A Window to Toxoplasma gondii Egress


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Toxoplasma and Toxoplasmosis
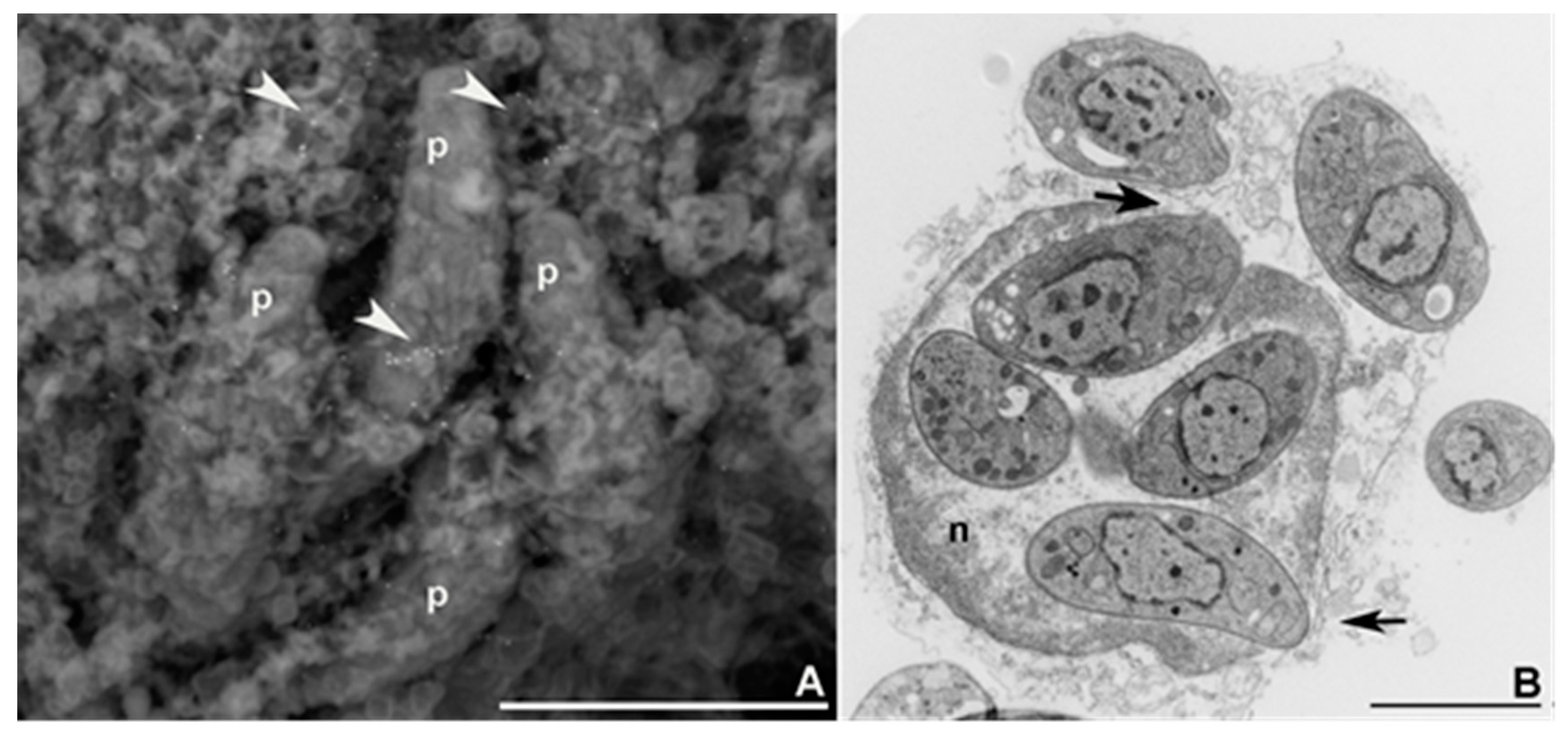
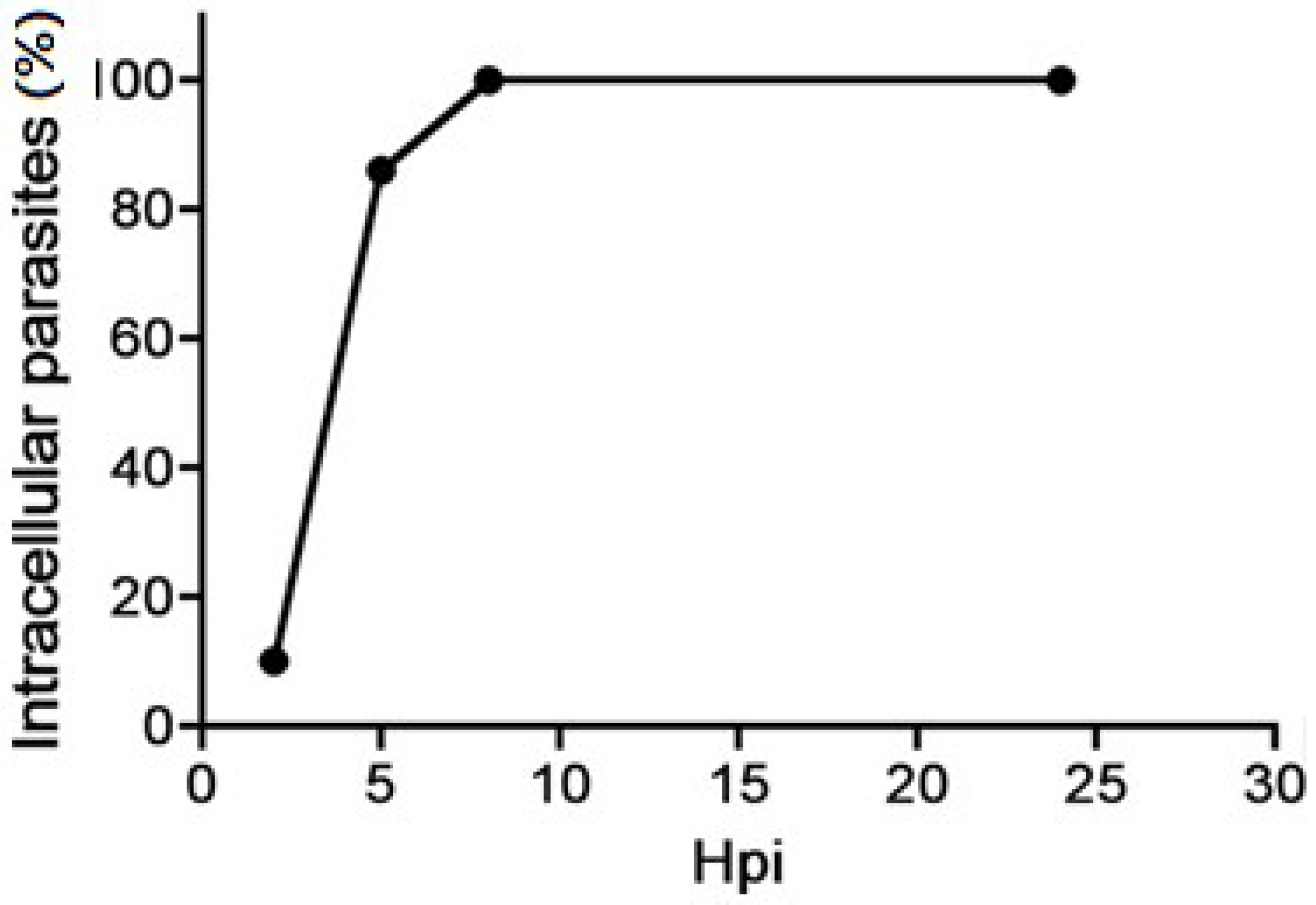
2. Tachyzoite’s Cellular Cycle
3. Egress Signalling
3.1. Micronemes
3.2. Dense Granules
3.3. Perforin-Like Proteins
3.4. Calpain
3.5. Kinases
3.6. Abscisic Acid Response
3.7. Potassium Fluxes and pH
3.8. External and Inflammatory Factors Triggering Cells
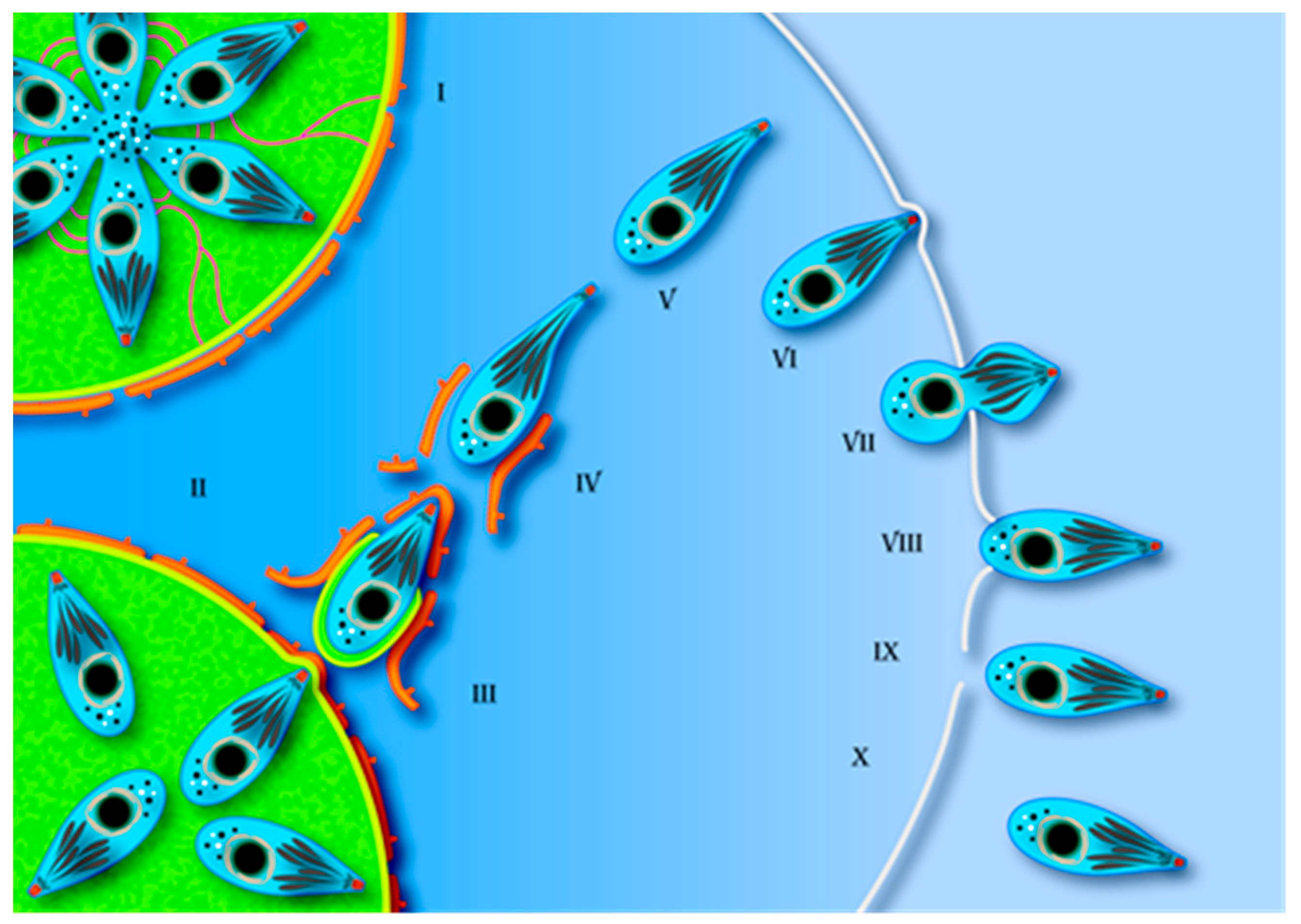
4. Egress Structure and Route
5. Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Furtado, J.M.; Smith, J.R.; Belfort, R.; Gattey, D.; Winthrop, K.L. Toxoplasmosis: A global threat. J. Glob. Infect. Dis. 2011, 3, 281–284. [Google Scholar] [CrossRef] [PubMed]
- Jones, J.L.; Lopez, A.; Wilson, M.; Schulkin, J.; Gibbs, R. Congenital toxoplasmosis: A review. Obstet. Gynecol. Surv. 2001, 56, 296–305. [Google Scholar] [CrossRef] [PubMed]
- Carellos, E.V.; Caiaffa, W.T.; Andrade, G.M.; Abreu, M.N.; Januário, J.N. Congenital toxoplasmosis in the state of Minas Gerais, Brazil: A neglected infectious disease? Epidemiol. Infect. 2014, 142, 644–655. [Google Scholar] [CrossRef] [PubMed]
- Foroutan-Rad, M.; Majidiani, H.; Dalvand, S.; Daryani, A.; Kooti, W.; Saki, J.; Hedayati-Rad, F.; Ahmadpour, E. Toxoplasmosis in Blood Donors: A Systematic Review and Meta-Analysis. Transfus. Med. Rev. 2016, 30, 116–122. [Google Scholar] [CrossRef] [PubMed]
- Montoya, J.G.; Liesenfeld, O. Toxoplasmosis. Lancet 2004, 363, 1965–1976. [Google Scholar] [CrossRef]
- Assolini, J.P.; Concato, V.M.; Gonçalves, M.D.; Carloto, A.C.M.; Conchon-Costa, I.; Pavanelli, W.R.; Melanda, F.N.; Costa, I.N. Nanomedicine advances in toxoplasmosis: Diagnostic, treatment, and vaccine applications. Parasitol. Res. 2017, 116, 1603–1615. [Google Scholar] [CrossRef] [PubMed]
- Alday, P.H.; Doggett, J.S. Drugs in development for toxoplasmosis: Advances, challenges, and current status. Drug Des. Dev. Ther. 2017, 11, 273–293. [Google Scholar] [CrossRef] [PubMed]
- Radke, J.R.; Gubbels, M.J.; Jerome, M.E.; Radke, J.B.; Striepen, B.; White, M.W. Identification of a sporozoite-specific member of the Toxoplasma SAG superfamily via genetic complementation. Mol. Microbiol. 2004, 52, 93–105. [Google Scholar] [CrossRef] [PubMed]
- Carruthers, V.; Boothroyd, J.C. Pulling together: An integrated model of Toxoplasma cell invasion. Curr. Opin. Microbiol. 2007, 10, 83–89. [Google Scholar] [CrossRef] [PubMed]
