Prion Strains and Transmission Barrier Phenomena


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Experimental Prion Transmission
2.1. Early Cases of Prion Interspecies Transmission
2.2. Emergence of the Prion Strain Concept
3. From Prion Strain Characterization
3.1. First Approaches
3.1.1. Incubation Time, Clinical Signs, Vacuolation Tissue Tropism
3.1.2. Biophysical Parameters (Circular Dichroism, Infra Red, …)
3.1.3. Biochemical Methods (Western Blot, Resistance to Chaotropic Agents, Conformation-Dependent Immunoassays…)
3.2. New Insights
3.2.1. Templating Activity
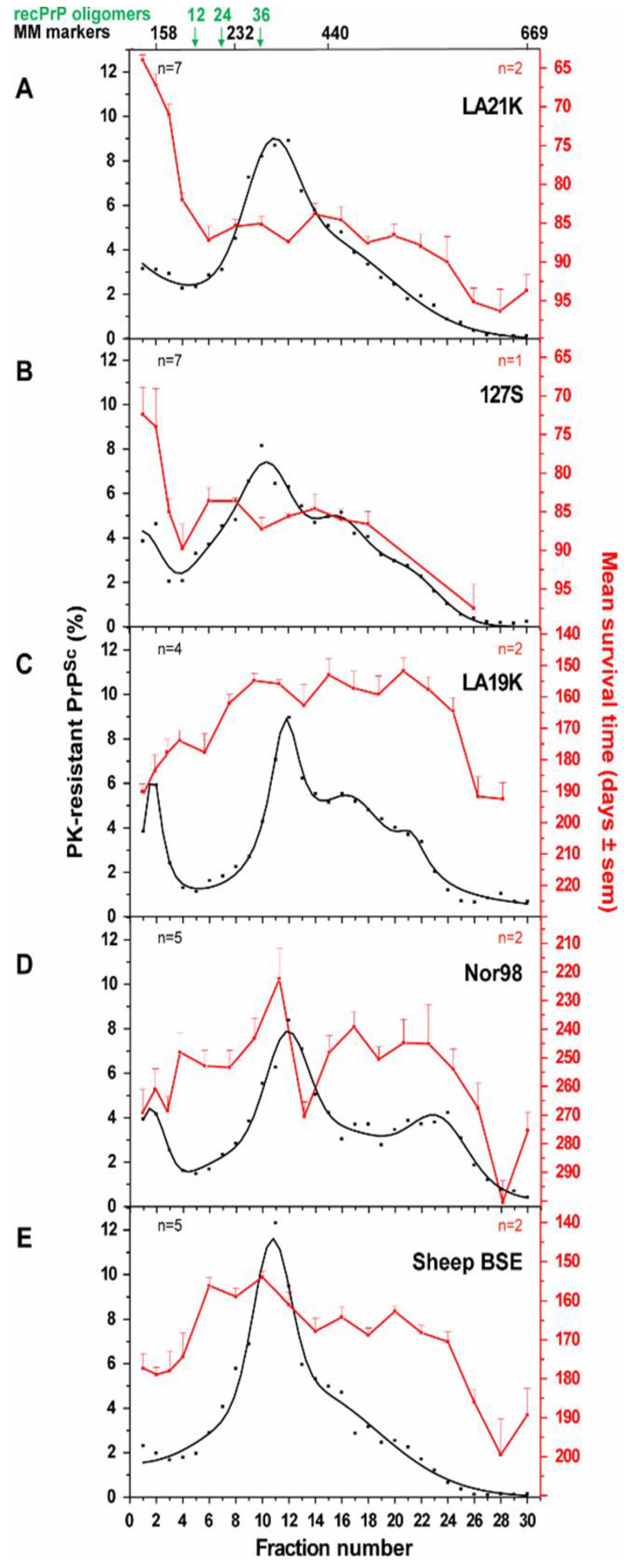
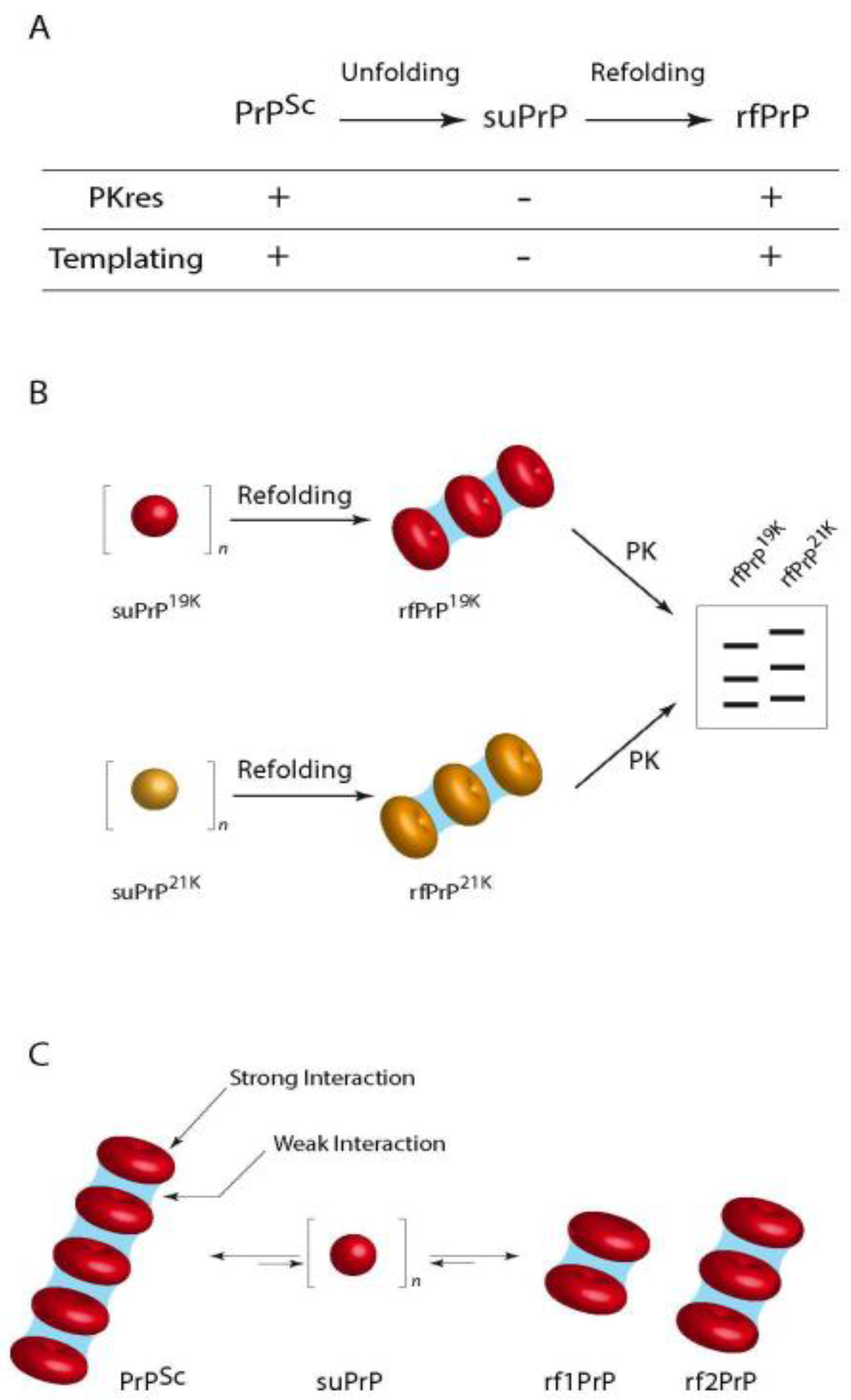
3.2.2. Size Distribution of Aggregates (Quaternary Structure)
3.3. When Two Strains Look the Same
Of prion isolates, strains and types
- Isolate: we refer here as to biological material that has been obtained through sampling of infected individuals;
- Strain: the term corresponds to a defined prion population isolated from one specified animal, with regards to the precision of the investigation technique: from basic observations (clinical signs incubation time and so on) to fine biochemical and biophysical parameters that are now becoming precise enough to allow for the discrimination of quasi-species within one strain; for the sake of simplicity, one regularly and erroneously omit the name of the host from which the strain has been originally isolated, even though a totally different prion population may have been selected when passed to the new host.
- Type: refers more particularly to a combination of biochemical parameters (mainly to the size of the unglycosylated PrPSc fragment after proteinase K partial digestion) that are independent from the host.
