Evolution of Diagnostic Tests for Chronic Wasting Disease, a Naturally Occurring Prion Disease of Cervids



Abstract
:1. Background and Introduction
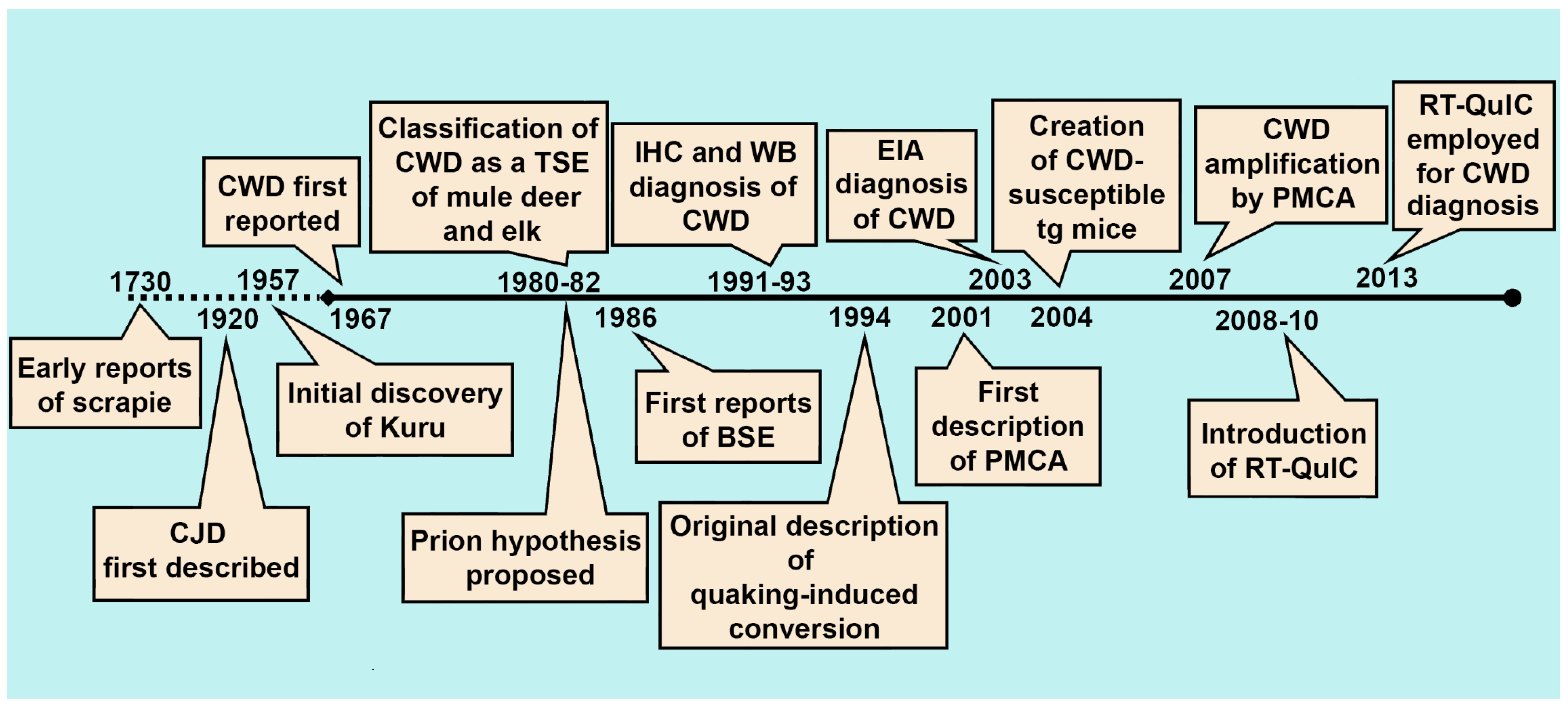
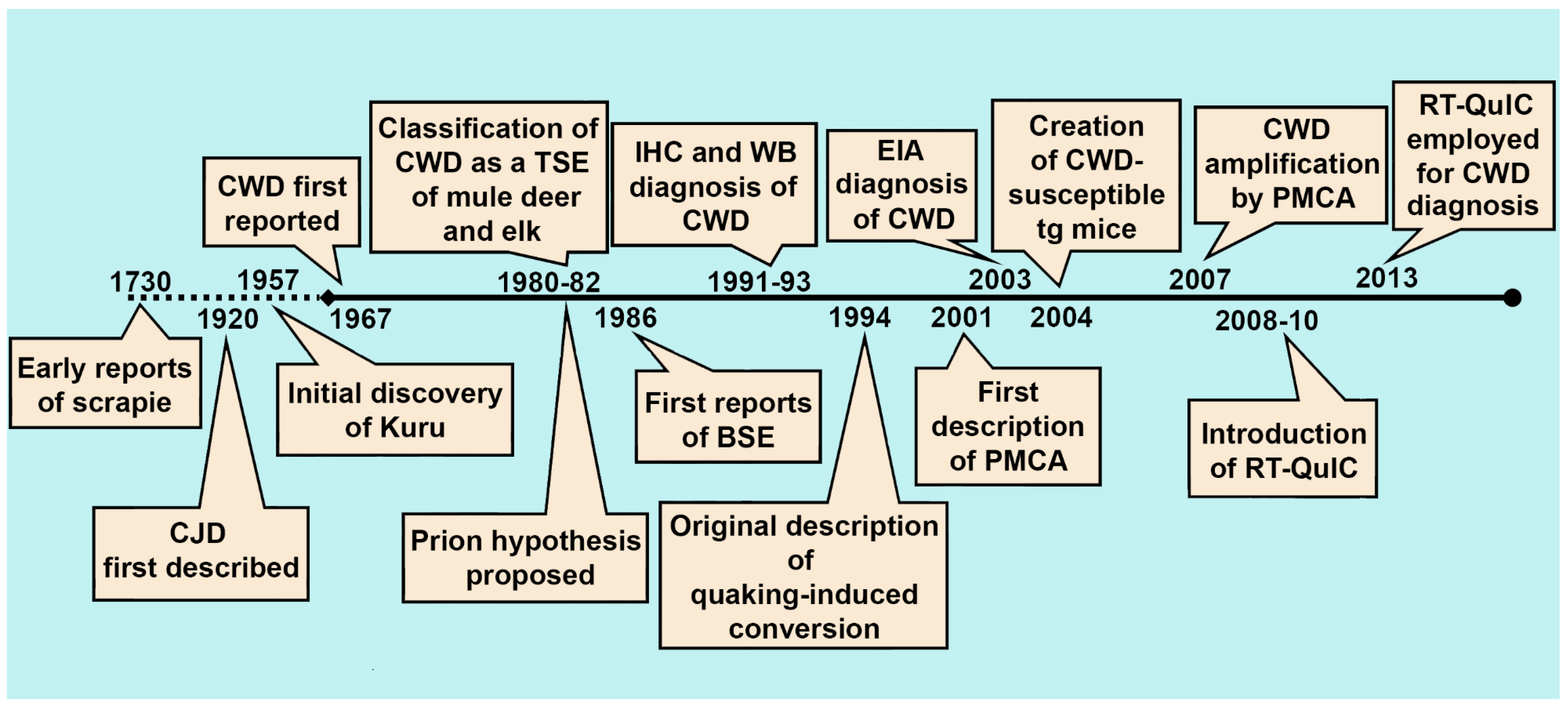
2. The History of CWD Diagnostics
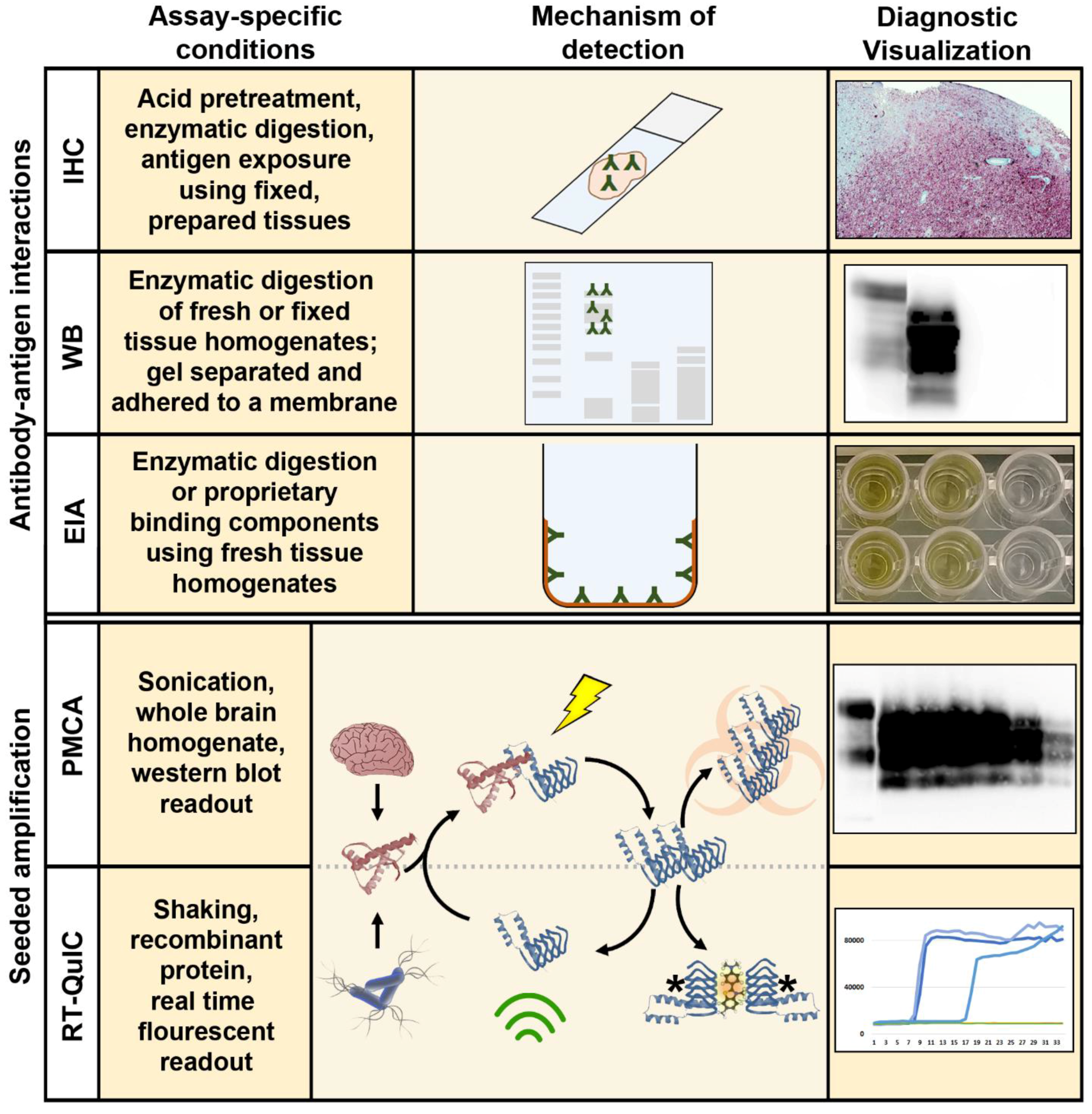
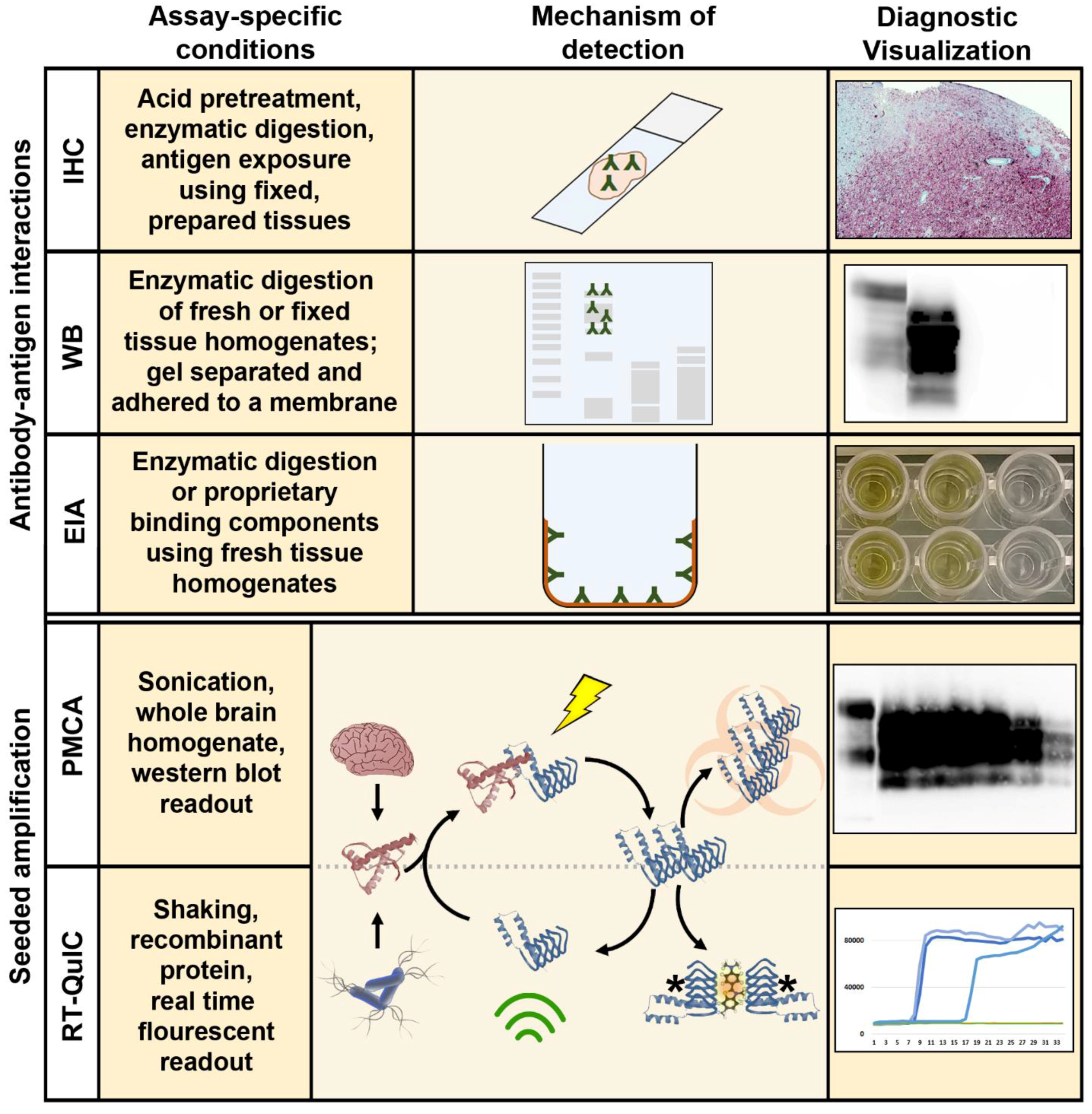
2.1. Immunohistochemistry, Western Blotting, and Enzyme Immunoassay
2.2. Bioassay
3. The Present State of CWD Diagnostics
3.1. Amplification Assays for the Detection of Ultra-Low Levels of CWD Prions
3.2. Protein Misfolding Cyclic Amplification
3.3. Quaking Induced Conversion
3.4. Tyramide Signal Amplification
3.5. Cervid Prion Cell Assay
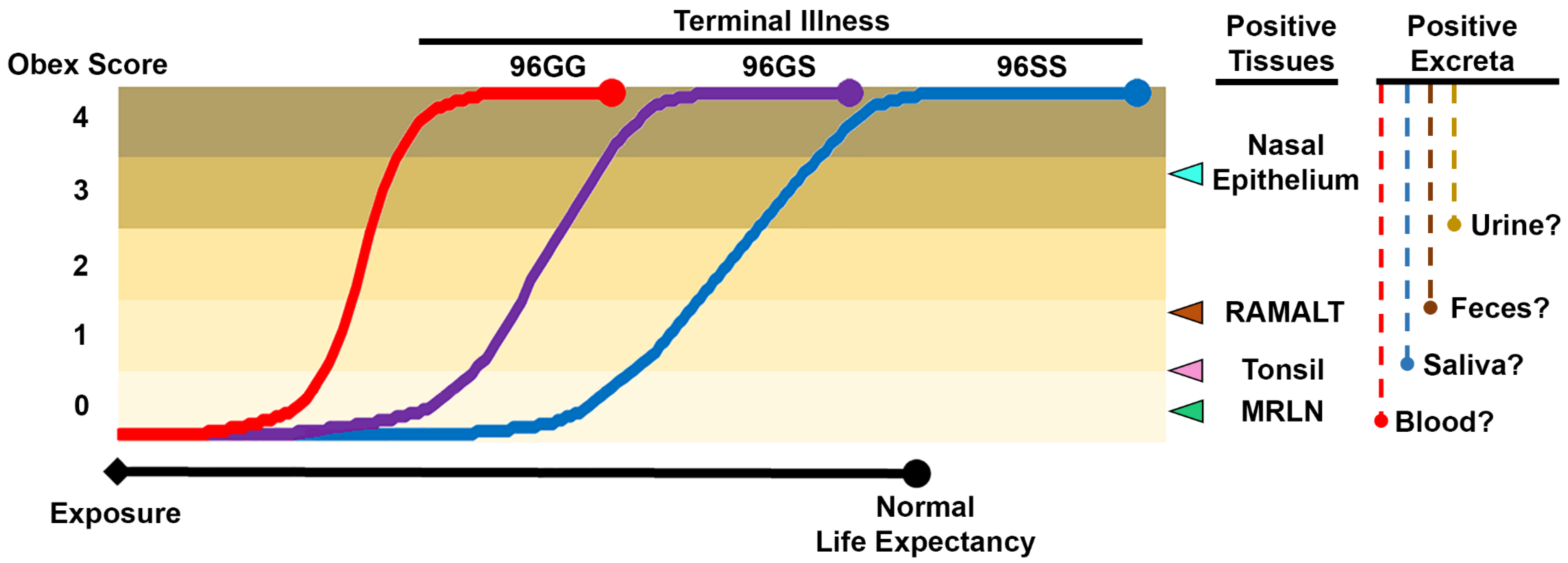
3.6. Sample Selection for Antemortem Testing
3.7. Bodily Fluids and Excreta
3.8. Accessible Peripheral Tissues
3.9. Sample Collection in Farmed and Free-Ranging Cervids
3.10. Genetic Background and Antemortem CWD Diagnostic Sensitivity
4. The Future of CWD Diagnostics
4.1. Improving Current CWD Diagnostic Approaches
4.2. Exploring New Frontiers in CWD Diagnostics
4.3. Realms beyond CWD Diagnosis
5. Conclusions
Acknowledgments
Conflicts of Interest
References
- Williams, E.S.; Young, S. Chronic wasting disease of captive mule deer: A spongiform encephalopathy. J. Wildl. Dis. 1980, 16, 89–98. [Google Scholar] [CrossRef] [PubMed]
- Haley, N.J.; Hoover, E.A. Chronic Wasting Disease of Cervids: Current Knowledge and Future Perspectives. Annu. Rev. Anim. Biosci. 2015, 3, 305–325. [Google Scholar] [CrossRef] [PubMed]
- Benestad, S.L.; Mitchell, G.; Simmons, M.; Ytrehus, B.; Vikoren, T. First case of chronic wasting disease in Europe in a Norwegian free-ranging reindeer. Vet. Res. 2016, 47, 88. [Google Scholar] [CrossRef] [PubMed]
- Belay, E.D.; Maddox, R.A.; Williams, E.S.; Miller, M.W.; Gambetti, P.; Schonberger, L.B. Chronic wasting disease and potential transmission to humans. Emerg. Infect. Dis. 2004, 10, 977–984. [Google Scholar] [CrossRef] [PubMed]
- Collins, S.J.; Lawson, V.A.; Masters, C.L. Transmissible spongiform encephalopathies. Lancet 2004, 363, 51–61. [Google Scholar] [CrossRef]
- Kong, Q.; Huang, S.; Zou, W.; Vanegas, D.; Wang, M.; Wu, D.; Yuan, J.; Zheng, M.; Bai, H.; Deng, H.; et al. Chronic wasting disease of elk: Transmissibility to humans examined by transgenic mouse models. J. Neurosci. 2005, 25, 7944–7949. [Google Scholar] [CrossRef] [PubMed]
- Wilson, R.; Plinston, C.; Hunter, N.; Casalone, C.; Corona, C.; Tagliavini, F.; Suardi, S.; Ruggerone, M.; Moda, F.; Graziano, S.; et al. Chronic wasting disease and atypical forms of bovine spongiform encephalopathy and scrapie are not transmissible to mice expressing wild-type levels of human prion protein. J. Gen. Virol. 2012, 93 Pt 7, 1624–1629. [Google Scholar] [CrossRef] [PubMed]
- Perrott, M.R.; Sigurdson, C.J.; Mason, G.L.; Hoover, E.A. Mucosal transmission and pathogenesis of chronic wasting disease in ferrets. J. Gen. Virol. 2012, 94 Pt 2, 432–442. [Google Scholar] [CrossRef] [PubMed]
- Heisey, D.M.; Mickelsen, N.A.; Schneider, J.R.; Johnson, C.J.; Langenberg, J.A.; Bochsler, P.N.; Keane, D.P.; Barr, D.J. Chronic wasting disease (CWD) susceptibility of several North American rodents that are sympatric with cervid CWD epidemics. J. Virol. 2009, 4, 210–215. [Google Scholar] [CrossRef] [PubMed]
- Sohn, H.J.; Kim, J.H.; Choi, K.S.; Nah, J.J.; Joo, Y.S.; Jean, Y.H.; Ahn, S.W.; Kim, O.K.; Kin, D.Y.; Balachandran, A. A case of chronic wasting disease in an elk imported to Korea from Canada. J. Vet. Med. Sci. 2002, 64, 855–858. [Google Scholar] [CrossRef] [PubMed]
- Williams, E.S.; Miller, M.W.; Kreeger, T.J.; Kahn, R.H.; Thorne, E.T. Chronic wasting disease of deer and elk: A review with recommendations for management. J. Wildl. Manag. 2002, 66, 551–563. [Google Scholar] [CrossRef]
- Williams, E.S. Chronic wasting disease. Vet. Pathol. 2005, 42, 530–549. [Google Scholar] [CrossRef] [PubMed]
- Stokstad, E. Norway plans to exterminate a large reindeer herd to stop a fatal infectious brain disease. Science Magazine, 3 August 2017. [Google Scholar]
