entomol. 2020, 34, 69–73. [Google Scholar] [CrossRef] [PubMed]
- Hoare, C.A. The Trypanosomes of Mammals: A Zoological Monograph; Blackwell Scientific Publications: Oxford, UK, 1972. [Google Scholar]
- Rodrigues, A.C.; Garcia, H.A.; Batista, J.S.; Minervino, A.H.H.; Góes-Cavalcante, G.; Maia Da Silva, F.; Ferreira, R.C.; Campaner, M.; Paiva, F.; Teixeira, M.M.G. Characterization of spliced leader genes of Trypanosoma (Megatrypanum) theileri: Phylogeographical analysis of Brazilian isolates from cattle supports spatial clustering of genotypes and parity with ribosomal markers. Parasitology 2010, 137, 111–122. [Google Scholar] [CrossRef]
- Hamilton, P.B.; Teixeira, M.M.; Stevens, J.R. The Evolution of Trypanosoma cruzi: The ‘bat seeding’ hypothesis. Trends Parasitol. 2012, 28, 136–141. [Google Scholar] [CrossRef]
- Jakes, K.A.; O’Donoghue, P.J.; Adlard, R.D. Phylogenetic relationships of Trypanosoma chelodina and Trypanosoma binneyi from Australian tortoises and platypuses inferred from small subunit rRNA analyses. Parasitology 2001, 123, 483–487. [Google Scholar] [CrossRef] [PubMed]
- Paparini, A.; Macgregor, J.; Irwin, P.J.; Warren, K.; Ryan, U.M. Novel genotypes of Trypanosoma binneyi from wild platypuses (Ornithorhynchus anatinus) and identification of a leech as a potential vector. Exp. Parasitol. 2014, 145, 42–50. [Google Scholar] [CrossRef]
- Bettiol, S.S.; Jakes, K.; Le, D.D.; Goldsmid, J.M.; Hocking, G. First record of trypanosomes in Tasmanian bandicoots. J. Parasitol. 1998, 84, 538–541. [Google Scholar] [CrossRef]
- Lukeš, J.; Skalický, T.; Týč, J.; Votýpka, J.; Yurchenko, V. Evolution of parasitism in kinetoplastid flagellates. Mol. Biochem. Parasitol. 2014, 195, 115–122. [Google Scholar] [CrossRef]
- Portas, T.J.; Evans, M.J.; Spratt, D.; Vaz, P.K.; Devlin, J.M.; Barbosa, A.D.; Wilson, B.A.; Rypalski, A.; Wimpenny, C.; Fletcher, D.; et al. Baseline health and disease assessment of founder eastern quolls (Dasyurus viverrinus) during a conservation translocation to mainland Australia. J. Wildl. Dis. 2020, 56, 547–559. [Google Scholar] [CrossRef]
- Paguem, A.; Abanda, B.; Ndjonka, D.; Weber, J.S.; Ngomtcho, S.C.H.; Manchang, K.T.; Adoulmoumini, M.; Eisenbarth, A.; Renz, A.; Kelm, S.; et al. Widespread co-endemicity of Trypanosoma species infecting cattle in the Sudano-Sahelian and Guinea Savannah zones of Cameroon. BMC Vet. Res. 2019, 15, 344. [Google Scholar] [CrossRef]
- Rodrigues, M.S.; Lima, L.; Xavier, S.C.d.C.; Herrera, H.M.; Rocha, F.L.; Roque, A.L.R.; Teixeira, M.M.G.; Jansen, A.M. Uncovering Trypanosoma spp. diversity of wild mammals by the use of DNA from blood clots. Int. J. Parasitol. Parasites Wildl. 2019, 8, 171–181. [Google Scholar] [CrossRef]
- Brandão, E.M.V.; Xavier, S.C.C.; Carvalhaes, J.G.; D’Andrea, P.S.; Lemos, F.G.; Azevedo, F.C.; Cássia-Pires, R.; Jansen, A.M.; Roque, A.L.R. Trypanosomatids in small mammals of an Agroecosystem in Central Brazil: Another piece in the puzzle of parasite transmission in an anthropogenic landscape. Pathogens 2019, 8, 190. [Google Scholar] [CrossRef]
- Roellig, D.M.; Ellis, A.E.; Yabsley, M.J. Oral transmission of Trypanosoma cruzi with opposing evidence for the theory of carnivory. J. Parasitol. 2009, 95, 360–364. [Google Scholar] [CrossRef]