4. To the Study of Species Barrier in Prion Transmission
4.1. Some Great Examples
4.2. Importance of Primary Sequence, Aminoacid Polymorphism
4.3. Co-Infections
4.4. Influence of Expression Level and Post Translational Modifications
4.4.1. PrP Expression Level
4.4.2. Secondary Modifications: Glycosylation, Sialylation, Protease Digestion…
4.5. Prion Route May Influence Prion Transmission Fate
4.6. Immune Status, Age of the Host
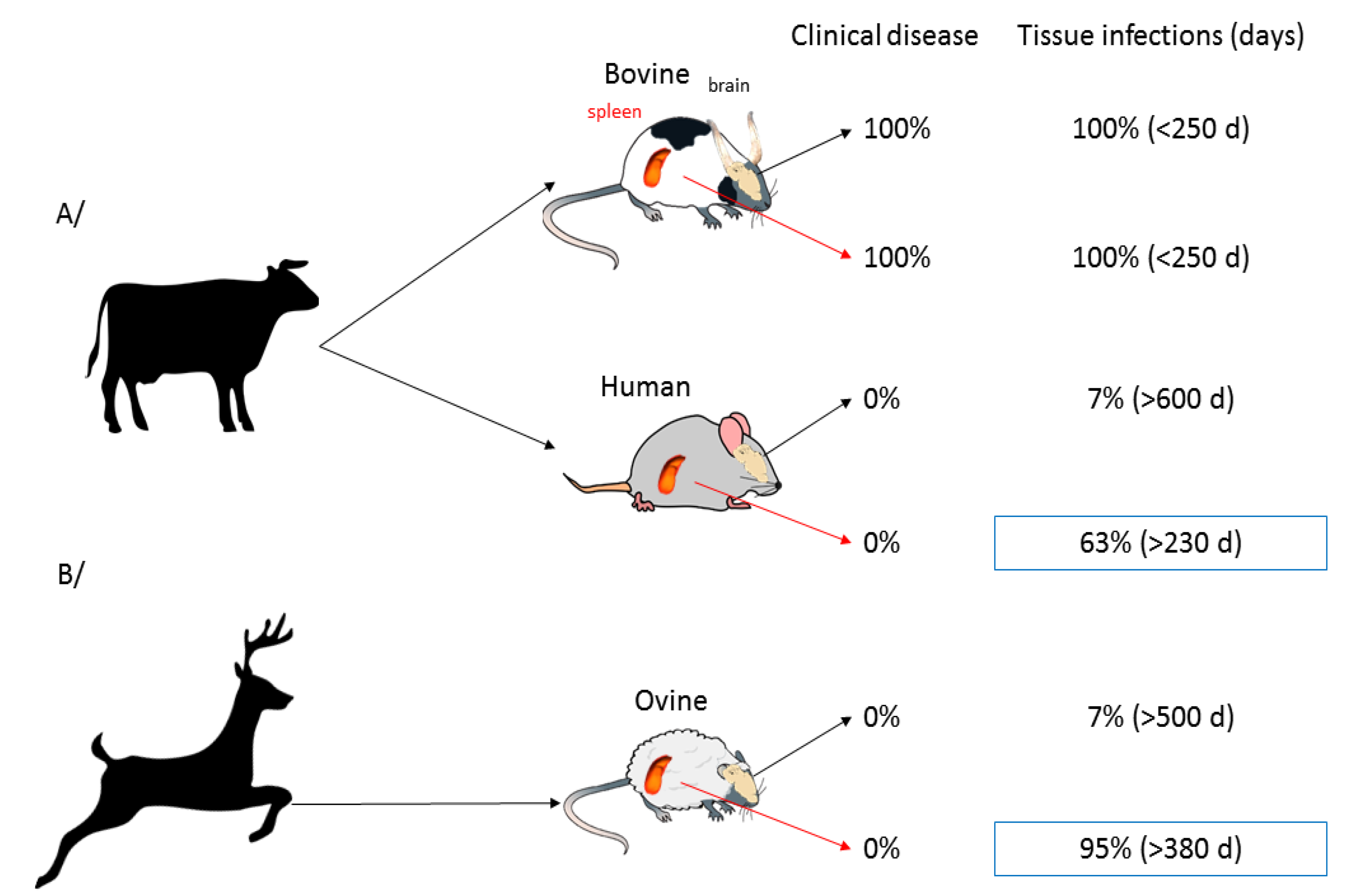
4.7. Cell/organ Selectivity
5. Consequences on Prion Adaptation to New Host
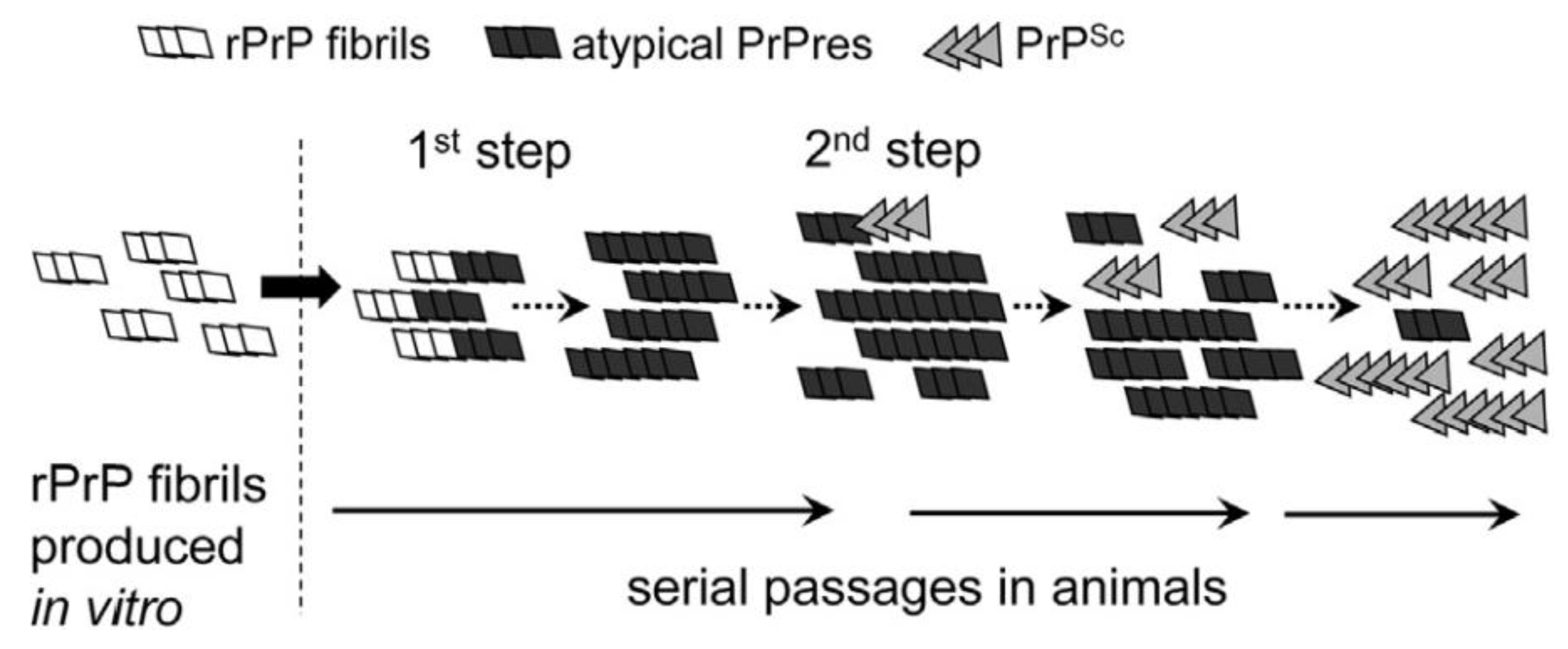
5.1. De Novo Synthesis and In Vitro Assessment of Species Barrier
5.2. In Vitro Assessment of High Species Barrier
6. Proposed Mechanisms for PrP Conversion, Strain Determination and Species Barrier Crossing
6.1. Prion Diversity from a Structural Portfolio and Selection of Mutants upon Species Barrier Crossing
6.2. Deformed Templating
7. Conclusions
Conflicts of Interest
References
- Collinge, J.; Palmer, M.S.; Sidle, K.C.; Hill, A.F.; Gowland, I.; Meads, J.; Asante, E.; Bradley, R.; Doey, L.J.; Lantos, P.L. Unaltered susceptibility to BSE in transgenic mice expressing human prion protein. Nature 1995, 378, 779–783. [Google Scholar] [CrossRef] [PubMed]
- Collinge, J.; Sidle, K.C.; Meads, J.; Ironside, J.; Hill, A.F. Molecular analysis of prion strain variation and the aetiology of “new variant” CJD. Nature 1996, 383, 685–690. [Google Scholar] [CrossRef] [PubMed]
- Gajdusek, D.C.; Gibbs, C.J.; Alpers, M. Experimental transmission of a Kuru-like syndrome to chimpanzees. Nature 1966, 209, 794–796. [Google Scholar] [CrossRef] [PubMed]
- Griffith, J.S. Self-replication and scrapie. Nature 1967, 215, 1043–1044. [Google Scholar] [CrossRef] [PubMed]
- Prusiner, S.B. Novel proteinaceous infectious particles cause scrapie. Science 1982, 216, 136–144. [Google Scholar] [CrossRef] [PubMed]
- Cuillé, J. Chelle La tremblante du mouton est bien inoculable. CR Séances Acad. Sci. Paris 1936, 206, 78–79. [Google Scholar]
- Plummer, P.J. Scrapie—A Disease of Sheep: A Review of the literature. Can. J. Comp. Med. Vet. Sci. 1946, 10, 49–54. [Google Scholar] [PubMed]
- Pattison, I.H.; Gordon, W.S.; Millson, G.C. Experimental Production of Scrapie in Goats. J. Comp. Pathol. Ther. 1959, 69, 300IN19–312IN20. [Google Scholar] [CrossRef]
- Pattison, I.H.; Millson, G.C. Scrapie produced experimentally in goats with special reference to the clinical syndrome. J. Comp. Pathol. 1961, 71, 101–109. [Google Scholar] [CrossRef]
- Hadlow, W.J.; Race, R.E.; Kennedy, R.C. Experimental infection of sheep and goats with transmissible mink encephalopathy virus. Can. J. Vet. Res. 1987, 51, 135–144. [Google Scholar] [PubMed]
- Nonno, R.; Bari, M.A.D.; Cardone, F.; Vaccari, G.; Fazzi, P.; Dell’Omo, G.; Cartoni, C.; Ingrosso, L.; Boyle, A.; Galeno, R.; et al. Efficient Transmission and Characterization of Creutzfeldt–Jakob Disease Strains in Bank Voles. PLoS Pathog. 2006, 2. [Google Scholar] [CrossRef] [PubMed]
- Watts, J.C.; Giles, K.; Patel, S.; Oehler, A.; DeArmond, S.J.; Prusiner, S.B. Evidence That Bank Vole PrP Is a Universal Acceptor for Prions. PLoS Pathog. 2014, 10. [Google Scholar] [CrossRef] [PubMed]
- Orrú, C.D.; Groveman, B.R.; Raymond, L.D.; Hughson, A.G.; Nonno, R.; Zou, W.; Ghetti, B.; Gambetti, P.; Caughey, B. Bank Vole Prion Protein As an Apparently Universal Substrate for RT-QuIC-Based Detection and Discrimination of Prion Strains. PLoS Pathog. 2015, 11, e1004983. [Google Scholar] [CrossRef]
- Robinson, M.M.; Hadlow, W.J.; Knowles, D.P.; Huff, T.P.; Lacy, P.A.; Marsh, R.F.; Gorham, J.R. Experimental infection of cattle with the agents of transmissible mink encephalopathy and scrapie. J. Comp. Pathol. 1995, 113, 241–251. [Google Scholar] [CrossRef]
- Büeler, H.; Aguzzi, A.; Sailer, A.; Greiner, R.-A.; Autenried, P.; Aguet, M.; Weissmann, C. Mice devoid of PrP are resistant to scrapie. Cell 1993, 73, 1339–1347. [Google Scholar] [CrossRef]
- Telling, G.C.; Scott, M.; Hsiao, K.K.; Foster, D.; Yang, S.L.; Torchia, M.; Sidle, K.C.; Collinge, J.; DeArmond, S.J.; Prusiner, S.B. Transmission of Creutzfeldt-Jakob disease from humans to transgenic mice expressing chimeric human-mouse prion protein. Proc. Natl. Acad. Sci. USA 1994, 91, 9936–9940. [Google Scholar] [CrossRef] [PubMed]
- Brandner, S.; Isenmann, S.; Raeber, A.; Fischer, M.; Sailer, A.; Kobayashi, Y.; Marino, S.; Weissmann, C.; Aguzzi, A. Normal host prion protein necessary for scrapie-induced neurotoxicity. Nature 1996, 379, 339–343. [Google Scholar] [CrossRef] [PubMed]
- Fraser, H.; Dickinson, A.G. Scrapie in mice: Agent-strain differences in the distribution and intensity of grey matter vacuolation. J. Comp. Pathol. 1973, 83, 29–40. [Google Scholar] [CrossRef]
- Asher, D.M.; Gibbs, C.J.; Gajdusek, D.C. Pathogenesis of subacute spongiform encephalopathies. Ann. Clin. Lab. Sci. 1976, 6, 84–103. [Google Scholar] [PubMed]
- Lowenstein, D.H.; Butler, D.A.; Westaway, D.; McKinley, M.P.; DeArmond, S.J.; Prusiner, S.B. Three hamster species with different scrapie incubation times and neuropathological features encode distinct prion proteins. Mol. Cell. Biol. 1990, 10, 1153–1163. [Google Scholar] [CrossRef] [PubMed]
- Kimberlin, R.H.; Walker, C.A.; Fraser, H. The Genomic Identity of Different Strains of Mouse Scrapie Is Expressed in Hamsters and Preserved on Reisolation in Mice. J. Gen. Virol. 1989, 70, 2017–2025. [Google Scholar] [CrossRef] [PubMed]
- Kimberlin, R.H.; Walker, C.A. Pathogenesis of mouse scrapie: Dynamics of agent replication in spleen, spinal cord and brain after infection by different routes. J. Comp. Pathol. 1979, 89, 551–562. [Google Scholar] [CrossRef]
- Prusiner, S.B.; Groth, D.F.; Cochran, S.P.; Masiarz, F.R.; McKinley, M.P.; Martinez, H.M. Molecular properties, partial purification and assay by incubation period measurements of the hamster scrapie agent. Biochemistry 1980, 19, 4883–4891. [Google Scholar] [CrossRef] [PubMed]
- Mould, D.L.; Dawson, A.M.; Rennie, J.C. Very early replication of scrapie in lymphocytic tissue. Nature 1970, 228, 779–780. [Google Scholar] [CrossRef] [PubMed]
- Béringue, V.; Andreoletti, O.; Le Dur, A.; Essalmani, R.; Vilotte, J.-L.; Lacroux, C.; Reine, F.; Herzog, L.; Biacabe, A.-G.; Baron, T.; et al. A Bovine Prion Acquires an Epidemic Bovine Spongiform Encephalopathy Strain-Like Phenotype on Interspecies Transmission. J. Neurosci. 2007, 27, 6965–6971. [Google Scholar] [CrossRef] [PubMed]
- Caughey, B.; Raymond, G.J.; Bessen, R.A. Strain-dependent differences in beta-sheet conformations of abnormal prion protein. J. Biol. Chem. 1998, 273, 32230–32235. [Google Scholar] [CrossRef] [PubMed]
- Ostapchenko, V.G.; Sawaya, M.R.; Makarava, N.; Savtchenko, R.; Nilsson, K.P.R.; Eisenberg, D.; Baskakov, I.V. Two amyloid States of the prion protein display significantly different folding patterns. J. Mol. Biol. 2010, 400, 908–921. [Google Scholar] [CrossRef] [PubMed]