- Frénal, K.; Dubremetz, J.F.; Lebrun, M.; Soldati-Favre, D. Gliding motility powers invasion and egress in Apicomplexa. Nat. Rev. Microbiol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Alexander, D.L.; Mital, J.; Ward, G.E.; Bradley, P.; Boothroyd, J.C. Identification of the moving junction complex of Toxoplasma gondii: A collaboration between distinct secretory organelles. PLoS Pathog. 2005, 1, 137–149. [Google Scholar] [CrossRef] [PubMed]
- Mercier, C.; Dubremetz, J.F.; Rauscher, B.; Lecoedier, L.; Sibley, L.D.; Cesbron-DeLauw, M.F. Biogenesis of nanotubular network in Toxoplasma parasitophorous vacuole induced by parasite proteins. Mol. Biol. Cell 2002, 13, 2397–2409. [Google Scholar] [CrossRef] [PubMed]
- Arrizabalaga, G.; Boothroyd, J.C. Role of calcium during Toxoplasma gondii invasion and egress. Int. J. Parasitol. 2004, 34, 361–368. [Google Scholar] [CrossRef] [PubMed]
- Hoff, E.F.; Carruthers, V.B. Is Toxoplasma egress the first step in invasion? Trends Parasitol. 2002, 18, 251–255. [Google Scholar] [CrossRef]
- Moudy, R.; Manning, T.J.; Beckers, C.J. The loss of cytoplasmic potassium upon host cell breakdown triggers egress of Toxoplasma gondii. J. Biol. Chem. 2001, 276, 41492–41501. [Google Scholar] [CrossRef] [PubMed]
- Lavine, M.D.; Arrizabalaga, G. Invasion and egress by the obligate intracellular parasite Toxoplasma gondii: Potential targets for the development of new antiparasitic drugs. Curr. Pharm. Des. 2007, 13, 641–651. [Google Scholar] [CrossRef] [PubMed]
- Miranda, K.; Pace, D.A.; Cintron, R.; Rodrigues, J.C.; Fang, J.; Smith, A.; Rohloff, P.; Coelho, E.; de Haas, F.; de Souza, W.; et al. Characterization of a novel organelle in Toxoplasma gondii with similar composition and function to the plant vacuole. Mol. Microbiol. 2010, 76, 1358–1375. [Google Scholar] [CrossRef] [PubMed]
- Bonhomme, A.; Pingret, L.; Bonhomme, P.; Michel, J.; Balossier, G.; Lhotel, M.; Pinon, J.M. Subcellular calcium localization in Toxoplasma gondii by electron microscopy and by X-ray and electron energy loss spectroscopies. Microsc. Res. Tech. 1993, 25, 276–285. [Google Scholar] [CrossRef] [PubMed]
- Borges-Pereira, L.; Budu, A.; McKnight, C.A.; Moore, C.A.; Vella, S.A.; Hortua Triana, M.A.; Liu, J.; Garcia, C.R.; Pace, D.A.; Moreno, S.N. Calcium signaling throughout the Toxoplasma gondii lytic cycle: A study using genetically encoded calcium indicators. J. Biol. Chem. 2015, 290, 26914–26926. [Google Scholar] [CrossRef] [PubMed]