- Picard, J. Experts explain deer disease. Oneida Daily Dispatch News, 13 May 2005. [Google Scholar]
- Watts, T. CWD leads to new regulations for taxidermists. Oakland Press News, 28 May 2009. [Google Scholar]
- Angers, R.C.; Browning, S.R.; Seward, T.S.; Sigurdson, C.J.; Miller, M.W.; Hoover, E.A.; Telling, G.C. Prions in skeletal muscles of deer with chronic wasting disease. Science 2006, 311, 1117. [Google Scholar] [CrossRef] [PubMed]
- Race, B.; Meade-White, K.; Race, R.; Chesebro, B. Prion infectivity in fat of deer with chronic wasting disease. J. Virol. 2009, 83, 9608–9610. [Google Scholar] [CrossRef] [PubMed]
- Angers, R.C.; Seward, T.S.; Napier, D.; Green, M.; Hoover, E.; Spraker, T.; O'Rourke, K.; Balachandran, A.; Telling, G.C. Chronic wasting disease prions in elk antler velvet. Emerg. Infect. Dis. 2009, 15, 696–703. [Google Scholar] [CrossRef] [PubMed]
- Haley, N.J.; Seelig, D.M.; Zabel, M.D.; Telling, G.C.; Hoover, E.A. Detection of CWD prions in urine and saliva of deer by transgenic mouse bioassay. PLoS ONE 2009, 4, e4848. [Google Scholar] [CrossRef] [PubMed]
- Mathiason, C.K.; Powers, J.G.; Dahmes, S.J.; Osborn, D.A.; Miller, K.V.; Warren, R.J.; Mason, G.L.; Hays, S.A.; Hayes-Klug, J.; Seelig, D.M.; et al. Infectious prions in the saliva and blood of deer with chronic wasting disease. Science 2006, 314, 133–136. [Google Scholar] [CrossRef] [PubMed]
- Tamguney, G.; Miller, M.W.; Wolfe, L.L.; Sirochman, T.M.; Glidden, D.V.; Palmer, C.; Lemus, A.; DeArmond, S.J.; Prusiner, S.B. Asymptomatic deer excrete infectious prions in faeces. Nature 2009, 461, 529–532. [Google Scholar] [CrossRef] [PubMed]
- Bahmanyar, S.; Williams, E.S.; Johnson, F.B.; Young, S.; Gajdusek, D.C. Amyloid plaques in spongiform encephalopathy of mule deer. J. Comp. Pathol. 1985, 95, 1–5. [Google Scholar] [CrossRef]
- Guiroy, D.C.; Williams, E.S.; Song, K.J.; Yanagihara, R.; Gajdusek, D.C. Fibrils in brain of Rocky Mountain elk with chronic wasting disease contain scrapie amyloid. Acta Neuropathol. 1993, 86, 77–80. [Google Scholar] [CrossRef] [PubMed]
- Guiroy, D.C.; Williams, E.S.; Yanagihara, R.; Gajdusek, D.C. Immunolocalization of scrapie amyloid (PrP27-30) in chronic wasting disease of Rocky Mountain elk and hybrids of captive mule deer and white-tailed deer. Neurosci. Lett. 1991, 126, 195–198. [Google Scholar] [CrossRef]
- Guiroy, D.C.; Williams, E.S.; Yanagihara, R.; Gajdusek, D.C. Topographic distribution of scrapie amyloid-immunoreactive plaques in chronic wasting disease in captive mule deer (Odocoileus hemionus hemionus). Acta Neuropathol. 1991, 81, 475–478. [Google Scholar] [CrossRef] [PubMed]
- Hibler, C.P.; Wilson, K.L.; Spraker, T.R.; Miller, M.W.; Zink, R.R.; DeBuse, L.L.; Anderson, E.; Schweitzer, D.; Kennedy, J.A.; Baeten, L.A.; et al. Field validation and assessment of an enzyme-linked immunosorbent assay for detecting chronic wasting disease in mule deer (Odocoileus hemionus), white-tailed deer (Odocoileus virginianus), and Rocky Mountain elk (Cervus elaphus nelsoni). J. Vet. Diagn. Investig. 2003, 15, 311–319. [Google Scholar] [CrossRef] [PubMed]
- Bian, J.; Napier, D.; Khaychuck, V.; Angers, R.; Graham, C.; Telling, G. Cell-based quantification of chronic wasting disease prions. J. Virol. 2010, 84, 8322–8326. [Google Scholar] [CrossRef] [PubMed]
- Soto, C.; Saborio, G.P.; Anderes, L. Cyclic amplification of protein misfolding: Application to prion-related disorders and beyond. Trends Neurosci. 2002, 25, 390–394. [Google Scholar] [CrossRef]
- Kurt, T.D.; Perrott, M.R.; Wilusz, C.J.; Wilusz, J.; Supattapone, S.; Telling, G.C.; Hoover, E.A. Efficient in vitro amplification of chronic wasting disease PrPRES. J. Virol. 2007, 81, 9605–9608. [Google Scholar] [CrossRef] [PubMed]
- Wilham, J.M.; Orru, C.D.; Bessen, R.A.; Atarashi, R.; Sano, K.; Race, B.; Meade-White, K.D.; Taubner, L.M.; Timmes, A.; Caughey, B. Rapid end-point quantitation of prion seeding activity with sensitivity comparable to bioassays. PLoS Pathog. 2010, 6, e1001217. [Google Scholar] [CrossRef] [PubMed]