- Cobb, N.J.; Apostol, M.I.; Chen, S.; Smirnovas, V.; Surewicz, W.K. Conformational Stability of Mammalian Prion Protein Amyloid Fibrils Is Dictated by a Packing Polymorphism within the Core Region. J. Biol. Chem. 2014, 289, 2643–2650. [Google Scholar] [CrossRef] [PubMed]
- Silva, C.J.; Erickson-Beltran, M.L.; Dynin, I.C. Covalent Surface Modification of Prions: A Mass Spectrometry-Based Means of Detecting Distinctive Structural Features of Prion Strains. Biochemistry 2016, 55, 894–902. [Google Scholar] [CrossRef] [PubMed]
- Langeveld, J.P.; Jacobs, J.G.; Erkens, J.H.; Bossers, A.; van Zijderveld, F.G.; van Keulen, L.J. Rapid and discriminatory diagnosis of scrapie and BSE in retro-pharyngeal lymph nodes of sheep. BMC Vet. Res. 2006, 2, 19. [Google Scholar] [CrossRef] [PubMed]
- Cali, I.; Castellani, R.; Alshekhlee, A.; Cohen, Y.; Blevins, J.; Yuan, J.; Langeveld, J.P.M.; Parchi, P.; Safar, J.G.; Zou, W.-Q.; et al. Co-existence of scrapie prion protein types 1 and 2 in sporadic Creutzfeldt–Jakob disease: Its effect on the phenotype and prion-type characteristics. Brain 2009, 132, 2643–2658. [Google Scholar] [CrossRef] [PubMed]
- Bessen, R.A.; Marsh, R.F. Biochemical and physical properties of the prion protein from two strains of the transmissible mink encephalopathy agent. J. Virol. 1992, 66, 2096–2101. [Google Scholar] [PubMed]
- Korth, C.; Stierli, B.; Streit, P.; Moser, M.; Schaller, O.; Fischer, R.; Schulz-Schaeffer, W.; Kretzschmar, H.; Raeber, A.; Braun, U.; et al. Prion (PrPSc)-specific epitope defined by a monoclonal antibody. Nature 1997, 390, 36337. [Google Scholar] [CrossRef] [PubMed]
- Safar, J.; Wille, H.; Itri, V.; Groth, D.; Serban, H.; Torchia, M.; Cohen, F.E.; Prusiner, S.B. Eight prion strains have PrP(Sc) molecules with different conformations. Nat. Med. 1998, 4, 1157–1165. [Google Scholar] [CrossRef] [PubMed]
- Safar, J.G.; Geschwind, M.D.; Deering, C.; Didorenko, S.; Sattavat, M.; Sanchez, H.; Serban, A.; Vey, M.; Baron, H.; Giles, K.; et al. Diagnosis of human prion disease. Proc. Natl. Acad. Sci. USA 2005, 102, 3501–3506. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.P.; Peden, A.H.; Gröner, A.; Ironside, J.W.; Head, M.W. Distinct Stability States of Disease-Associated Human Prion Protein Identified by Conformation-Dependent Immunoassay. J. Virol. 2010, 84, 12030–12038. [Google Scholar] [CrossRef] [PubMed]
- Mays, C.E.; Kim, C.; Haldiman, T.; van der Merwe, J.; Lau, A.; Yang, J.; Grams, J.; Di Bari, M.A.; Nonno, R.; Telling, G.C.; et al. Prion disease tempo determined by host-dependent substrate reduction. J. Clin. Investig. 2014, 124, 847–858. [Google Scholar] [CrossRef] [PubMed]
- Neuendorf, E.; Weber, A.; Saalmueller, A.; Schatzl, H.; Reifenberg, K.; Pfaff, E.; Groschup, M.H. Glycosylation Deficiency at Either One of the Two Glycan Attachment Sites of Cellular Prion Protein Preserves Susceptibility to Bovine Spongiform Encephalopathy and Scrapie Infections. J. Biol. Chem. 2004, 279, 53306–53316. [Google Scholar] [CrossRef] [PubMed]
- Moudjou, M.; Chapuis, J.; Mekrouti, M.; Reine, F.; Herzog, L.; Sibille, P.; Laude, H.; Vilette, D.; Andréoletti, O.; Rezaei, H.; et al. Glycoform-independent prion conversion by highly efficient, cell-based, protein misfolding cyclic amplification. Sci. Rep. 2016, 6. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, S.; Katorcha, E.; Daus, M.L.; Lasch, P.; Beekes, M.; Baskakov, I.V. Sialylation controls prion fate in vivo. J. Biol. Chem. 2016. [Google Scholar] [CrossRef]
- Katorcha, E.; Makarava, N.; Savtchenko, R.; d’Azzo, A.; Baskakov, I.V. Sialylation of Prion Protein Controls the Rate of Prion Amplification, the Cross-Species Barrier, the Ratio of PrPSc Glycoform and Prion Infectivity. PLoS Pathog. 2014, 10, e1004366. [Google Scholar] [CrossRef] [PubMed]
- Telling, G.C.; Parchi, P.; DeArmond, S.J.; Cortelli, P.; Montagna, P.; Gabizon, R.; Mastrianni, J.; Lugaresi, E.; Gambetti, P.; Prusiner, S.B. Evidence for the Conformation of the Pathologic Isoform of the Prion Protein Enciphering and Propagating Prion Diversity. Science 1996, 274, 2079–2082. [Google Scholar] [CrossRef] [PubMed]
- Makarava, N.; Ostapchenko, V.G.; Savtchenko, R.; Baskakov, I.V. Conformational Switching within Individual Amyloid Fibrils. J. Biol. Chem. 2009, 284, 14386–14395. [Google Scholar] [CrossRef] [PubMed]
- Mahal, S.P.; Baker, C.A.; Demczyk, C.A.; Smith, E.W.; Julius, C.; Weissmann, C. Prion strain discrimination in cell culture: The cell panel assay. Proc. Natl. Acad. Sci. USA 2007, 104, 20908–20913. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Browning, S.; Mahal, S.P.; Oelschlegel, A.M.; Weissmann, C. Darwinian Evolution of Prions in Cell Culture. Science 2010, 327, 869–872. [Google Scholar] [CrossRef] [PubMed]
- Weissmann, C.; Li, J.; Mahal, S.P.; Browning, S. Prions on the move. EMBO Rep. 2011, 12, 1109–1117. [Google Scholar] [CrossRef] [PubMed]
- Weissmann, C. Mutation and Selection of Prions. PLoS Pathog. 2012, 8, e1002582. [Google Scholar] [CrossRef] [PubMed]
- Oelschlegel, A.M.; Weissmann, C. Acquisition of Drug Resistance and Dependence by Prions. PLoS Pathog. 2013, 9, e1003158. [Google Scholar] [CrossRef] [PubMed]
- Collinge, J.; Clarke, A.R. A General Model of Prion Strains and Their Pathogenicity. Science 2007, 318, 930–936. [Google Scholar] [CrossRef] [PubMed]
- Saá, P.; Cervenakova, L. Protein misfolding cyclic amplification (PMCA): Current status and future directions. Virus Res. 2015. [Google Scholar] [CrossRef] [PubMed]
- Saborio, G.P.; Permanne, B.; Soto, C. Sensitive detection of pathological prion protein by cyclic amplification of protein misfolding. Nature 2001, 411, 810–813. [Google Scholar] [CrossRef] [PubMed]
- Moudjou, M.; Sibille, P.; Fichet, G.; Reine, F.; Chapuis, J.; Herzog, L.; Jaumain, E.; Laferrière, F.; Richard, C.-A.; Laude, H.; et al. Highly Infectious Prions Generated by a Single Round of Microplate-Based Protein Misfolding Cyclic Amplification. mBio 2014, 5, e00829-13. [Google Scholar] [CrossRef] [PubMed]
- Haldiman, T.; Kim, C.; Cohen, Y.; Chen, W.; Blevins, J.; Qing, L.; Cohen, M.L.; Langeveld, J.; Telling, G.C.; Kong, Q.; et al. Co-existence of Distinct Prion Types Enables Conformational Evolution of Human PrPSc by Competitive Selection. J. Biol. Chem. 2013, 288, 29846–29861. [Google Scholar] [CrossRef] [PubMed]
- Castilla, J.; Saá, P.; Hetz, C.; Soto, C. In vitro generation of infectious scrapie prions. Cell 2005, 121, 195–206. [Google Scholar] [CrossRef] [PubMed]
- Castilla, J.; Morales, R.; Saá, P.; Barria, M.; Gambetti, P.; Soto, C. Cell-free propagation of prion strains. EMBO J. 2008, 27, 2557–2566. [Google Scholar] [CrossRef] [PubMed]
- Green, K.M.; Castilla, J.; Seward, T.S.; Napier, D.L.; Jewell, J.E.; Soto, C.; Telling, G.C. Accelerated High Fidelity Prion Amplification Within and Across Prion Species Barriers. PLOS Pathog. 2008, 4, e1000139. [Google Scholar] [CrossRef] [PubMed]