- Pressman, B.C. Biological applications of ionophores. Annu. Rev. Biochem. 1976, 45, 501–530. [Google Scholar] [CrossRef] [PubMed]
- Endo, T.; Sethi, K.K.; Piekarski, G. Toxoplasma gondii: Calcium ionophore A23187-mediated exit of trophozoites from infected murine macrophages. Exp. Parasitol. 1982, 53, 179–188. [Google Scholar] [CrossRef]
- Pingret, L.; Millot, J.M.; Sharonov, S.; Bonhomme, A.; Manfait, M.; Pinon, J.M. Relationship between intracellular free calcium concentrations and the intracellular development of Toxoplasma gondii. J. Histochem. Cytochem. 1996, 44, 1123–1229. [Google Scholar] [CrossRef] [PubMed]
- Black, M.W.; Arrizabalaga, G.; Boothroyd, J.C. Ionophore-resistant mutants of Toxoplasma gondii reveal host cell permeabilization as an early event in egress. Mol. Cell. Biol. 2000, 20, 9399–9408. [Google Scholar] [CrossRef] [PubMed]
- Caldas, L.A.; de Souza, W.; Attias, M. Microscopic analysis of calcium ionophore activated egress of Toxoplasma gondii from the host cell. Vet. Parasitol. 2010, 167, 8–18. [Google Scholar] [CrossRef] [PubMed]
- Coleman, B.I.; Gubbels, M. A genetic screen to isolate Toxoplasma gondii host-cell egress mutants. J. Vis. Exp. 2012, 3807. [Google Scholar] [CrossRef] [PubMed]
- Carruthers, V.B.; Sibley, L.D. Mobilization of intracellular calcium stimulates microneme discharge in Toxoplasma Gondii. Mol. Microbiol. 1999, 31, 421–428. [Google Scholar] [CrossRef] [PubMed]
- Moreno, S.N.; Zhong, L. Acidocalcisomes in Toxoplasma gondii tachyzoites. Biochem. J. 1996, 313, 655–659. [Google Scholar] [CrossRef] [PubMed]
- Bullen, H.E.; Jia, Y.; Yamaryo-Botté, Y.; Bisio, H.; Zhang, O.; Jemelin, N.K.; Marq, J.B.; Carruthers, V.; Botté, C.Y.; Soldati-Favre, D. Phosphatidic acid-mediated signalling regulates microneme secretion in Toxoplasma. Cell Host Microbe 2016, 19, 349–360. [Google Scholar] [CrossRef] [PubMed]
- Gras, S.; Jackson, A.; Woods, S.; Pall, G.; Whitelaw, J.; Leung, J.M.; Ward, G.E.; Roberts, C.W.; Meissner, M. Parasites lacking the micronemal protein MIC2 are deficient in surface attachment and host cell egress, but remain virulent in vivo. Wellcome Open Res. 2017, 2, 32. [Google Scholar] [CrossRef] [PubMed]