- Rocha, F.L.; Roque, A.L.R.; de Lima, J.S.; Cheida, C.C.; Lemos, F.G.; de Azevedo, F.C.; Arrais, R.C.; Bilac, D.; Herrera, H.M.; Mourão, G.; et al. Trypanosoma cruzi infection in neotropical wild carnivores (Mammalia: Carnivora): At the top of the T. cruzi transmission chain. PLoS ONE 2013, 8, e67463. [Google Scholar] [CrossRef]
- Hamilton, P.B.; Gibson, W.C.; Stevens, J.R. Patterns of co-evolution between trypanosomes and their hosts deduced from ribosomal RNA and protein-coding gene phylogenies. Mol. Phylogenet. Evol. 2007, 44, 15–25. [Google Scholar] [CrossRef]
- Lemos, M.; Fermino, B.R.; Simas-Rodrigues, C.; Hoffmann, L.; Silva, R.; Camargo, E.P.; Teixeira, M.M.G.; Souto-Padrón, T. Phylogenetic and morphological characterization of trypanosomes from Brazilian armoured catfishes and leeches reveal high species diversity, mixed infections and a new fish trypanosome species. Parasites Vectors 2015, 8. [Google Scholar] [CrossRef]
- Barratt, J.; Kaufer, A.; Peters, B.; Craig, D.; Lawrence, A.; Roberts, T.; Lee, R.; McAuliffe, G.; Stark, D.; Ellis, J. Isolation of novel trypanosomatid, Zelonia australiensis sp. nov. (Kinetoplastida: Trypanosomatidae) provides support for a Gondwanan origin of dixenous parasitism in the Leishmaniinae. PLoS Negl. Trop. Dis. 2017, 11, e0005215. [Google Scholar] [CrossRef]
- Rose, K.; Curtis, J.; Baldwin, T.; Mathis, A.; Kumar, B.; Sakthianandeswaren, A.; Spurck, T.; Low Choy, J.; Handman, E. Cutaneous leishmaniasis in red Kangaroos: Isolation and characterisation of the causative organisms. Int. J. Parasitol. 2004, 34, 655–664. [Google Scholar] [CrossRef]
- Humberg, R.M.P.; Oshiro, E.T.; e Cruz, M.d.S.P.; Ribolla, P.E.M.; Alonso, D.P.; Ferreira, A.M.T.; Bonamigo, R.A.; Tasso, N.; de Oliveira, A.G. Leishmania chagasi in opossums (Didelphis albiventris) in an urban area endemic for visceral leishmaniasis, Campo Grande, Mato Grosso Do Sul, Brazil. Am. J. Trop. Med. Hyg. 2012, 87, 470–472. [Google Scholar] [CrossRef][Green Version]
- Medkour, H.; Laidoudi, Y.; Lafri, I.; Davoust, B.; Mekroud, A.; Bitam, I.; Mediannikov, O. Canine vector-borne protozoa: Molecular and serological investigation for Leishmania spp., Trypanosoma spp., Babesia spp., and Hepatozoon spp. in dogs from Northern Algeria. Vet. Parasitol. Reg. Stud. Rep. 2020, 19, 100353. [Google Scholar] [CrossRef]
- Paparini, A.; Irwin, P.J.; Warren, K.; McInnes, L.M.; de Tores, P.; Ryan, U.M. Identification of novel trypanosome genotypes in bative Australian marsupials. Vet. Parasitol. 2011, 183, 21–30. [Google Scholar] [CrossRef]
- Dougall, A.; Shilton, C.; Low Choy, J.; Alexander, B.; Walton, S. New reports of Australian cutaneous leishmaniasis in northern Australian macropods. Epidemiol. Infect. 2009, 137, 1516. [Google Scholar] [CrossRef]
- Dougall, A.M.; Alexander, B.; Holt, D.C.; Harris, T.; Sultan, A.H.; Bates, P.A.; Rose, K.; Walton, S.F. Evidence incriminating midges (Diptera: Ceratopogonidae) as potential vectors of Leishmania in Australia. Int. J. Parasitol. 2011, 41, 571–579. [Google Scholar] [CrossRef]