- Munoz-Montesino, C.; Sizun, C.; Moudjou, M.; Herzog, L.; Reine, F.; Chapuis, J.; Ciric, D.; Igel-Egalon, A.; Laude, H.; Béringue, V.; et al. Generating Bona Fide Mammalian Prions with Internal Deletions. J. Virol. 2016, 90, 6963–6975. [Google Scholar] [CrossRef] [PubMed]
- Munoz-Montesino, C.; Sizun, C.; Moudjou, M.; Herzog, L.; Reine, F.; Igel-Egalon, A.; Barbereau, C.; Chapuis, J.; Ciric, D.; Laude, H.; et al. A stretch of residues within the protease-resistant core is not necessary for prion structure and infectivity. Prion 2017, 11, 25–30. [Google Scholar] [CrossRef] [PubMed]
- Deleault, N.R.; Lucassen, R.W.; Supattapone, S. RNA molecules stimulate prion protein conversion. Nature 2003, 425, 717–720. [Google Scholar] [CrossRef] [PubMed]
- Deleault, N.R.; Geoghegan, J.C.; Nishina, K.; Kascsak, R.; Williamson, R.A.; Supattapone, S. Protease-resistant Prion Protein Amplification Reconstituted with Partially Purified Substrates and Synthetic Polyanions. J. Biol. Chem. 2005, 280, 26873–26879. [Google Scholar] [CrossRef] [PubMed]
- Deleault, N.R.; Harris, B.T.; Rees, J.R.; Supattapone, S. Formation of native prions from minimal components in vitro. Proc. Natl. Acad. Sci. USA 2007, 104, 9741–9746. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Wang, X.; Yuan, C.-G.; Ma, J. Generating a Prion with Bacterially Expressed Recombinant Prion Protein. Science 2010, 327, 1132–1135. [Google Scholar] [CrossRef] [PubMed]
- Wilham, J.M.; Orrú, C.D.; Bessen, R.A.; Atarashi, R.; Sano, K.; Race, B.; Meade-White, K.D.; Taubner, L.M.; Timmes, A.; Caughey, B. Rapid End-Point Quantitation of Prion Seeding Activity with Sensitivity Comparable to Bioassays. PLoS Pathog. 2010, 6. [Google Scholar] [CrossRef] [PubMed]
- Christina, D.; Orrú, J.M.W. Prion disease blood test using immunoprecipitation and improved quaking-induced conversion. mBio 2011, 2, e00078-11. [Google Scholar] [CrossRef]
- Henderson, D.M.; Davenport, K.A.; Haley, N.J.; Denkers, N.D.; Mathiason, C.K.; Hoover, E.A. Quantitative Assessment of Prion Infectivity in Tissues and Body Fluids by RT-QuIC. J. Gen. Virol. 2014. [Google Scholar] [CrossRef]
- Espinosa, J.C.; Nonno, R.; Di Bari, M.; Aguilar-Calvo, P.; Pirisinu, L.; Fernández-Borges, N.; Vanni, I.; Vaccari, G.; Marín-Moreno, A.; Frassanito, P.; et al. PrPc Governs Susceptibility to Prion Strains in Bank Vole, While Other Host Factors Modulate Strain Features. J. Virol. 2016, 90, 10660–10669. [Google Scholar] [CrossRef] [PubMed]
- Orrú, C.D.; Favole, A.; Corona, C.; Mazza, M.; Manca, M.; Groveman, B.R.; Hughson, A.G.; Acutis, P.L.; Caramelli, M.; Zanusso, G.; et al. Detection and Discrimination of Classical and Atypical L-Type Bovine Spongiform Encephalopathy by Real-Time Quaking-Induced Conversion. J. Clin. Microbiol. 2015, 53, 1115–1120. [Google Scholar] [CrossRef] [PubMed]
- Magnusson, K.; Simon, R.; Sjölander, D.; Sigurdson, C.J.; Hammarström, P.; Nilsson, K.P.R. Multimodal fluorescence microscopy of prion strain specific PrP deposits stained by thiophene-based amyloid ligands. Prion 2014, 8, 319–329. [Google Scholar] [CrossRef] [PubMed]
- Bessen, R.A.; Marsh, R.F. Distinct PrP properties suggest the molecular basis of strain variation in transmissible mink encephalopathy. J. Virol. 1994, 68, 7859–7868. [Google Scholar] [PubMed]
- Bessen, R.A.; Kocisko, D.A.; Raymond, G.J.; Nandan, S.; Lansbury, P.T.; Caughey, B. Non-genetic propagation of strain-specific properties of scrapie prion protein. Nature 1995, 375, 698–700. [Google Scholar] [CrossRef] [PubMed]
- Tixador, P.; Herzog, L.; Reine, F.; Jaumain, E.; Chapuis, J.; Le Dur, A.; Laude, H.; Béringue, V. The Physical Relationship between Infectivity and Prion Protein Aggregates Is Strain-Dependent. PLoS Pathog. 2010, 6, e1000859. [Google Scholar] [CrossRef] [PubMed]
- Laferrière, F.; Tixador, P.; Moudjou, M.; Chapuis, J.; Sibille, P.; Herzog, L.; Reine, F.; Jaumain, E.; Laude, H.; Rezaei, H.; et al. Quaternary Structure of Pathological Prion Protein as a Determining Factor of Strain-Specific Prion Replication Dynamics. PLoS Pathog. 2013, 9, e1003702. [Google Scholar] [CrossRef] [PubMed]
- Baron, T.G.M.; Madec, J.-Y.; Calavas, D. Similar Signature of the Prion Protein in Natural Sheep Scrapie and Bovine Spongiform Encephalopathy-Linked Diseases. J. Clin. Microbiol. 1999, 37, 3701–3704. [Google Scholar] [PubMed]
- Casalone, C.; Zanusso, G.; Acutis, P.; Ferrari, S.; Capucci, L.; Tagliavini, F.; Monaco, S.; Caramelli, M. Identification of a second bovine amyloidotic spongiform encephalopathy: Molecular similarities with sporadic Creutzfeldt-Jakob disease. Proc. Natl. Acad. Sci. USA 2004, 101, 3065–3070. [Google Scholar] [CrossRef] [PubMed]
- Bartz, J.C.; Bessen, R.A.; McKenzie, D.; Marsh, R.F.; Aiken, J.M. Adaptation and Selection of Prion Protein Strain Conformations following Interspecies Transmission of Transmissible Mink Encephalopathy. J. Virol. 2000, 74, 5542–5547. [Google Scholar] [CrossRef] [PubMed]
- Le Dur, A.; Laï, T.L.; Stinnakre, M.-G.; Laisné, A.; Chenais, N.; Rakotobe, S.; Passet, B.; Reine, F.; Soulier, S.; Herzog, L.; et al. Divergent prion strain evolution driven by PrP(c) expression level in transgenic mice. Nat. Commun. 2017, 8, 14170. [Google Scholar] [CrossRef] [PubMed]
- Kimberlin, R.H.; Walker, C.A. Evidence that the Transmission of One Source of Scrapie Agent to Hamsters Involves Separation of Agent Strains from a Mixture. J. Gen. Virol. 1978, 39, 487–496. [Google Scholar] [CrossRef] [PubMed]
- Race, R.; Chesebro, B. Scrapie infectivity found in resistant species. Nature 1998, 392, 770. [Google Scholar] [CrossRef] [PubMed]
- Hill, A.F.; Joiner, S.; Linehan, J.; Desbruslais, M.; Lantos, P.L.; Collinge, J. Species-barrier-independent prion replication in apparently resistant species. Proc. Natl. Acad. Sci. USA 2000, 97, 10248–10253. [Google Scholar] [CrossRef] [PubMed]
- Prusiner, S.B.; Scott, M.; Foster, D.; Pan, K.-M.; Groth, D.; Mirenda, C.; Torchia, M.; Yang, S.-L.; Serban, D.; Carlson, G.A.; et al. Transgenetic studies implicate interactions between homologous PrP isoforms in scrapie prion replication. Cell 1990, 63, 673–686. [Google Scholar] [CrossRef]
- Castilla, J.; Gonzalez-Romero, D.; Saá, P.; Morales, R.; De Castro, J.; Soto, C. Crossing the Species Barrier by PrPSc Replication In Vitro Generates Unique Infectious Prions. Cell 2008, 134, 757–768. [Google Scholar] [CrossRef] [PubMed]
- Bruce, M.E.; Will, R.G.; Ironside, J.W.; McConnell, I.; Drummond, D.; Suttie, A.; McCardle, L.; Chree, A.; Hope, J.; Birkett, C.; et al. Transmissions to mice indicate that “new variant” CJD is caused by the BSE agent. Nature 1997, 389, 498–501. [Google Scholar] [CrossRef] [PubMed]
- Scott, M.R.; Will, R.; Ironside, J.; Nguyen, H.-O.B.; Tremblay, P.; DeArmond, S.J.; Prusiner, S.B. Compelling transgenetic evidence for transmission of bovine spongiform encephalopathy prions to humans. Proc. Natl. Acad. Sci. USA 1999, 96, 15137–15142. [Google Scholar] [CrossRef] [PubMed]