- Ravindran, S.; Lodoen, M.B.; Verhelst, S.H.; Bogyo, M.; Boothroyd, J.C. 4-Bromophenacyl bromide specifically inhibits rhoptry secretion during Toxoplasma invasion. PLoS ONE 2009, 4, e8143. [Google Scholar] [CrossRef] [PubMed]
- Beck, J.R.; Fung, C.; Straub, K.W.; Coppens, I.; Vashisht, A.A.; Wohlschlegel, J.A.; Bradley, P.J. A Toxoplasma palmitoyl acyl transferase and the palmitoylated armadillo repeat protein TgARO govern apical rhoptry tethering and reveal a critical role for the rhoptries in host cell invasion but not egress. PLoS Pathog. 2013, 9, e1003162. [Google Scholar] [CrossRef] [PubMed]
- Cesbron-Delauw, M.F.; Guy, B.; Torpier, G.; Pierce, R.J.; Lenzen, G.; Cesbron, J.Y.; Charif, H.; Lepage, P.; Darcy, F.; Lecqoc, J.P. Molecular characterization of a 23-kilodalton major antigen secreted by Toxoplasma gondii. Proc. Natl. Acad. Sci. USA 1989, 86, 7537–7541. [Google Scholar] [CrossRef] [PubMed]
- LaFavers, K.A.; Márquez-Nogueras, K.M.; Coppens, I.; Moreno, S.N.J.; Arrizabalaga, G. A novel dense granule protein, GRA41, regulates timing of egress and calcium sensitivity in Toxoplasma gondii. Cell Microbiol. 2017, 19. [Google Scholar] [CrossRef] [PubMed]
- Pszenny, V.; Ehrenman, K.; Romano, J.D.; Kennard, A.; Schultz, A.; Roos, D.A.; Grigg, E.; Carruthers, V.B.; Coppens, I. A Lipolytic Lecithin: Cholesterol Acyltransferase Secreted by Toxoplasma Facilitates Parasite Replication and Egress. J. Biol. Chem. 2016, 291, 3725–3746. [Google Scholar] [CrossRef] [PubMed]
- Dramsi, S.; Cossart, P. Listeriolysin O: A genuine cytolysin optimized for an intracellular parasite. J. Cell Biol. 2002, 156, 943–946. [Google Scholar] [CrossRef] [PubMed]
- Kafsack, B.F.; Pena, J.D.; Coppens, I.; Ravindran, S.; Boothroyd, J.C.; Carruthers, V.B. Rapid membrane disruption by a perforin-like protein facilitates parasite exit from host cells. Science 2009, 323, 530–533. [Google Scholar] [CrossRef] [PubMed]
- Kafsack, B.F.; Carruthers, V.B. Apicomplexan perforin-like proteins. Commun. Integr. Biol. 2010, 3, 18–23. [Google Scholar] [CrossRef] [PubMed]
- Chandramohanadas, R.; Davis, P.H.; Beiting, D.P.; Harbut, M.B.; Darling, C.; Velmourougane, G.; Lee, M.Y.; Greer, P.A.; Roos, D.S.; Greenbaum, D.C. Apicomplexan parasites co-opt host calpains to facilitate their escape from infected cells. Science 2009, 324, 794–797. [Google Scholar] [CrossRef] [PubMed]
- Billker, O.; Lourido, S.; Sibley, L.D. Calciumdependent signalling and kinases in apicomplexan parasites. Cell Host Microbe 2009, 5, 612–622. [Google Scholar] [CrossRef] [PubMed]