- Rong, J.; Bunce, M.; Wayne, A.; Pacioni, C.; Ryan, U.; Irwin, P. A high prevalence of Theileria penicillata in woylies (Bettongia penicillata). Exp. Parasitol. 2012, 131, 157–161. [Google Scholar] [CrossRef]
- Greay, T.L.; Zahedi, A.; Krige, A.S.; Owens, J.M.; Rees, R.L.; Ryan, U.M.; Oskam, C.L.; Irwin, P.J. Endemic, exotic and novel apicomplexan parasites detected during a national study of ticks from companion animals in Australia. Parasites Vectors 2018, 11, 197. [Google Scholar] [CrossRef]
- Loh, S.M.; Egan, S.; Gillett, A.; Banks, P.B.; Ryan, U.M.; Irwin, P.J.; Oskam, C.L. Molecular surveillance of piroplasms in ticks from small and medium-sized urban and peri-urban mammals in Australia. Int. J. Parasitol. Parasites Wildl. 2018, 7, 197–203. [Google Scholar] [CrossRef]
- Storey-Lewis, B.; Mitrovic, A.; McParland, B. Molecular detection and characterisation of Babesia and Theileria in Australian hard ticks. Ticks Tick Borne Dis. 2018, 9, 471–478. [Google Scholar] [CrossRef]
- Murdoch, F.A.; Spratt, D.M. Ecology of the common marsupial tick (Ixodes tasmani Neumann) (Acarina: Ixodidae), in eastern Australia. Aus. J. Zool. 2005, 53, 383–388. [Google Scholar] [CrossRef]
- Bettiol, S.S.; Goldsmid, J.M.; Le, D.D.; Driessen, M. The first record of a member of the genus Hepatozoon in the eastern barred bandicoot (Perameles gunnii) in Tasmania. J. Parasitol. 1996, 82, 829. [Google Scholar] [CrossRef]
- Portas, T.; Fletcher, D.; Spratt, D.; Reiss, A.; Holz, P.; Stalder, K.; Devlin, J.; Taylor, D.; Dobroszczyk, D.; Manning, A.D. Health evaluation of free-ranging eastern bettongs (Bettongia gaimardi) during translocation for reintroduction in Australia. J. Wildl. Dis. 2014, 50, 210–223. [Google Scholar] [CrossRef] [PubMed]
- Vilcins, I.M.E.; Old, J.M.; Deane, E. Detection of a Hepatozoon and spotted fever group Rickettsia species in the common marsupial tick (Ixodes tasmani) collected from wild Tasmanian devils (Sarcophilus harrisii), Tasmania. Vet. Parasitol. 2009, 162, 23–31. [Google Scholar] [CrossRef]
- Barbosa, A.D.; Gofton, A.W.; Paparini, A.; Codello, A.; Greay, T.; Gillett, A.; Warren, K.; Irwin, P.; Ryan, U. Increased genetic diversity and prevalence of co-infection with Trypanosoma spp. in koalas (Phascolarctos cinereus) and their ticks identified using next-generation sequencing (NGS). PLoS ONE 2017, 12, e0181279. [Google Scholar] [CrossRef] [PubMed]
- Reis-Cunha, J.L.; Baptista, R.P.; Rodrigues-Luiz, G.F.; Coqueiro-dos-Santos, A.; Valdivia, H.O.; de Almeida, L.V.; Cardoso, M.S.; D’Ávila, D.A.; Dias, F.H.C.; Fujiwara, R.T.; et al. Whole genome sequencing of Trypanosoma cruzi field isolates reveals extensive genomic variability and complex aneuploidy patterns within TcII DTU. BMC Genom. 2018, 19, 816. [Google Scholar] [CrossRef] [PubMed]
- Northover, A.S.; Elliot, A.D.; Keatley, S.; Lim, Z.; Botero, A.; Ash, A.; Lymbery, A.J.; Wayne, A.F.; Godfrey, S.S.; Thompson, R.C.A. Debilitating disease in a polyparasitised woylie (Bettongia penicillata): A diagnostic investigation. Int. J. Parasitol. Parasites. Wildl. 2018, 7, 274–279. [Google Scholar] [CrossRef]