- Asante, E.A.; Linehan, J.M.; Desbruslais, M.; Joiner, S.; Gowland, I.; Wood, A.L.; Welch, J.; Hill, A.F.; Lloyd, S.E.; Wadsworth, J.D.F.; et al. BSE prions propagate as either variant CJD-like or sporadic CJD-like prion strains in transgenic mice expressing human prion protein. EMBO J. 2002, 21, 6358–6366. [Google Scholar] [CrossRef] [PubMed]
- Béringue, V.; Herzog, L.; Jaumain, E.; Reine, F.; Sibille, P.; Le Dur, A.; Vilotte, J.-L.; Laude, H. Facilitated Cross-Species Transmission of Prions in Extraneural Tissue. Science 2012, 335, 472–475. [Google Scholar] [CrossRef] [PubMed]
- Williams, E.S.; Young, S. Spongiform encephalopathies in Cervidae. Rev. Sci. Tech. Int. Off. Epizoot. 1992, 11, 551–567. [Google Scholar] [CrossRef]
- Watts, J.C.; Balachandran, A.; Westaway, D. The expanding universe of prion diseases. PLoS Pathog. 2006, 2, e26. [Google Scholar] [CrossRef] [PubMed]
- Benestad, S.L.; Mitchell, G.; Simmons, M.; Ytrehus, B.; Vikøren, T. First case of chronic wasting disease in Europe in a Norwegian free-ranging reindeer. Vet. Res. 2016, 47. [Google Scholar] [CrossRef] [PubMed]
- Sigurdson, C.J.; Manco, G.; Schwarz, P.; Liberski, P.; Hoover, E.A.; Hornemann, S.; Polymenidou, M.; Miller, M.W.; Glatzel, M.; Aguzzi, A. Strain Fidelity of Chronic Wasting Disease upon Murine Adaptation. J. Virol. 2006, 80, 12303–12311. [Google Scholar] [CrossRef] [PubMed]
- Herbst, A.; Velásquez, C.D.; Triscott, E.; Aiken, J.M.; McKenzie, D. Chronic Wasting Disease Prion Strain Emergence and Host Range Expansion. Emerg. Infect. Dis. 2017, 23, 1598–1600. [Google Scholar] [CrossRef] [PubMed]
- Kurt, T.D.; Jiang, L.; Fernández-Borges, N.; Bett, C.; Liu, J.; Yang, T.; Spraker, T.R.; Castilla, J.; Eisenberg, D.; Kong, Q.; et al. Human prion protein sequence elements impede cross-species chronic wasting disease transmission. J. Clin. Investig. 2015, 125, 1485. [Google Scholar] [CrossRef] [PubMed]
- Czub, S.; Schulz-Schaeffer, W.; Stahl-Hennig, C.; Beekes, M.; Schaetzl, H.; Motzkus, D. Chronic Wasting Disease: PRION 2017 CONFERENCE ABSTRACT First Evidence of Intracranial and Peroral Transmission of Chronic Wasting Disease (CWD) into Cynomolgus Macaques: A Work in Progress. Available online: http://chronic-wasting-disease.blogspot.hk/2017/06/prion-2017-conference-abstract-first.html (accessed on 26 December 2017).
- Di Bari, M.A.; Chianini, F.; Vaccari, G.; Esposito, E.; Conte, M.; Eaton, S.L.; Hamilton, S.; Finlayson, J.; Steele, P.J.; Dagleish, M.P.; et al. The bank vole (Myodes glareolus) as a sensitive bioassay for sheep scrapie. J. Gen. Virol. 2008, 89, 2975–2985. [Google Scholar] [CrossRef] [PubMed]
- Peretz, D.; Williamson, R.A.; Legname, G.; Matsunaga, Y.; Vergara, J.; Burton, D.R.; DeArmond, S.J.; Prusiner, S.B.; Scott, M.R. A change in the conformation of prions accompanies the emergence of a new prion strain. Neuron 2002, 34, 921–932. [Google Scholar] [CrossRef]
- Gao, C.; Han, J.; Zhang, J.; Wei, J.; Zhang, B.-Y.; Tian, C.; Zhang, J.; Shi, Q.; Dong, X.-P. Protein Misfolding Cyclic Amplification Cross-Species Products of Mouse-Adapted Scrapie Strain 139A and Hamster-Adapted Scrapie Strain 263K with Brain and Muscle Tissues of Opposite Animals Generate Infectious Prions. Mol. Neurobiol. 2017, 54, 3771–3782. [Google Scholar] [CrossRef] [PubMed]
- Watts, J.C.; Giles, K.; Saltzberg, D.J.; Dugger, B.N.; Patel, S.; Oehler, A.; Bhardwaj, S.; Sali, A.; Prusiner, S.B. Guinea Pig Prion Protein Supports Rapid Propagation of Bovine Spongiform Encephalopathy and Variant Creutzfeldt-Jakob Disease Prions. J. Virol. 2016, 90, 9558–9569. [Google Scholar] [CrossRef] [PubMed]
- Davenport, K.A.; Henderson, D.M.; Bian, J.; Telling, G.C.; Mathiason, C.K.; Hoover, E.A. Insights into Chronic Wasting Disease and Bovine Spongiform Encephalopathy Species Barriers by Use of Real-Time Conversion. J. Virol. 2015, 89, 9524–9531. [Google Scholar] [CrossRef] [PubMed]
- Barria, M.A.; Telling, G.C.; Gambetti, P.; Mastrianni, J.A.; Soto, C. Generation of a New Form of Human PrPSc in Vitro by Interspecies Transmission from Cervid Prions. J. Biol. Chem. 2011, 286, 7490–7495. [Google Scholar] [CrossRef] [PubMed]
- Marsh, R.F.; Kincaid, A.E.; Bessen, R.A.; Bartz, J.C. Interspecies Transmission of Chronic Wasting Disease Prions to Squirrel Monkeys (Saimiri sciureus). J. Virol. 2005, 79, 13794–13796. [Google Scholar] [CrossRef] [PubMed]
- Race, B.; Meade-White, K.D.; Miller, M.W.; Barbian, K.D.; Rubenstein, R.; LaFauci, G.; Cervenakova, L.; Favara, C.; Gardner, D.; Long, D.; et al. Susceptibilities of Nonhuman Primates to Chronic Wasting Disease. Emerg. Infect. Dis. 2009, 15, 1366–1376. [Google Scholar] [CrossRef] [PubMed]
- Scott, M.; Foster, D.; Mirenda, C.; Serban, D.; Coufal, F.; Wälchli, M.; Torchia, M.; Groth, D.; Carlson, G.; DeArmond, S.J.; et al. Transgenic mice expressing hamster prion protein produce species-specific scrapie infectivity and amyloid plaques. Cell 1989, 59, 847–857. [Google Scholar] [CrossRef]
- Race, R.E.; Priola, S.A.; Bessen, R.A.; Ernst, D.; Dockter, J.; Rall, G.F.; Mucke, L.; Chesebro, B.; Oldstone, M.B. Neuron-specific expression of a hamster prion protein minigene in transgenic mice induces susceptibility to hamster scrapie agent. Neuron 1995, 15, 1183–1191. [Google Scholar] [CrossRef]
- Priola, S.A. Prion protein and species barriers in the transmissible spongiform encephalopathies. Biomed. Pharmacother. 1999, 53, 27–33. [Google Scholar] [CrossRef]
- Sarradin, P.; Viglietta, C.; Limouzin, C.; Andréoletti, O.; Daniel-Carlier, N.; Barc, C.; Leroux-Coyau, M.; Berthon, P.; Chapuis, J.; Rossignol, C.; et al. Transgenic Rabbits Expressing Ovine PrP Are Susceptible to Scrapie. PLoS Pathog. 2015, 11, e1005077. [Google Scholar] [CrossRef] [PubMed]
- Tranulis, M.A. Influence of the prion protein gene, Prnp, on scrapie susceptibility in sheep. APMIS 2002, 110, 33–43. [Google Scholar] [CrossRef] [PubMed]
- Goldmann, W. PrP genetics in ruminant transmissible spongiform encephalopathies. Vet. Res. 2008, 39, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Dong, X.-P. Epidemiological characteristics of human prion diseases. Infect. Dis. Poverty 2016, 5. [Google Scholar] [CrossRef] [PubMed]
- Watts, J.C.; Giles, K.; Stöhr, J.; Oehler, A.; Bhardwaj, S.; Grillo, S.K.; Patel, S.; DeArmond, S.J.; Prusiner, S.B. Spontaneous Generation of Rapidly Transmissible Prions in Transgenic Mice Expressing Wild-Type Bank Vole Prion Protein. Proc. Natl. Acad. Sci. USA 2012, 109, 3498–3503. [Google Scholar] [CrossRef] [PubMed]
- Tateishi, J.; Kitamoto, T.; Hoque, M.Z.; Furukawa, H. Experimental transmission of Creutzfeldt-Jakob disease and related diseases to rodents. Neurology 1996, 46, 532–537. [Google Scholar] [CrossRef] [PubMed]
- Palmer, M.S.; Dryden, A.J.; Hughes, J.T.; Collinge, J. Homozygous prion protein genotype predisposes to sporadic Creutzfeldt-Jakob disease. Nature 1991, 352, 340–342. [Google Scholar] [CrossRef] [PubMed]
- Zeidler, M.; Stewart, G.E.; Barraclough, C.R.; Bateman, D.E.; Bates, D.; Burn, D.J.; Colchester, A.C.; Durward, W.; Fletcher, N.A.; Hawkins, S.A.; et al. New variant Creutzfeldt-Jakob disease: Neurological features and diagnostic tests. Lancet 1997, 350, 903–907. [Google Scholar] [CrossRef]