- Lourido, S.; Shuman, J.; Zhang, C.; Shokat, K.M.; Hui, R.; Sibley, L.D. Calcium-dependent protein kinase 1 is an essential regulator of exocytosis in Toxoplasma. Nature 2010, 465, 359–362. [Google Scholar] [CrossRef] [PubMed]
- Gaji, R.Y.; Johnson, D.E.; Treeck, M.; Wang, M.; Hudmon, A.; Arrizabalaga, G. Phosphorylation of a myosin motor by TgCDPK3 facilitates rapid initiation of motility during Toxoplasma gondii egress. PLoS Pathog. 2015, 11, e1005268. [Google Scholar] [CrossRef] [PubMed]
- Garrison, E.; Treeck, M.; Ehret, E.; Butz, H.; Garbuz, T.; Oswald, B.P.; Settles, M.; Boothroyd, J.; Arrizabalaga, G. A forward genetic screen reveals that calcium-dependent protein kinase 3 regulates egress in Toxoplasma. PLoS Pathog. 2012, 8, e1003049. [Google Scholar] [CrossRef] [PubMed]
- McCoy, J.M.; Whitehead, L.; van Dooren, G.G.; Tonkin, C.J. TgCDPK3 regulates calcium-dependent egress of Toxoplasma gondii from host cells. PLoS Pathog. 2012, 8, e1003066. [Google Scholar] [CrossRef] [PubMed]
- Lourido, S.; Tang, K.; Sibley, L.D. Distinct signalling pathways control Toxoplasma egress and hostcell invasion. EMBO J. 2012, 31, 4524–4534. [Google Scholar] [CrossRef] [PubMed]
- Blackman, M.J.; Carruthers, V.B. Recent insights into apicomplexan parasite egress provide new views to a kill. Curr. Opin. Microbiol. 2013, 16, 459–464. [Google Scholar] [CrossRef] [PubMed]
- Lavine, M.D.; Knoll, L.J.; Rooney, P.J.; Arrizabalaga, G. A Toxoplasma gondii mutant defective in responding to calcium fluxes shows reduced in vivo pathogenicity. Mol. Biochem. Parasitol. 2007, 155, 113–122. [Google Scholar] [CrossRef] [PubMed]
- Treeck, M.; Sanders, J.L.; Gaji, R.Y.; LaFavers, K.A.; Child, M.A.; Arrizabalaga, G.; Elias, J.E.; Boothroyd, J.C. The calcium-dependent protein kinase 3 of Toxoplasma influences basal calcium levels and functions beyond egress as revealed by quantitative phosphoproteome analysis. PLoS Pathog. 2014, 10, e1004197. [Google Scholar] [CrossRef] [PubMed]
- Brown, K.M.; Long, S.; Sibley, L.D. Plasma membrane association by N-acylation governs PKG function in Toxoplasma gondii. mBio 2017, 8, e00375-17. [Google Scholar] [CrossRef] [PubMed]
- Heaslip, A.T.; Nishi, M.; Stein, B.; Hu, K. The motility of a human parasite, Toxoplasma gondii, is regulated by a novel lysine methyltransferase. PLoS Pathog. 2011, 7, e1002201. [Google Scholar] [CrossRef] [PubMed]
