Epigenetic Landscape during Coronavirus Infection

Abstract
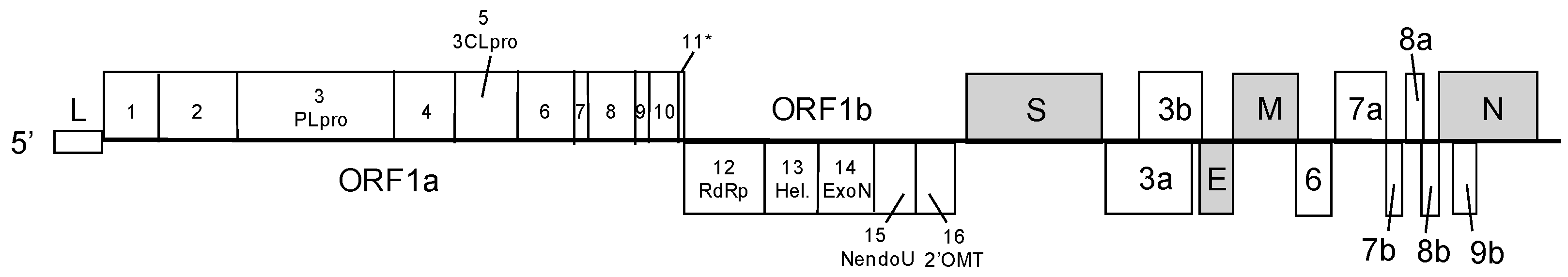
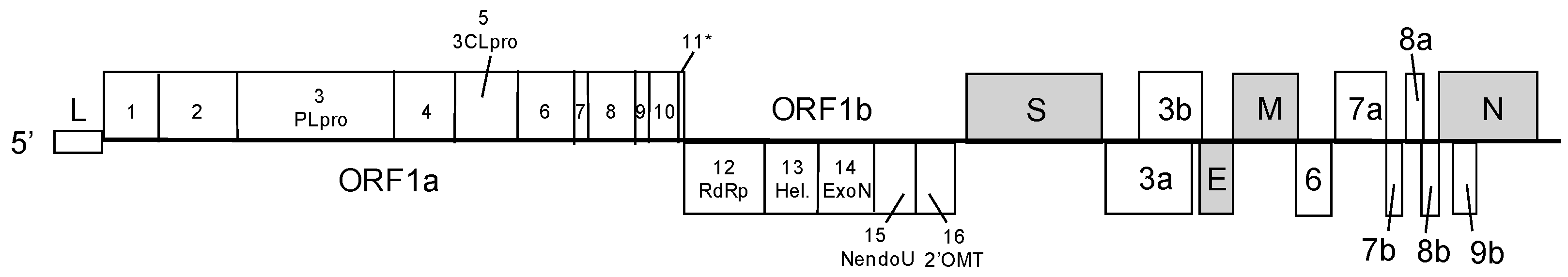
:1. Coronaviruses