- Atarashi, R.; Moore, R.A.; Sim, V.L.; Hughson, A.G.; Dorward, D.W.; Onwubiko, H.A.; Priola, S.A.; Caughey, B. Ultrasensitive detection of scrapie prion protein using seeded conversion of recombinant prion protein. Nat. Methods 2007, 4, 645–650. [Google Scholar] [CrossRef] [PubMed]
- Haley, N.J.; Carver, S.; Hoon-Hanks, L.L.; Henderson, D.M.; Davenport, K.A.; Bunting, E.; Gray, S.; Trindle, B.; Galeota, J.; LeVan, I.; et al. Detection of chronic wasting disease in the lymph nodes of free-ranging cervids by real-time quaking-induced conversion. J. Clin. Microbiol. 2014, 52, 3237–3243. [Google Scholar] [CrossRef] [PubMed]
- Miller, M.W.; Williams, E.S. Chronic wasting disease of cervids. Curr. Top. Microbiol. Immunol. 2004, 284, 193–214. [Google Scholar] [PubMed]
- Sigurdson, C.J.; Aguzzi, A. Chronic wasting disease. Biochim. Biophys. Acta 2007, 1772, 610–618. [Google Scholar] [CrossRef] [PubMed]
- Bolton, D.C.; McKinley, M.P.; Prusiner, S.B. Identification of a protein that purifies with the scrapie prion. Science 1982, 218, 1309–1311. [Google Scholar] [CrossRef] [PubMed]
- Prusiner, S.B. Novel proteinaceous infectious particles cause scrapie. Science 1982, 216, 136–144. [Google Scholar] [CrossRef] [PubMed]
- Masujin, K.; Okada, H.; Miyazawa, K.; Matsuura, Y.; Imamura, M.; Iwamaru, Y.; Murayama, Y.; Yokoyama, T. Emergence of a novel bovine spongiform encephalopathy (BSE) prion from an atypical H-type BSE. Sci. Rep. 2016, 6, 22753. [Google Scholar] [CrossRef] [PubMed]
- Peters, J.; Miller, J.M.; Jenny, A.L.; Peterson, T.L.; Carmichael, K.P. Immunohistochemical diagnosis of chronic wasting disease in preclinically affected elk from a captive herd. J. Vet. Diagn. Investig. 2000, 12, 579–582. [Google Scholar] [CrossRef] [PubMed]
- Sigurdson, C.J.; Williams, E.S.; Miller, M.W.; Spraker, T.R.; O’Rourke, K.I.; Hoover, E.A. Oral transmission and early lymphoid tropism of chronic wasting disease PrPres in mule deer fawns (Odocoileus hemionus). J. Gen. Virol. 1999, 80 Pt 10, 2757–2764. [Google Scholar] [CrossRef] [PubMed]
- Keane, D.P.; Barr, D.J.; Keller, J.E.; Hall, S.M.; Langenberg, J.A.; Bochsler, P.N. Comparison of retropharyngeal lymph node and obex region of the brainstem in detection of chronic wasting disease in white-tailed deer (Odocoileus virginianus). J. Vet. Diagn. Investig. 2008, 20, 58–60. [Google Scholar] [CrossRef] [PubMed]
- Keane, D.P.; Barr, D.J.; Bochsler, P.N.; Hall, S.M.; Gidlewski, T.; O’Rourke, K.I.; Spraker, T.R.; Samuel, M.D. Chronic wasting disease in a Wisconsin white-tailed deer farm. J. Vet. Diagn. Investig. 2008, 20, 698–703. [Google Scholar] [CrossRef] [PubMed]
- Spraker, T.R.; Balachandran, A.; Zhuang, D.; O’Rourke, K.I. Variable patterns of distribution of PrP(CWD) in the obex and cranial lymphoid tissues of Rocky Mountain elk (Cervus elaphus nelsoni) with subclinical chronic wasting disease. Vet. Rec. 2004, 155, 295–302. [Google Scholar] [CrossRef] [PubMed]
- Fox, K.A.; Jewell, J.E.; Williams, E.S.; Miller, M.W. Patterns of PrPCWD accumulation during the course of chronic wasting disease infection in orally inoculated mule deer (Odocoileus hemionus). J. Gen. Virol. 2006, 87 Pt 11, 3451–3461. [Google Scholar] [CrossRef] [PubMed]
- Haley, N.J.; Siepker, C.; Hoon-Hanks, L.L.; Mitchell, G.; Walter, W.D.; Manca, M.; et al. Seeded Amplification of Chronic Wasting Disease Prions in Nasal Brushings and Recto-anal Mucosa-Associated Lymphoid Tissues from Elk by Real-Time Quaking-Induced Conversion. J. Clin. Microbiol. 2016, 54, 1117–1126. [Google Scholar] [CrossRef] [PubMed]
- Haley, N.J.; Siepker, C.; Walter, W.D.; Thomsen, B.V.; Greenlee, J.J.; Lehmkuhl, A.D.; Richt, J.A. Antemortem Detection of Chronic Wasting Disease Prions in Nasal Brush Collections and Rectal Biopsy Specimens from White-Tailed Deer by Real-Time Quaking-Induced Conversion. J. Clin. Microbiol. 2016, 54, 1108–1116. [Google Scholar] [CrossRef] [PubMed]