- Uboldi, A.; Stewart, R.; Tonkin, C. cAMP signalling acts as a negative regulator of Toxoplasma motility. In Proceedings of the Toxo 14—The 14th Biennial Conference of the Toxoplasma gondii Research Community, Tomar, Portugal, 31 May–4 June 2017. [Google Scholar]
- Jia, Y.; Marq, J.B.; Bisio, H.; Jacot, D.; Mueller, C.; Yu, L.; Choudhary, J.; Brochet, M.; Soldati-Favre, D. Crosstalk between PKA and PKG controls pH-dependent host cell egress of Toxoplasma gondii. EMBO J. 2017, 36, 3250–3267. [Google Scholar] [CrossRef] [PubMed]
- Caldas, L.A.; de Souza, W.; Attias, M. Calcium ionophore-induced egress of Toxoplasma gondii shortly after host cell invasion. Vet. Parasitol. 2007, 147, 210–220. [Google Scholar] [CrossRef] [PubMed]
- Caldas, L.A.; Seabra, S.H.; Attias, M.; de Souza, W. The effect of kinase, actin, myosin and dynamin inhibitors on host cell egress by Toxoplasma Gondii. Parasitol. Int. 2013, 62, 475–482. [Google Scholar] [CrossRef] [PubMed]
- Nagamune, K.; Hicks, L.M.; Fux, B.; Brossier, F.; Chini, E.N.; Sibley, L.D. Abscisic acid controls calcium-dependent egress and development in Toxoplasma gondii. Nature 2008, 451, 207–210. [Google Scholar] [CrossRef] [PubMed]
- Nagamune, K.; Xiong, L.; Chini, E.; Sibley, L.D. Plants, endosymbionts and parasites: Abscisic acid and calcium signaling. Commun. Integr. Biol. 2008, 1, 62–65. [Google Scholar] [CrossRef] [PubMed]
- Fruth, I.A.; Arrizabalaga, G. Toxoplasma gondii: Induction of egress by the potassium ionophore nigericin. Int. J. Parasitol. 2007, 37, 1559–1567. [Google Scholar] [CrossRef] [PubMed]
- Roiko, M.S.; Svezhova, N.; Carruthers, V.B. Acidification Activates Toxoplasma gondii Motility and Egress by Enhancing Protein Secretion and Cytolytic Activity. PLoS Pathog. 2014, 10, e1004488. [Google Scholar] [CrossRef] [PubMed]
- Tomita, T.; Yamada, T.; Weiss, L.M.; Orlofsky, A. Externally triggered egress is the major fate of Toxoplasma gondii during acute infection. J. Immunol. 2009, 183, 6667–6680. [Google Scholar] [CrossRef] [PubMed]
- Persson, C.M.; Lambert, H.; Vutova, P.P.; Dellacasa-Lindberg, I.; Nederby, J.; Yagita, H.; Ljunggren, H.G.; Grandien, A.; Barragan, A.; Chambers, B.J. Transmission of Toxoplasma gondii from infected dendritic cells to natural killer cells. Infect. Immun. 2009, 77, 970–976. [Google Scholar] [CrossRef] [PubMed]