- Northover, A.S.; Lymbery, A.J.; Wayne, A.F.; Godfrey, S.S.; Thompson, R.C.A. The hidden consequences of altering host-parasite relationships during fauna translocations. Biol. Conserv. 2018, 220, 140–148. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target | Primer | Sequence | Size | Ref. |
|---|---|---|---|---|
| Trypanosomes | ||||
| 18S primary SSU | SLF | GCTTGTTTCAAGGACTTAGC | ~1.5 kb | [32] |
| S762R | GACTTTTGCTTCCTCTAATG | [33] | ||
| 18S secondary SSU2 | S823F | CGAACAACTGCCCTATCAGC | ~904 bp | [33] |
| S662R | GACTACAATGGTCTCTAATC | [33] | ||
| 18S secondary SSU1 | S825F | ACCGTTTCGGCTTTTGTTGG | ~959 bp | [33] |
| SLIR | ACATTGTAGTGCGCGTGTC | [32] | ||
| GAPDH primary | GAPDGF | CTYMTCGGNAMKGAGATYGAYG | ~900 bp | [32] |
| GAPDHR | GRTKSGARTADCCCCACTCG | [32] | ||
| GAPDH secondary | GAPDGF | CTYMTCGGNAMKGAGATYGAYG | ~880 bp | [32] |
| Ga4 | GTTYTGCAGSGTCGCCTTGG | [34] | ||
| Piroplasms | ||||
| 18S primary | BT1F | GGCTCATTACAACAGTTATAG | ~903 bp | [35] |
| BT1R | CCCAAAGACTTTGATTTCTCTC | [35] | ||
| 18S secondary | BT2F | CCGTGCTAATTGTAGGGCTAATAC | ~800 bp | [35] |
| BT2R | GGACTACGACGGTATCTGATCG | [35] | ||
| Hepatozoon spp. | ||||
| 18S | HepF300 | GTTTCTGACCTATCAGCTTTCGACG | ~600 bp | [36] |
| Hep900 | CAAATCTAAGAATTTCACCTCTGAC | [36] | ||
| Haemosporidia | ||||
| Cytb primary | HaemNF | CATATATTAAGAGAATTATGGAG | ~580 bp | [37] |
| HaemNR2 | AGAGGTGTAGCATATCTATCTAC | [37] | ||
| Cytb secondary | HaemF | ATGGTGCTTCGATATATGCATG | ~520 bp | [38] |
| HaemR2 | GCATTATCTGGATGTGATAATGGT | [38] |
| Site | n | Trypanosoma spp. | T. copemani | T. cyclops-Like |
|---|---|---|---|---|
| Black River | 31 | 11 | 2 | 9 |
| Takone | 23 | 7 | 5 | 2 |
| West Pencil Pine | 14 | 11 | 0 | 11 |
| Freycinet | 27 | 3 | 3 | 0 |
| Overall | 95 | 32 | 10 | 22 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Egan, S.L.; Ruiz-Aravena, M.; Austen, J.M.; Barton, X.; Comte, S.; Hamilton, D.G.; Hamede, R.K.; Ryan, U.M.; Irwin, P.J.; Jones, M.E.; et al. Blood Parasites in Endangered Wildlife-Trypanosomes Discovered during a Survey of Haemoprotozoa from the Tasmanian Devil. Pathogens 2020, 9, 873. https://doi.org/10.3390/pathogens9110873
Egan SL, Ruiz-Aravena M, Austen JM, Barton X, Comte S, Hamilton DG, Hamede RK, Ryan UM, Irwin PJ, Jones ME, et al. Blood Parasites in Endangered Wildlife-Trypanosomes Discovered during a Survey of Haemoprotozoa from the Tasmanian Devil. Pathogens. 2020; 9(11):873. https://doi.org/10.3390/pathogens9110873
Chicago/Turabian StyleEgan, Siobhon L., Manuel Ruiz-Aravena, Jill M. Austen, Xavier Barton, Sebastien Comte, David G. Hamilton, Rodrigo K. Hamede, Una M. Ryan, Peter J. Irwin, Menna E. Jones, and et al. 2020. "Blood Parasites in Endangered Wildlife-Trypanosomes Discovered during a Survey of Haemoprotozoa from the Tasmanian Devil" Pathogens 9, no. 11: 873. https://doi.org/10.3390/pathogens9110873
APA StyleEgan, S. L., Ruiz-Aravena, M., Austen, J. M., Barton, X., Comte, S., Hamilton, D. G., Hamede, R. K., Ryan, U. M., Irwin, P. J., Jones, M. E., & Oskam, C. L. (2020). Blood Parasites in Endangered Wildlife-Trypanosomes Discovered during a Survey of Haemoprotozoa from the Tasmanian Devil. Pathogens, 9(11), 873. https://doi.org/10.3390/pathogens9110873