- Thomsen, B.V.; Schneider, D.A.; O’Rourke, K.I.; Gidlewski, T.; McLane, J.; Allen, R.W.; McIsaac, A.A.; Mitchell, G.B.; Keane, D.P.; Spraker, T.R.; et al. Diagnostic accuracy of rectal mucosa biopsy testing for chronic wasting disease within white-tailed deer (Odocoileus virginianus) herds in North America: Effects of age, sex, polymorphism at PRNP codon 96, and disease progression. J. Vet. Diagn. Investig. 2012, 24, 878–887. [Google Scholar] [CrossRef] [PubMed]
- Sigurdson, C.J.; Barillas-Mury, C.; Miller, M.W.; Oesch, B.; van Keulen, L.J.; Langeveld, J.P.; Hoover,, E.A. PrP(CWD) lymphoid cell targets in early and advanced chronic wasting disease of mule deer. J. Gen. Virol. 2002, 83 Pt 10, 2617–2628. [Google Scholar] [CrossRef] [PubMed]
- Wild, M.A.; Spraker, T.R.; Sigurdson, C.J.; O’Rourke, K.I.; Miller, M.W. Preclinical diagnosis of chronic wasting disease in captive mule deer (Odocoileus hemionus) and white-tailed deer (Odocoileus virginianus) using tonsillar biopsy. J. Gen. Virol. 2002, 83 Pt 10, 2629–2634. [Google Scholar] [CrossRef] [PubMed]
- Spraker, T.R.; O’Rourke, K.I.; Balachandran, A.; Zink, R.R.; Cummings, B.A.; Miller, M.W.; Powers, B.E. Validation of monoclonal antibody F99/97.6.1 for immunohistochemical staining of brain and tonsil in mule deer (Odocoileus hemionus) with chronic wasting disease. J. Vet. Diagn. Investig. 2002, 14, 3–7. [Google Scholar] [CrossRef] [PubMed]
- Spraker, T.R.; VerCauteren, K.C.; Gidlewski, T.; Schneider, D.A.; Munger, R.; Balachandran, A.; O'Rourke, K.I. Antemortem detection of PrPCWD in preclinical, ranch-raised Rocky Mountain elk (Cervus elaphus nelsoni) by biopsy of the rectal mucosa. J. Vet. Diagn. Investig. 2009, 21, 15–24. [Google Scholar] [CrossRef] [PubMed]
- Miller, M.W.; Williams, E.S.; McCarty, C.W.; Spraker, T.R.; Kreeger, T.J.; Larsen, C.T.; Thorne, E.T. Epizootiology of chronic wasting disease in free-ranging cervids in Colorado and Wyoming. J. Wildl. Dis. 2000, 36, 676–690. [Google Scholar] [CrossRef] [PubMed]
- Williams, E.S.; Miller, M.W. Chronic wasting disease in deer and elk in North America. Rev. Sci. Tech. 2002, 21, 305–316. [Google Scholar] [CrossRef] [PubMed]
- Joly, D.O.; Samuel, M.D.; Langenberg, J.A.; Blanchong, J.A.; Batha, C.A.; Rolley, R.E.; Keane, D.P.; Ribic, C.A. Spatial epidemiology of chronic wasting disease in Wisconsin white-tailed deer. J. Wildl. Dis. 2006, 42, 578–588. [Google Scholar] [CrossRef] [PubMed]
- Jennelle, C.S.; Samuel, M.D.; Nolden, C.A.; Keane, D.P.; Barr, D.J.; Johnson, C.; Vanderloo, J.P.; Aiken, J.M.; Hamir, A.N.; Hoover, E.A. Surveillance for transmissible spongiform encephalopathy in scavengers of white-tailed deer carcasses in the chronic wasting disease area of Wisconsin. J. Toxicol. Environ. Health A 2009, 72, 1018–1024. [Google Scholar] [CrossRef] [PubMed]
- Saunders, S.E.; Bartelt-Hunt, S.L.; Bartz, J.C. Occurrence, transmission, and zoonotic potential of chronic wasting disease. Emerg. Infect. Dis. 2012, 18, 369–376. [Google Scholar] [CrossRef] [PubMed]
- Johnson, C.; Johnson, J.; Vanderloo, J.P.; Keane, D.; Aiken, J.M.; McKenzie, D. Prion protein polymorphisms in white-tailed deer influence susceptibility to chronic wasting disease. J. Gen. Virol. 2006, 87 Pt 7, 2109–2114. [Google Scholar] [CrossRef] [PubMed]
- O’Rourke, K.I.; Spraker, T.R.; Zhuang, D.; Greenlee, J.J.; Gidlewski, T.E.; Hamir, A.N. Elk with a long incubation prion disease phenotype have a unique PrPd profile. Neuroreport 2007, 18, 1935–1938. [Google Scholar] [CrossRef] [PubMed]
- Kelly, A.C.; Mateus-Pinilla, N.E.; Diffendorfer, J.; Jewell, E.; Ruiz, M.O.; Killefer, J.; Shelton, P.; Beissel, T.; Novakofski, J. Prion sequence polymorphisms and chronic wasting disease resistance in Illinois white-tailed deer (Odocoileus virginianus). Prion 2008, 2, 28–36. [Google Scholar] [CrossRef] [PubMed]
- Robinson, S.J.; Samuel, M.D.; O’Rourke, K.I.; Johnson, C.J. The role of genetics in chronic wasting disease of North American cervids. Prion 2012, 6, 153–162. [Google Scholar] [CrossRef] [PubMed]
- Jewell, J.E.; Conner, M.M.; Wolfe, L.L.; Miller, M.W.; Williams, E.S. Low frequency of PrP genotype 225SF among free-ranging mule deer (Odocoileus hemionus) with chronic wasting disease. J. Gen. Virol. 2005, 86 Pt 8, 2127–2134. [Google Scholar] [CrossRef] [PubMed]