- Fernández-Borges, N.; Espinosa, J.C.; Marín-Moreno, A.; Aguilar-Calvo, P.; Asante, E.A.; Kitamoto, T.; Mohri, S.; Andréoletti, O.; Torres, J.M. Protective Effect of Val129-PrP against Bovine Spongiform Encephalopathy but not Variant Creutzfeldt-Jakob Disease. Emerg. Infect. Dis. 2017, 23, 1522–1530. [Google Scholar] [CrossRef] [PubMed]
- Shibuya, S.; Higuchi, J.; Shin, R.-W.; Tateishi, J.; Kitamoto, T. Protective prion protein polymorphisms against sporadic Creutzfeldt-Jakob disease. Lancet 1998, 351, 419. [Google Scholar] [CrossRef]
- Asante, E.A.; Smidak, M.; Grimshaw, A.; Houghton, R.; Tomlinson, A.; Jeelani, A.; Jakubcova, T.; Hamdan, S.; Richard-Londt, A.; Linehan, J.M.; et al. A naturally occurring variant of the human prion protein completely prevents prion disease. Nature 2015, 522, 478–481. [Google Scholar] [CrossRef] [PubMed]
- Green, K.M.; Browning, S.R.; Seward, T.S.; Jewell, J.E.; Ross, D.L.; Green, M.A.; Williams, E.S.; Hoover, E.A.; Telling, G.C. The elk PRNP codon 132 polymorphism controls cervid and scrapie prion propagation. J. Gen. Virol. 2008, 89, 598–608. [Google Scholar] [CrossRef] [PubMed]
- Cartoni, C.; Schininà, M.E.; Maras, B.; Nonno, R.; Vaccari, G.; Di Bari, M.; Conte, M.; De Pascalis, A.; Principe, S.; Cardone, F.; et al. Quantitative profiling of the pathological prion protein allotypes in bank voles by liquid chromatography–mass spectrometry. J. Chromatogr. B 2007, 849, 302–306. [Google Scholar] [CrossRef] [PubMed]
- Kimberlin, R.H.; Walker, C.A. Competition between strains of scrapie depends on the blocking agent being infectious. Intervirology 1985, 23, 74–81. [Google Scholar] [CrossRef] [PubMed]
- Kimberlin, R.H.; Cole, S.; Walker, C.A. Transmissible mink encephalopathy (TME) in Chinese hamsters: Identification of two strains of TME and comparisons with scrapie. Neuropathol. Appl. Neurobiol. 1986, 12, 197–206. [Google Scholar] [CrossRef] [PubMed]
- Kimberlin, R.H.; Cole, S.; Walker, C.A. Temporary and Permanent Modifications to a Single Strain of Mouse Scrapie on Transmission to Rats and Hamsters. J. Gen. Virol. 1987, 68, 1875–1881. [Google Scholar] [CrossRef] [PubMed]
- Priola, S.A.; Caughey, B.; Race, R.E.; Chesebro, B. Heterologous PrP molecules interfere with accumulation of protease-resistant PrP in scrapie-infected murine neuroblastoma cells. J. Virol. 1994, 68, 4873–4878. [Google Scholar] [PubMed]
- Priola, S.A.; Chesebro, B. A single hamster PrP amino acid blocks conversion to protease-resistant PrP in scrapie-infected mouse neuroblastoma cells. J. Virol. 1995, 69, 7754–7758. [Google Scholar] [PubMed]
- Telling, G.C.; Scott, M.; Mastrianni, J.; Gabizon, R.; Torchia, M.; Cohen, F.E.; DeArmond, S.J.; Prusiner, S.B. Prion propagation in mice expressing human and chimeric PrP transgenes implicates the interaction of cellular PrP with another protein. Cell 1995, 83, 79–90. [Google Scholar] [CrossRef]
- Abid, K.; Morales, R.; Soto, C. Cellular factors implicated in prion replication. FEBS Lett. 2010, 584, 2409–2414. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.I.; Yang, Q.; Perrier, V.; Baskakov, I.V. The dominant-negative effect of the Q218K variant of the prion protein does not require protein X. Protein Sci. Publ. Protein Soc. 2007, 16, 2166–2173. [Google Scholar] [CrossRef] [PubMed]
- Dickinson, A.G.; Fraser, H.; Meikle, V.M.; Outram, G.W. Competition between different scrapie agents in mice. Nat. New Biol. 1972, 237, 244–245. [Google Scholar] [CrossRef] [PubMed]
- Bartz, J.C.; Aiken, J.M.; Bessen, R.A. Delay in onset of prion disease for the HY strain of transmissible mink encephalopathy as a result of prior peripheral inoculation with the replication-deficient DY strain. J. Gen. Virol. 2004, 85, 265–273. [Google Scholar] [CrossRef] [PubMed]
- Shikiya, R.A.; Ayers, J.I.; Schutt, C.R.; Kincaid, A.E.; Bartz, J.C. Coinfecting Prion Strains Compete for a Limiting Cellular Resource. J. Virol. 2010, 84, 5706–5714. [Google Scholar] [CrossRef] [PubMed]
- Bartz, J.C.; Kramer, M.L.; Sheehan, M.H.; Hutter, J.A.L.; Ayers, J.I.; Bessen, R.A.; Kincaid, A.E. Prion Interference Is Due to a Reduction in Strain-Specific PrPSc Levels. J. Virol. 2007, 81, 689–697. [Google Scholar] [CrossRef] [PubMed]
- Sandberg, M.K.; Al-Doujaily, H.; Sharps, B.; De Oliveira, M.W.; Schmidt, C.; Richard-Londt, A.; Lyall, S.; Linehan, J.M.; Brandner, S.; Wadsworth, J.D.F.; et al. Prion neuropathology follows the accumulation of alternate prion protein isoforms after infective titre has peaked. Nat. Commun. 2014, 5. [Google Scholar] [CrossRef]
- Wiseman, F.K.; Cancellotti, E.; Piccardo, P.; Iremonger, K.; Boyle, A.; Brown, D.; Ironside, J.W.; Manson, J.C.; Diack, A.B. The glycosylation status of PrPc is a key factor in determining transmissible spongiform encephalopathy transmission between species. J. Virol. 2015, 89, 4738–4747. [Google Scholar] [CrossRef] [PubMed]
- Cancellotti, E.; Wiseman, F.; Tuzi, N.L.; Baybutt, H.; Monaghan, P.; Aitchison, L.; Simpson, J.; Manson, J.C. Altered Glycosylated PrP Proteins Can Have Different Neuronal Trafficking in Brain but Do Not Acquire Scrapie-like Properties. J. Biol. Chem. 2005, 280, 42909–42918. [Google Scholar] [CrossRef] [PubMed]
- Priola, S.A.; Lawson, V.A. Glycosylation influences cross-species formation of protease-resistant prion protein. EMBO J. 2001, 20, 6692–6699. [Google Scholar] [CrossRef] [PubMed]
- Katorcha, E.; Srivastava, S.; Klimova, N.; Baskakov, I.V. Sialylation of Glycosylphosphatidylinositol (GPI) Anchors of Mammalian Prions Is Regulated in a Host-, Tissue- and Cell-specific Manner. J. Biol. Chem. 2016, 291, 17009–17019. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, S.; Makarava, N.; Katorcha, E.; Savtchenko, R.; Brossmer, R.; Baskakov, I.V. Post-conversion sialylation of prions in lymphoid tissues. Proc. Natl. Acad. Sci. USA 2015, 112, E6654–E6662. [Google Scholar] [CrossRef] [PubMed]
- Aguilar-Calvo, P.; Xiao, X.; Bett, C.; Eraña, H.; Soldau, K.; Castilla, J.; Nilsson, K.P.R.; Surewicz, W.K.; Sigurdson, C.J. Post-translational modifications in PrP expand the conformational diversity of prions in vivo. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef] [PubMed]
- McCulloch, L.; Brown, K.L.; Bradford, B.M.; Hopkins, J.; Bailey, M.; Rajewsky, K.; Manson, J.C.; Mabbott, N.A. Follicular Dendritic Cell-Specific Prion Protein (PrPc) Expression Alone Is Sufficient to Sustain Prion Infection in the Spleen. PLoS Pathog. 2011, 7, e1002402. [Google Scholar] [CrossRef] [PubMed]
- Krautler, N.J.; Kana, V.; Kranich, J.; Tian, Y.; Perera, D.; Lemm, D.; Schwarz, P.; Armulik, A.; Browning, J.L.; Tallquist, M.; et al. Follicular Dendritic Cells Emerge from Ubiquitous Perivascular Precursors. Cell 2012, 150, 194–206. [Google Scholar] [CrossRef] [PubMed]