- Melzer, T.; Duffy, A.; Weiss, L.M.; Halonen, S.K. The gamma interferon (IFN-γ)-inducible GTP-binding protein IGTP is necessary for toxoplasma vacuolar disruption and induces parasite egression in IFN-γ-stimulated astrocytes. Infect. Immun. 2008, 76, 4883–4894. [Google Scholar] [CrossRef] [PubMed]
- Niedelman, W.; Sprokholt, J.K.; Clough, B.; Frickel, E.; Saeij, J.P.J. Cell Death of Gamma Interferon-Stimulated Human Fibroblasts upon Toxoplasma gondii Infection Induces Early Parasite Egress and Limits Parasite Replication. Infect. Immun. 2013, 81, 4341–4349. [Google Scholar] [CrossRef] [PubMed]
- Ji, Y.S.; Sun, X.M.; Liu, X.Y.; Suo, X. Toxoplasma gondii: Effects of exogenous nitric oxide on egress of tachyzoites from infected macrophages. Exp. Parasitol. 2012, 133, 70–74. [Google Scholar] [CrossRef] [PubMed]
- Yan, X.; Ji, Y.; Liu, X.; Suo, X. Nitric oxide stimulates early egress of Toxoplasma gondii tachyzoites from Human foreskin fibroblast cells. Parasites Vectors 2015, 8, 420. [Google Scholar] [CrossRef] [PubMed]
- Yao, Y.; Liu, M.; Ren, C.; Shen, J.; Ji, Y. Exogenous tumor necrosis factor-alpha could induce egress of Toxoplasma gondii from human foreskin fibroblast cells. Parasite 2017, 24, 45. [Google Scholar] [CrossRef] [PubMed]
- Carruthers, V.B.; Moreno, S.N.; Sibley, L.D. Ethanol and acetaldehyde elevate intracellular [Ca2+] and stimulate microneme discharge in Toxoplasma gondii. Biochem. J. 1999, 342, 379–386. [Google Scholar] [CrossRef] [PubMed]
- Lovett, J.L.; Marchesini, N.; Moreno, S.N.; Sibley, L.D. Toxoplasma gondii microneme secretion involves intracellular Ca2+ release from inositol 1,4,5-triphosphate (IP(3))/ryanodine-sensitive stores. J. Biol. Chem. 2002, 277, 25870–25876. [Google Scholar] [CrossRef] [PubMed]
- Stommel, E.W.; Ely, K.H.; Schwartzman, J.D.; Kasper, L.H. Toxoplasma gondii: Dithiol-induced Ca2+ flux causes egress of parasites from the parasitophorous vacuole. Exp. Parasitol. 1997, 87, 88–97. [Google Scholar] [CrossRef] [PubMed]
- Gaji, R.Y.; Behnke, M.S.; Lehmann, M.M.; White, M.W.; Carruthers, V.B. Cell cycle-dependent, intercellular transmission of Toxoplasma gondii is accompanied by marked changes in parasite gene expression. Mol. Microbiol. 2011, 79, 192–204. [Google Scholar] [CrossRef] [PubMed]
- Carey, K.L.; Westwood, N.J.; Mitchison, T.J.; Ward, G.E. A small-molecule approach to studying invasive mechanisms of Toxoplasma gondii. Proc. Natl. Acad. Sci. USA 2004, 101, 7433–7438. [Google Scholar] [CrossRef] [PubMed]