- O’Rourke, K.I.; Besser, T.E.; Miller, M.W.; Cline, T.F.; Spraker, T.R.; Jenny, A.L.; Wild, M.A.; Sebarth, G.L.; Williams, E.S. PrP genotypes of captive and free-ranging Rocky Mountain elk (Cervus elaphus nelsoni) with chronic wasting disease. J. Gen. Virol. 1999, 80 Pt 10, 2765–2769. [Google Scholar]
- Johnson, C.; Johnson, J.; Clayton, M.; McKenzie, D.; Aiken, J. Prion protein gene heterogeneity in free-ranging white-tailed deer within the chronic wasting disease affected region of Wisconsin. J. Wildl. Dis. 2003, 39, 576–581. [Google Scholar] [CrossRef] [PubMed]
- Miller, M.W.; Williams, E.S.; Hobbs, N.T.; Wolfe, L.L. Environmental sources of prion transmission in mule deer. Emerg. Infect. Dis. 2004, 10, 1003–1006. [Google Scholar] [CrossRef] [PubMed]
- Miller, M.W.; Wild, M.A.; Williams, E.S. Epidemiology of chronic wasting disease in captive Rocky Mountain elk. J. Wildl. Dis. 1998, 34, 532–538. [Google Scholar] [CrossRef] [PubMed]
- Browning, S.R.; Mason, G.L.; Seward, T.; Green, M.; Eliason, G.A.; Mathiason, C.; Miller, M.W.; Williams, E.S.; Hoover, E.; Telling, G.C. Transmission of prions from mule deer and elk with chronic wasting disease to transgenic mice expressing cervid PrP. J. Virol. 2004, 78, 13345–13350. [Google Scholar] [CrossRef] [PubMed]
- Mathiason, C.K.; Hayes-Klug, J.; Hays, S.A.; Powers, J.; Osborn, D.A.; Dahmes, S.J.; Miller, K.V.; Warren, R.J.; Mason, G.L.; Telling, G.C.; et al. B cells and platelets harbor prion infectivity in the blood of deer infected with chronic wasting disease. J. Virol. 2010, 84, 5097–5107. [Google Scholar] [CrossRef] [PubMed]
- Haley, N.; Mathiason, C.; Zabel, M.D.; Telling, G.C.; Hoover, E. Detection of sub-clinical CWD infection in conventional test-negative deer long after oral exposure to urine and feces from CWD+ deer. PLoS ONE 2009, 4, e7990. [Google Scholar] [CrossRef] [PubMed]
- Haley, N.J.; Mathiason, C.; Carver, S.; Telling, G.C.; Zabel, M.C.; Hoover, E.A. Sensitivity of protein misfolding cyclic amplification vs. immunohistochemistry in antemortem detection of CWD infection. J. Gen. Virol. 2012, 93, 1141–1150. [Google Scholar] [CrossRef] [PubMed]
- Chronic Wasting Disease Program Standards; United States Department of Agriculture: Ames, IA, USA, 2014.
- Canadian Food Inspection Agency. Accredited Veterinarian’s Manual; Canadian Food Inspection Agency: Ottawa, ON, Canada, 2017. [Google Scholar]
- Henderson, D.M.; Davenport, K.A.; Haley, N.J.; Denkers, N.D.; Mathiason, C.K.; Hoover, E.A. Quantitative assessment of prion infectivity in tissues and body fluids by real-time quaking-induced conversion. J. Gen. Virol. 2015, 96 Pt 1, 210–219. [Google Scholar] [CrossRef] [PubMed]
- Henderson, D.; Tennant, J.; Haley, N.; Denkers, N.D.; Mathiason, C.; Hoover, E. Detection of CWD Prion-seeding Activity in Deer and Elk Feces by Real-time Quaking Induced Conversion. J. Gen. Virol. 2017, in press. [Google Scholar]
- Haley, N.J.; Mathiason, C.K.; Carver, S.; Zabel, M.; Telling, G.C.; Hoover, E.A. Detection of CWD prions in salivary, urinary, and intestinal tissues of deer: Potential mechanisms of prion shedding and transmission. J. Virol. 2011, 85, 6309–6318. [Google Scholar] [CrossRef] [PubMed]
- Hoover, C.E.; Davenport, K.A.; Henderson, D.M.; Denkers, N.D.; Mathiason, C.K.; Soto, C.; Zabel, M.D.; Hoover, E.A. Pathways of Prion Spread during Early Chronic Wasting Disease in Deer. J. Virol. 2017, 91, e00077-17. [Google Scholar] [CrossRef] [PubMed]
- Castilla, J.; Saa, P.; Morales, R.; Abid, K.; Maundrell, K.; Soto, C. Protein misfolding cyclic amplification for diagnosis and prion propagation studies. Methods Enzymol. 2006, 412, 3–21. [Google Scholar] [PubMed]
- Gonzalez-Montalban, N.; Makarava, N.; Ostapchenko, V.G.; Savtchenk, R.; Alexeeva, I.; Rohwer, R.G.; Baskakov, I.V. Highly efficient protein misfolding cyclic amplification. PLoS Pathog. 2011, 7, e1001277. [Google Scholar] [CrossRef] [PubMed]