- Béringue, V.; Demoy, M.; Lasmézas, C.I.; Gouritin, B.; Weingarten, C.; Deslys, J.-P.; Andreux, J.-P.; Couvreur, P.; Dormont, D. Role of spleen macrophages in the clearance of scrapie agent early in pathogenesis. J. Pathol. 2000, 190, 495–502. [Google Scholar] [CrossRef]
- Montrasio, F.; Cozzio, A.; Flechsig, E.; Rossi, D.; Klein, M.A.; Rülicke, T.; Raeber, A.J.; Vosshenrich, C.A.J.; Proft, J.; Aguzzi, A.; et al. B lymphocyte-restricted expression of prion protein does not enable prion replication in prion protein knockout mice. Proc. Natl. Acad. Sci. USA 2001, 98, 4034–4037. [Google Scholar] [CrossRef] [PubMed]
- Prinz, M.; Heikenwalder, M.; Junt, T.; Schwarz, P.; Glatzel, M.; Heppner, F.L.; Fu, Y.-X.; Lipp, M.; Aguzzi, A. Positioning of follicular dendritic cells within the spleen controls prion neuroinvasion. Nature 2003, 425, 957–962. [Google Scholar] [CrossRef] [PubMed]
- Shikiya, R.A.; Langenfeld, K.A.; Eckland, T.E.; Trinh, J.; Holec, S.A.M.; Mathiason, C.K.; Kincaid, A.E.; Bartz, J.C. PrPSc formation and clearance as determinants of prion tropism. PLoS Pathog. 2017, 13, e1006298. [Google Scholar] [CrossRef] [PubMed]
- Ironside, J.W.; Head, M.W.; McCardle, L.; Knight, R. Neuropathology of variant Creutzfeldt-Jakob disease. Acta Neurobiol. Exp. 2002, 62, 175–182. [Google Scholar] [CrossRef]
- Brandner, S.; Whitfield, J.; Boone, K.; Puwa, A.; O’Malley, C.; Linehan, J.M.; Joiner, S.; Scaravilli, F.; Calder, I.; Alpers, M.P.; et al. Central and peripheral pathology of kuru: Pathological analysis of a recent case and comparison with other forms of human prion disease. Philos. Trans. R. Soc. B Biol. Sci. 2008, 363, 3755–3763. [Google Scholar] [CrossRef] [PubMed]
- Rubenstein, R.; Chang, B. Re-Assessment of PrPSc Distribution in Sporadic and Variant CJD. PLoS ONE 2013, 8, e66352. [Google Scholar] [CrossRef] [PubMed]
- Bons, N.; Lehmann, S.; Nishida, N.; Mestre-Frances, N.; Dormont, D.; Belli, P.; Delacourte, A.; Grassi, J.; Brown, P. BSE infection of the small short-lived primate Microcebus murinus. C. R. Biol. 2002, 325, 67–74. [Google Scholar] [CrossRef]
- Herzog, C.; Salès, N.; Etchegaray, N.; Charbonnier, A.; Freire, S.; Dormont, D.; Deslys, J.-P.; Lasmézas, C.I. Tissue distribution of bovine spongiform encephalopathy agent in primates after intravenous or oral infection. Lancet Lond. Engl. 2004, 363, 422–428. [Google Scholar] [CrossRef]
- Lotscher, M.; Recher, M.; Hunziker, L.; Klein, M.A. Immunologically Induced, Complement-Dependent Up-Regulation of the Prion Protein in the Mouse Spleen: Follicular Dendritic Cells Versus Capsule and Trabeculae. J. Immunol. 2003, 170, 6040–6047. [Google Scholar] [CrossRef] [PubMed]
- Heikenwalder, M.; Zeller, N.; Seeger, H.; Prinz, M.; Klöhn, P.-C.; Schwarz, P.; Ruddle, N.H.; Weissmann, C.; Aguzzi, A. Chronic lymphocytic inflammation specifies the organ tropism of prions. Science 2005, 307, 1107–1110. [Google Scholar] [CrossRef] [PubMed]
- Khalifé, M.; Young, R.; Passet, B.; Halliez, S.; Vilotte, M.; Jaffrezic, F.; Marthey, S.; Béringue, V.; Vaiman, D.; Le Provost, F.; et al. Transcriptomic Analysis Brings New Insight into the Biological Role of the Prion Protein during Mouse Embryogenesis. PLoS ONE 2011, 6, e23253. [Google Scholar] [CrossRef] [PubMed]
- Hunter, N.; Houston, F.; Foster, J.; Goldmann, W.; Drummond, D.; Parnham, D.; Kennedy, I.; Green, A.; Stewart, P.; Chong, A. Susceptibility of Young Sheep to Oral Infection with Bovine Spongiform Encephalopathy Decreases Significantly after Weaning. J. Virol. 2012, 86, 11856–11862. [Google Scholar] [CrossRef] [PubMed]
- Avrahami, D.; Gabizon, R. Age-related alterations affect the susceptibility of mice to prion infection. Neurobiol. Aging 2011, 32, 2006–2015. [Google Scholar] [CrossRef] [PubMed]
- Brown, K.L.; Wathne, G.J.; Sales, J.; Bruce, M.E.; Mabbott, N.A. The Effects of Host Age on Follicular Dendritic Cell Status Dramatically Impair Scrapie Agent Neuroinvasion in Aged Mice. J. Immunol. 2009, 183, 5199–5207. [Google Scholar] [CrossRef] [PubMed]
- Brown, K.L.; Mabbott, N.A. Evidence of subclinical prion disease in aged mice following exposure to bovine spongiform encephalopathy. J. Gen. Virol. 2014, 95, 231–243. [Google Scholar] [CrossRef] [PubMed]
- Langevin, C.; Andréoletti, O.; Le Dur, A.; Laude, H.; Béringue, V. Marked influence of the route of infection on prion strain apparent phenotype in a scrapie transgenic mouse model. Neurobiol. Dis. 2011, 41, 219–225. [Google Scholar] [CrossRef] [PubMed]
- Gill, O.N.; Spencer, Y.; Richard-Loendt, A.; Kelly, C.; Dabaghian, R.; Boyes, L.; Linehan, J.; Simmons, M.; Webb, P.; Bellerby, P.; et al. Prevalent abnormal prion protein in human appendixes after bovine spongiform encephalopathy epizootic: Large scale survey. BMJ 2013, 347, f5675. [Google Scholar] [CrossRef] [PubMed]
- Carp, R.I.; Meeker, H.; Sersen, E. Scrapie strains retain their distinctive characteristics following passages of homogenates from different brain regions and spleen. J. Gen. Virol. 1997, 78 Pt 1, 283–290. [Google Scholar] [CrossRef] [PubMed]
- Privat, N.; Levavasseur, E.; Yildirim, S.; Hannaoui, S.; Brandel, J.-P.; Laplanche, J.-L.; Béringue, V.; Seilhean, D.; Haïk, S. Region-specific protein misfolding cyclic amplification reproduces brain tropism of prion strains. J. Biol. Chem. 2017. [Google Scholar] [CrossRef] [PubMed]
- Nishida, N.; Harris, D.A.; Vilette, D.; Laude, H.; Frobert, Y.; Grassi, J.; Casanova, D.; Milhavet, O.; Lehmann, S. Successful Transmission of Three Mouse-Adapted Scrapie Strains to Murine Neuroblastoma Cell Lines Overexpressing Wild-Type Mouse Prion Protein. J. Virol. 2000, 74, 320–325. [Google Scholar] [CrossRef] [PubMed]
- Race, R.; Raines, A.; Raymond, G.J.; Caughey, B.; Chesebro, B. Long-Term Subclinical Carrier State Precedes Scrapie Replication and Adaptation in a Resistant Species: Analogies to Bovine Spongiform Encephalopathy and Variant Creutzfeldt-Jakob Disease in Humans. J. Virol. 2001, 75, 10106–10112. [Google Scholar] [CrossRef] [PubMed]
- Race, R.; Meade-White, K.; Raines, A.; Raymond, G.J.; Caughey, B.; Chesebro, B. Subclinical Scrapie Infection in a Resistant Species: Persistence, Replication and Adaptation of Infectivity during Four Passages. J. Infect. Dis. 2002, 186, S166–S170. [Google Scholar] [CrossRef] [PubMed]
- Thackray, A.M.; Hopkins, L.; Lockey, R.; Spiropoulos, J.; Bujdoso, R. Propagation of ovine prions from “poor” transmitter scrapie isolates in ovine PrP transgenic mice. Exp. Mol. Pathol. 2012, 92, 167–174. [Google Scholar] [CrossRef] [PubMed]
- Capobianco, R.; Casalone, C.; Suardi, S.; Mangieri, M.; Miccolo, C.; Limido, L.; Catania, M.; Rossi, G.; Fede, G.D.; Giaccone, G.; et al. Conversion of the BASE Prion Strain into the BSE Strain: The Origin of BSE? PLoS Pathog. 2007, 3. [Google Scholar] [CrossRef] [PubMed]
- Baron, T.; Vulin, J.; Biacabe, A.-G.; Lakhdar, L.; Verchere, J.; Torres, J.-M.; Bencsik, A. Emergence of classical BSE strain properties during serial passages of H-BSE in wild-type mice. PLoS ONE 2011, 6, e15839. [Google Scholar] [CrossRef] [PubMed]