- Graindorge, A.; Frénal, K.; Jacot, D.; Salamun, J.; Marq, J.B.; Soldati-Favre, D. The conoid associated motor MyoH is indispensable for Toxoplasma gondii entry and exit from host cells. PLoS Pathog. 2016, 12, e1005388. [Google Scholar] [CrossRef] [PubMed]
- Meissner, M.; Schluter, D.; Soldati, D. Role of Toxoplasma gondii myosin A in powering parasite gliding and host cell invasion. Science 2012, 298, 837–840. [Google Scholar] [CrossRef] [PubMed]
- Periz, J.; Whitelaw, J.; Harding, C.; Gras, S.; Del Rosario Minina, M.I.; Latorre-Barragan, F.; Lemgruber, L.; Reimer, M.A.; Insall, R.; Heaslip, A.; et al. Toxoplasma gondii F-actin forms an extensive filamentous network required for material exchange and parasite maturation. eLife 2017, 6, e24119. [Google Scholar] [CrossRef] [PubMed]
- Barbosa, H.S.; Ferreira-Silva, M.F.; Guimarães, E.V.; Carvalho, L.; Rodrigues, L.M. Absence of vacuolar membrane involving Toxoplasma gondii during its intranuclear localization. J. Parasitol. 2005, 91, 182–184. [Google Scholar] [CrossRef] [PubMed]
- Condit, R.C. Surf and turf: Mechanism of enhanced virus spread during poxvirus infection. Viruses 2010, 2, 1050–1054. [Google Scholar] [CrossRef] [PubMed]
- Caldas, L.A.; Attias, M.; de Souza, W. A structural analysis of the natural egress of Toxoplasma gondii. Microbes Infect. 2018, 20, 57–62. [Google Scholar] [CrossRef] [PubMed]
- Dogga, S.K.; Mukherjee, B.; Jacot, D.; Kockmann, T.; Molino, L.; Hammoudi, P.; Hartkoorn, R.C.; Hehl, A.B.; Soldati-Favre, D. A druggable secretory protein maturase of Toxoplasma essential for invasion and egress. eLife 2017, 6, e27480. [Google Scholar] [CrossRef] [PubMed]




© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Caldas, L.A.; De Souza, W. A Window to Toxoplasma gondii Egress. Pathogens 2018, 7, 69. https://doi.org/10.3390/pathogens7030069
Caldas LA, De Souza W. A Window to Toxoplasma gondii Egress. Pathogens. 2018; 7(3):69. https://doi.org/10.3390/pathogens7030069
Chicago/Turabian StyleCaldas, Lucio Ayres, and Wanderley De Souza. 2018. "A Window to Toxoplasma gondii Egress" Pathogens 7, no. 3: 69. https://doi.org/10.3390/pathogens7030069
APA StyleCaldas, L. A., & De Souza, W. (2018). A Window to Toxoplasma gondii Egress. Pathogens, 7(3), 69. https://doi.org/10.3390/pathogens7030069