- Haley, N.J.; Van de Motter, A.; Carver, S.; Henderson, D.; Davenport, K.; Seelig, D.M.; Mathiason, C.; Hoover, E. Prion-seeding activity in cerebrospinal fluid of deer with chronic wasting disease. PLoS ONE 2013, 8, e81488. [Google Scholar] [CrossRef] [PubMed]
- Park, J.H.; Choi, Y.G.; Park, S.J.; Choi, H.S.; Choi, E.K.; Kim, Y.S. Ultra-efficient Amplification of Abnormal Prion Protein by Modified Protein Misfolding Cyclic Amplification with Electric Current. Mol. Neurobiol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Chang, B.; Gray, P.; Piltch, M.; Bulgin, M.S.; Sorensen-Melson, S.; Miller, M.W.; Davies, P.; Brown, D.R.; Coughlin, D.R.; Rubenstein, R.; et al. Surround optical fiber immunoassay (SOFIA): An ultra-sensitive assay for prion protein detection. J. Virol. Methods 2009, 159, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Rubenstein, R.; Chang, B.; Gray, P.; Piltch, M.; Bulgin, M.S.; Sorensen-Melson, S.; Miller, M.W. A novel method for preclinical detection of PrPSc in blood. J. Gen. Virol. 2010, 91 Pt 7, 1883–1892. [Google Scholar] [CrossRef] [PubMed]
- Pulford, B.; Spraker, T.R.; Wyckoff, A.C.; Meyerett, C.; Bender, H.; Ferguson, A.; Wyatt, B.; Lockwood, K.; Powers, J.; Telling, C.G.; et al. Detection of PrPCWD in feces from naturally exposed Rocky Mountain elk (Cervus elaphus nelsoni) using protein misfolding cyclic amplification. J. Wildl. Dis. 2012, 48, 425–434. [Google Scholar] [CrossRef] [PubMed]
- Nichols, T.A.; Spraker, T.R.; Gidlewski, T.; Powers, J.G.; Telling, G.C.; VerCauteren, K.C.; Zabel, M.D. Detection of prion protein in the cerebrospinal fluid of elk (Cervus canadensis nelsoni) with chronic wasting disease using protein misfolding cyclic amplification. J. Vet. Diagn. Investig. 2012, 24, 746–749. [Google Scholar] [CrossRef] [PubMed]
- Saunders, S.E.; Shikiya, R.A.; Langenfeld, K.; Bartelt-Hunt, S.L.; Bartz, J.C. Replication efficiency of soil-bound prions varies with soil type. J. Virol. 2011, 85, 5476–5482. [Google Scholar] [CrossRef] [PubMed]
- Nichols, T.A.; Pulford, B.; Wyckoff, A.C.; Meyerett, C.; Michel, B.; Gertig, K.; Hoover, E.A.; Jewell, J.E.; Telling, G.C.; Zabel, M.D. Detection of protease-resistant cervid prion protein in water from a CWD-endemic area. Prion 2009, 3, 171–183. [Google Scholar] [CrossRef] [PubMed]
- Pritzkow, S.; Morales, R.; Moda, F.; Khan, U.; Telling, G.C.; Hoover, E.; Soto, C. Grass plants bind, retain, uptake, and transport infectious prions. Cell Rep. 2015, 11, 1168–1175. [Google Scholar] [CrossRef] [PubMed]
- Meyerett, C.; Michel, B.; Pulford, B.; Spraker, T.R.; Nichols, T.A.; Johnson, T. In vitro strain adaptation of CWD prions by serial protein misfolding cyclic amplification. Virology 2008, 382, 267–276. [Google Scholar] [CrossRef] [PubMed]
- Kocisko, D.A.; Come, J.H.; Priola, S.A.; Chesebro, B.; Raymond, G.J.; Lansbury, P.T.; Caughey, B. Cell-free formation of protease-resistant prion protein. Nature 1994, 370, 471–474. [Google Scholar] [CrossRef] [PubMed]
- Atarashi, R.; Wilham, J.M.; Christensen, L.; Hughson, A.G.; Moore, R.A.; Johnson, L.M.; Onwubiko, H.A.; Priola, S.A.; Caughey, B. Simplified ultrasensitive prion detection by recombinant PrP conversion with shaking. Nat. Methods 2008, 5, 211–212. [Google Scholar] [CrossRef] [PubMed]
- Davenport, K.A.; Henderson, D.M.; Bian, J.; Telling, G.C.; Mathiason, C.K.; Hoover, E.A. Insights into Chronic Wasting Disease and Bovine Spongiform Encephalopathy Species Barriers by Use of Real-Time Conversion. J. Virol. 2015, 89, 9524–9531. [Google Scholar] [CrossRef] [PubMed]
- Seelig, D.M.; Mason, G.L.; Telling, G.C.; Hoover, E.A. Chronic Wasting Disease Prion Trafficking via the Autonomic Nervous System. Am. J. Pathol. 2011, 179, 1319–1328. [Google Scholar] [CrossRef] [PubMed]
- Chao, J.; DeBiasio, R.; Zhu, Z.; Giuliano, K.A.; Schmidt, B.F. Immunofluorescence signal amplification by the enzyme-catalyzed deposition of a fluorescent reporter substrate (CARD). Cytometry 1996, 23, 48–53. [Google Scholar] [CrossRef]
- Henderson, D.M.; Manca, M.; Haley, N.J.; Denkers, N.D.; Nalls, A.V.; Mathiason, C.K.; Caughey, B.; Hoover, E.A. Rapid Antemortem Detection of CWD Prions in Deer Saliva. PLoS ONE 2013, 8, e74377. [Google Scholar] [CrossRef] [PubMed]