- Plinston, C.; Hart, P.; Hunter, N.; Manson, J.C.; Barron, R.M. Increased susceptibility of transgenic mice expressing human PrP to experimental sheep bovine spongiform encephalopathy is not due to increased agent titre in sheep brain tissue. J. Gen. Virol. 2014, 95, 1855–1859. [Google Scholar] [CrossRef] [PubMed]
- Priem, J.; Langeveld, J.P.M.; van Keulen, L.J.M.; van Zijderveld, F.G.; Andreoletti, O.; Bossers, A. Enhanced Virulence of Sheep-Passaged Bovine Spongiform Encephalopathy Agent Is Revealed by Decreased Polymorphism Barriers in Prion Protein Conversion Studies. J. Virol. 2014, 88, 2903–2912. [Google Scholar] [CrossRef] [PubMed]
- Torres, J.-M.; Espinosa, J.-C.; Aguilar-Calvo, P.; Herva, M.-E.; Relaño-Ginés, A.; Villa-Diaz, A.; Morales, M.; Parra, B.; Alamillo, E.; Brun, A.; et al. Elements Modulating the Prion Species Barrier and Its Passage Consequences. PLoS ONE 2014, 9. [Google Scholar] [CrossRef] [PubMed]
- Chapuis, J.; Moudjou, M.; Reine, F.; Herzog, L.; Jaumain, E.; Chapuis, C.; Quadrio, I.; Boulliat, J.; Perret-Liaudet, A.; Dron, M.; et al. Emergence of two prion subtypes in ovine PrP transgenic mice infected with human MM2-cortical Creutzfeldt-Jakob disease prions. Acta Neuropathol. Commun. 2016, 4. [Google Scholar] [CrossRef] [PubMed]
- Edgeworth, J.A.; Gros, N.; Alden, J.; Joiner, S.; Wadsworth, J.D.F.; Linehan, J.; Brandner, S.; Jackson, G.S.; Weissmann, C.; Collinge, J. Spontaneous generation of mammalian prions. Proc. Natl. Acad. Sci. USA 2010. [Google Scholar] [CrossRef] [PubMed]
- Thackray, A.M.; Hopkins, L.; Lockey, R.; Spiropoulos, J.; Bujdoso, R. Emergence of multiple prion strains from single isolates of ovine scrapie. J. Gen. Virol. 2011, 92, 1482–1491. [Google Scholar] [CrossRef] [PubMed]
- Okada, H.; Masujin, K.; Miyazawa, K.; Yokoyama, T. Transmissibility of H-Type Bovine Spongiform Encephalopathy to Hamster PrP Transgenic Mice. PLoS ONE 2015, 10. [Google Scholar] [CrossRef] [PubMed]
- Angers, R.C.; Kang, H.-E.; Napier, D.; Browning, S.; Seward, T.; Mathiason, C.; Balachandran, A.; McKenzie, D.; Castilla, J.; Soto, C.; et al. Prion Strain Mutation Determined by Prion Protein Conformational Compatibility and Primary Structure. Science 2010, 328, 1154–1158. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Borges, N.; de Castro, J.; Castilla, J. In vitro studies of the transmission barrier. Prion 2009, 3, 220–223. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Montalban, N.; Lee, Y.J.; Makarava, N.; Savtchenko, R.; Baskakov, I.V. Changes in prion replication environment cause prion strain mutation. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2013. [Google Scholar] [CrossRef] [PubMed]
- Deleault, N.R.; Walsh, D.J.; Piro, J.R.; Wang, F.; Wang, X.; Ma, J.; Rees, J.R.; Supattapone, S. Cofactor Molecules Maintain Infectious Conformation and Restrict Strain Properties in Purified Prions. Proc. Natl. Acad. Sci. USA 2012. [Google Scholar] [CrossRef] [PubMed]
- Ghaemmaghami, S.; Colby, D.W.; Nguyen, H.-O.B.; Hayashi, S.; Oehler, A.; DeArmond, S.J.; Prusiner, S.B. Convergent replication of mouse synthetic prion strains. Am. J. Pathol. 2013, 182, 866–874. [Google Scholar] [CrossRef] [PubMed]
- Sano, K.; Atarashi, R.; Ishibashi, D.; Nakagaki, T.; Satoh, K.; Nishida, N. Conformational Properties of Prion Strains Can Be Transmitted to Recombinant Prion Protein Fibrils in Real-Time Quaking-Induced Conversion. J. Virol. 2014, 88, 11791–11801. [Google Scholar] [CrossRef] [PubMed]
- Sano, K.; Atarashi, R.; Nishida, N. Structural conservation of prion strain specificities in recombinant prion protein fibrils in real-time quaking-induced conversion. Prion 2015, 9, 237–243. [Google Scholar] [CrossRef] [PubMed]
- Makarava, N.; Savtchenko, R.; Baskakov, I.V. Selective Amplification of Classical and Atypical Prions Using Modified Protein Misfolding Cyclic Amplification. J. Biol. Chem. 2013, 288, 33–41. [Google Scholar] [CrossRef] [PubMed]
- Chianini, F.; Fernández-Borges, N.; Vidal, E.; Gibbard, L.; Pintado, B.; De Castro, J.; Priola, S.A.; Hamilton, S.; Eaton, S.L.; Finlayson, J.; et al. Rabbits Are Not Resistant to Prion Infection. Proc. Natl. Acad. Sci. USA 2012, 109, 5080–5085. [Google Scholar] [CrossRef] [PubMed]
- Vidal, E.; Fernández-Borges, N.; Pintado, B.; Eraña, H.; Ordóñez, M.; Márquez, M.; Chianini, F.; Fondevila, D.; Sánchez-Martín, M.A.; Andreoletti, O.; et al. Transgenic Mouse Bioassay: Evidence That Rabbits Are Susceptible to a Variety of Prion Isolates. PLoS Pathog. 2015, 11, e1004977. [Google Scholar] [CrossRef] [PubMed]
- Vidal, E.; Fernández-Borges, N.; Pintado, B.; Ordóñez, M.; Márquez, M.; Fondevila, D.; Torres, J.M.; Pumarola, M.; Castilla, J. Bovine Spongiform Encephalopathy Induces Misfolding of Alleged Prion-Resistant Species Cellular Prion Protein without Altering Its Pathobiological Features. J. Neurosci. Off. J. Soc. Neurosci. 2013, 33, 7778–7786. [Google Scholar] [CrossRef] [PubMed]
- Beck, K.E.; Thorne, L.; Lockey, R.; Vickery, C.M.; Terry, L.A.; Bujdoso, R.; Spiropoulos, J. Strain typing of classical scrapie by transgenic mouse bioassay using protein misfolding cyclic amplification to replace primary passage. PLoS ONE 2013, 8, e57851. [Google Scholar] [CrossRef] [PubMed]
- Cohen, F.E.; Prusiner, S.B. Pathologic Conformations of Prion Proteins. Annu. Rev. Biochem. 1998, 67, 793–819. [Google Scholar] [CrossRef] [PubMed]
- Makarava, N.; Kovacs, G.G.; Savtchenko, R.; Alexeeva, I.; Ostapchenko, V.G.; Budka, H.; Rohwer, R.G.; Baskakov, I.V. A new mechanism for transmissible prion diseases. J. Neurosci. Off. J. Soc. Neurosci. 2012, 32, 7345–7355. [Google Scholar] [CrossRef] [PubMed]
- Makarava, N.; Savtchenko, R.; Alexeeva, I.; Rohwer, R.G.; Baskakov, I.V. New Molecular Insight into Mechanism of Evolution of Mammalian Synthetic Prions. Am. J. Pathol. 2016, 186, 1006–1014. [Google Scholar] [CrossRef] [PubMed]
- Igel-Egalon, A.; Moudjou, M.; Martin, D.; Busley, A.; Knäpple, T.; Herzog, L.; Reine, F.; Lepejova, N.; Richard, C.-A.; Béringue, V.; et al. Reversible unfolding of infectious prion assemblies reveals the existence of an oligomeric elementary brick. PLoS Pathog. 2017, 13, e1006557. [Google Scholar] [CrossRef] [PubMed]




© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Igel-Egalon, A.; Béringue, V.; Rezaei, H.; Sibille, P. Prion Strains and Transmission Barrier Phenomena. Pathogens 2018, 7, 5. https://doi.org/10.3390/pathogens7010005
Igel-Egalon A, Béringue V, Rezaei H, Sibille P. Prion Strains and Transmission Barrier Phenomena. Pathogens. 2018; 7(1):5. https://doi.org/10.3390/pathogens7010005
Chicago/Turabian StyleIgel-Egalon, Angélique, Vincent Béringue, Human Rezaei, and Pierre Sibille. 2018. "Prion Strains and Transmission Barrier Phenomena" Pathogens 7, no. 1: 5. https://doi.org/10.3390/pathogens7010005
APA StyleIgel-Egalon, A., Béringue, V., Rezaei, H., & Sibille, P. (2018). Prion Strains and Transmission Barrier Phenomena. Pathogens, 7(1), 5. https://doi.org/10.3390/pathogens7010005