- Elder, A.M.; Henderson, D.M.; Nalls, A.V.; Wilham, J.M.; Caughey, B.W.; Hoover, E.A.; Kincaid, A.E.; Bartz, J.C.; Mathiason, C.K. In Vitro Detection of prionemia in TSE-Infected Cervids and Hamsters. PLoS ONE 2013, 8, e80203. [Google Scholar] [CrossRef] [PubMed]
- Elder, A.M.; Henderson, D.M.; Nalls, A.V.; Hoover, E.A.; Kincaid, A.E.; Bartz, J.C.; Mathiason, C.K. Immediate and Ongoing Detection of Prions in the Blood of Hamsters and Deer following Oral, Nasal, or Blood Inoculations. J. Virol. 2015, 89, 7421–7424. [Google Scholar] [CrossRef] [PubMed]
- Van Keulen, L.J.; Schreuder, B.E.; Meloen, R.H.; Mooij-Harkes, G.; Vromans, M.E.; Langeveld, J.P. Immunohistochemical detection of prion protein in lymphoid tissues of sheep with natural scrapie. J. Clin. Microbiol. 1996, 34, 1228–1231. [Google Scholar] [PubMed]
- Gonzalez, L.; Dagleish, M.P.; Bellworthy, S.J.; Siso, S.; Stack, M.J.; Chaplin, M.J.; Balachandran, A. Postmortem diagnosis of preclinical and clinical scrapie in sheep by the detection of disease-associated PrP in their rectal mucosa. Vet. Rec. 2006, 158, 325–331. [Google Scholar] [CrossRef] [PubMed]
- O’Rourke, K.I.; Baszler, T.V.; Besser, T.E.; Miller, J.M.; Cutlip, R.C.; Wells, G.A.; Ryder, S.J.; Parish, S.M.; Hamir, A.N.; Cockett, N.E.; et al. Preclinical diagnosis of scrapie by immunohistochemistry of third eyelid lymphoid tissue. J. Clin. Microbiol. 2000, 38, 3254–3259. [Google Scholar]
- Castilla, J.; Saa, P.; Soto, C. Detection of prions in blood. Nat. Med. 2005, 11, 982–985. [Google Scholar] [CrossRef] [PubMed]
- Saa, P.; Castilla, J.; Soto, C. Presymptomatic detection of prions in blood. Science 2006, 313, 92–94. [Google Scholar] [CrossRef] [PubMed]
- Spraker, T.R.; Gidlewski, T.L.; Balachandran, A.; VerCauteren, K.C.; Creekmore, L.; Munger, R.D. Detection of PrP(CWD) in postmortem rectal lymphoid tissues in Rocky Mountain elk (Cervus elaphus nelsoni) infected with chronic wasting disease. J. Vet. Diagn. Investig. 2006, 18, 553–557. [Google Scholar] [CrossRef] [PubMed]
- Wolfe, L.L.; Spraker, T.R.; Gonzalez, L.; Dagleish, M.P.; Sirochman, T.M.; Brown, J.C.; Jeffrey, M.; Miller, M.W. PrPCWD in rectal lymphoid tissue of deer (Odocoileus spp.). J. Gen. Virol. 2007, 88 Pt 7, 2078–2082. [Google Scholar] [CrossRef] [PubMed]
- Monello, R.J.; Powers, J.G.; Hobbs, N.T.; Spraker, T.R.; O’Rourke, K.I.; Wild, M.A. Efficacy of antemortem rectal biopsies to diagnose and estimate prevalence of chronic wasting disease in free-ranging cow elk (Cervus elaphus nelsoni). J. Wildl. Dis. 2013, 49, 270–278. [Google Scholar] [CrossRef] [PubMed]
- Dechen Quinn, A.C.; Williams, D.M.; Porter, W.F.; Fitzgerald, S.D.; Hynes, K. Effects of capture-related injury on postcapture movement of white-tailed deer. J. Wildl. Dis. 2014, 50, 250–258. [Google Scholar] [CrossRef] [PubMed]
- Sharma, K.; Mewara, A.; Gupta, N.; Sharma, A.; Varma, S. Multiplex PCR in diagnosis of M. tuberculosis and M. avium co-infection from lymph node in an AIDS patient. Indian J. Med. Microbiol. 2015, 33, 151–153. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.J.; Robertson, D.; Tang, M.K.; Jackson, M.S.; MacKenzie, D.; Bagg, J. Staphylococcus aureus in the oral cavity: A three-year retrospective analysis of clinical laboratory data. Br. Dent. J. 2003, 195, 701–703. [Google Scholar] [CrossRef] [PubMed]
- Warren, W.P.; Balcarek, K.; Smith, R.; Pass, R.F. Comparison of rapid methods of detection of cytomegalovirus in saliva with virus isolation in tissue culture. J. Clin. Microbiol. 1992, 30, 786–789. [Google Scholar] [PubMed]
- Perrott, M.R.; Sigurdson, C.J.; Mason, G.L.; Hoover, E.A. Evidence for distinct chronic wasting disease (CWD) strains in experimental CWD in ferrets. J. Gen. Virol. 2011, 93 Pt 1, 212–221. [Google Scholar] [CrossRef] [PubMed]
- Angers, R.C.; Kang, H.E.; Napier, D.; Browning, S.; Seward, T.; Mathiason, C.; Balachandran, A.; McKenzie, D.; Castilla, J.; Soto, C.; et al. Prion strain mutation determined by prion protein conformational compatibility and primary structure. Science 2010, 328, 1154–1158. [Google Scholar] [CrossRef] [PubMed]
- Orru, C.D.; Groveman, B.R.; Raymond, L.D.; Hughson, A.G.; Nonno, R.; Zou, W.; Ghetti, B.; Gambetti, P.; Caughey, B. Bank Vole Prion Protein As an Apparently Universal Substrate for RT-QuIC-Based Detection and Discrimination of Prion Strains. PLoS Pathog. 2015, 11, e1004983. [Google Scholar]
- Ayers, J.I.; Giasson, B.I.; Borchelt, D.R. Prion-like Spreading in Tauopathies. Biol. Psychiatry 2017. [Google Scholar] [CrossRef] [PubMed]
- Olanow, C.W. Do prions cause Parkinson disease?: The evidence accumulates. Ann. Neurol. 2014, 75, 331–333. [Google Scholar] [CrossRef] [PubMed]
- Stopschinski, B.E.; Diamond, M.I. The prion model for progression and diversity of neurodegenerative diseases. Lancet Neurol. 2017, 16, 323–332. [Google Scholar] [CrossRef]
- Edwards, G., 3rd; Moreno-Gonzalez, I.; Soto, C. Amyloid-beta and tau pathology following repetitive mild traumatic brain injury. Biochem. Biophys. Res. Commun. 2017, 483, 1137–1142. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, M.; Cramm, M.; Llorens, F.; Muller-Cramm, D.; Collins, S.; Atarashi, R.; Satoh, K.; Orru, C.D.; Groveman, B.R.; Zafar, S.; et al. The real-time quaking-induced conversion assay for detection of human prion disease and study of other protein misfolding diseases. Nat. Protoc. 2016, 11, 2233–2242. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
| Sample | Method | Number Positive Postmortem (Total Examined) | Sensitivity * | Specificity * | Reference | Sample Notes | |
|---|---|---|---|---|---|---|---|
| Tissues | Brainstem (obex) | IHC | NA | NA | NA | NA | IHC and ELISA of brainstem and RLN are considered the “gold standard” postmortem diagnostic approach for CWD. In deer, RLN are generally considered more sensitive, while in elk it is recommended both tissues be evaluated to confirm a diagnosis. |
| EIA | 53 (1986) | 92% | 100% | [26] | |||
| MRLN | IHC | NA | NA | NA | NA | ||
| EIA | 84 (2042) | 99% | >99% | [26] | |||
| RT-QuIC | 23 (1243) | 100% | 100% | [32] | Field samples, postmortem | ||
| Tonsil | IHC | 100 (1150) | 99% | 100% | [49] | Field samples, postmortem | |
| sPMCA | 30 (48) | ND † | ND † | [68] | Experimental animals, antemortem | ||
| RAMALT | IHC | 150 (561) | 68% | >99% | [46] | Field samples, postmortem | |
| RT-QuIC | 289 (409) | 70% | 94% | [45] | Field samples, antemortem | ||
| Nasal brushings | RT-QuIC | 289 (409) | 16% | 91% | [45] | Field samples, antemortem | |
| Body Fluids/Excreta | Blood | RT-QuIC | 16 (21) | 93% | 100% | [90] | Experimental animals, serial collection |
| CSF | sPMCA | 16 (37) | 19% | 100% | [79] | Field samples, postmortem | |
| RT-QuIC | 26 (48) | 50% | 96% | [74] | Experimental animals, postmortem | ||
| Saliva | RT-QuIC | 18 (22) | 78% | 98% | [88] | Experimental animals, antemortem | |
| Urine | RT-QuIC | 18 (22) | 39% | 100% | [88] | Experimental animals, antemortem | |
| Feces | sPMCA | 5 (36) | ND † | ND † | [78] | Field samples, ante- and postmortem | |
| RT-QuIC | 15 (25) | 53% | 100% | [97] | Field samples, antemortem |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Haley, N.J.; Richt, J.A. Evolution of Diagnostic Tests for Chronic Wasting Disease, a Naturally Occurring Prion Disease of Cervids. Pathogens 2017, 6, 35. https://doi.org/10.3390/pathogens6030035
Haley NJ, Richt JA. Evolution of Diagnostic Tests for Chronic Wasting Disease, a Naturally Occurring Prion Disease of Cervids. Pathogens. 2017; 6(3):35. https://doi.org/10.3390/pathogens6030035
Chicago/Turabian StyleHaley, Nicholas J., and Jürgen A. Richt. 2017. "Evolution of Diagnostic Tests for Chronic Wasting Disease, a Naturally Occurring Prion Disease of Cervids" Pathogens 6, no. 3: 35. https://doi.org/10.3390/pathogens6030035
APA StyleHaley, N. J., & Richt, J. A. (2017). Evolution of Diagnostic Tests for Chronic Wasting Disease, a Naturally Occurring Prion Disease of Cervids. Pathogens, 6(3), 35. https://doi.org/10.3390/pathogens6030035