2. Innate Immunity and Coronavirus Infections
3. Epigenetics
4. Chromatin
5. Epigenetic Regulation of Gene Expression
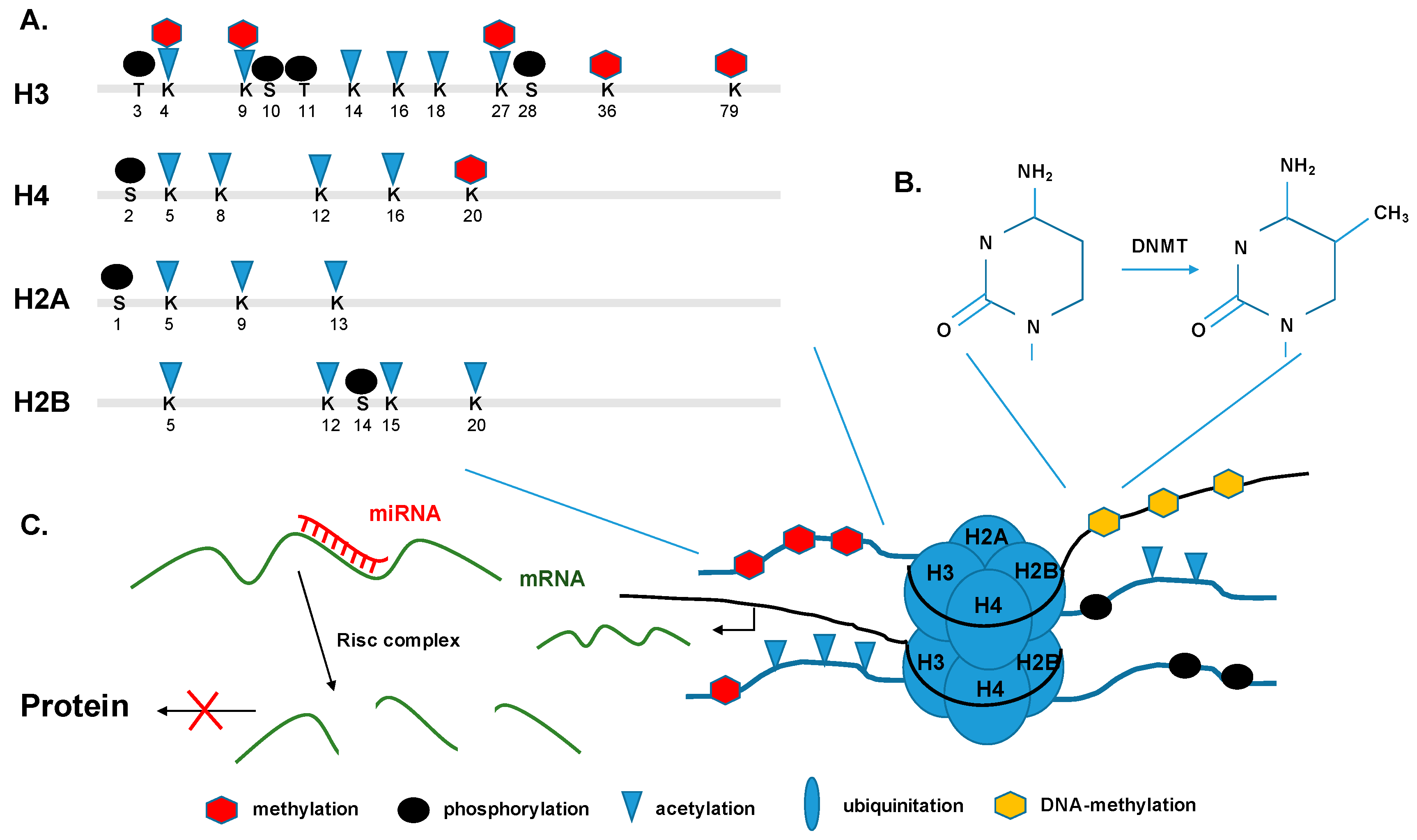
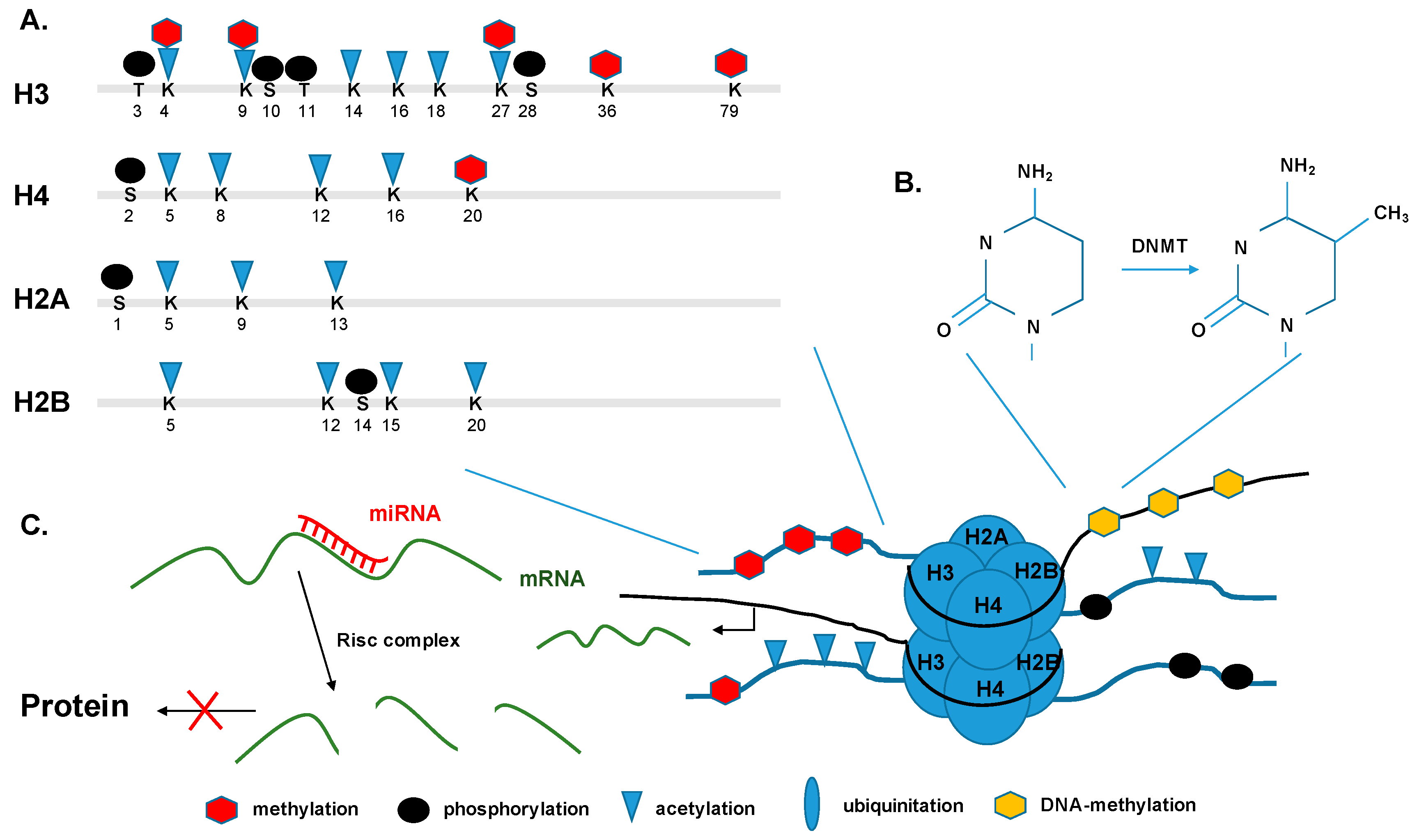
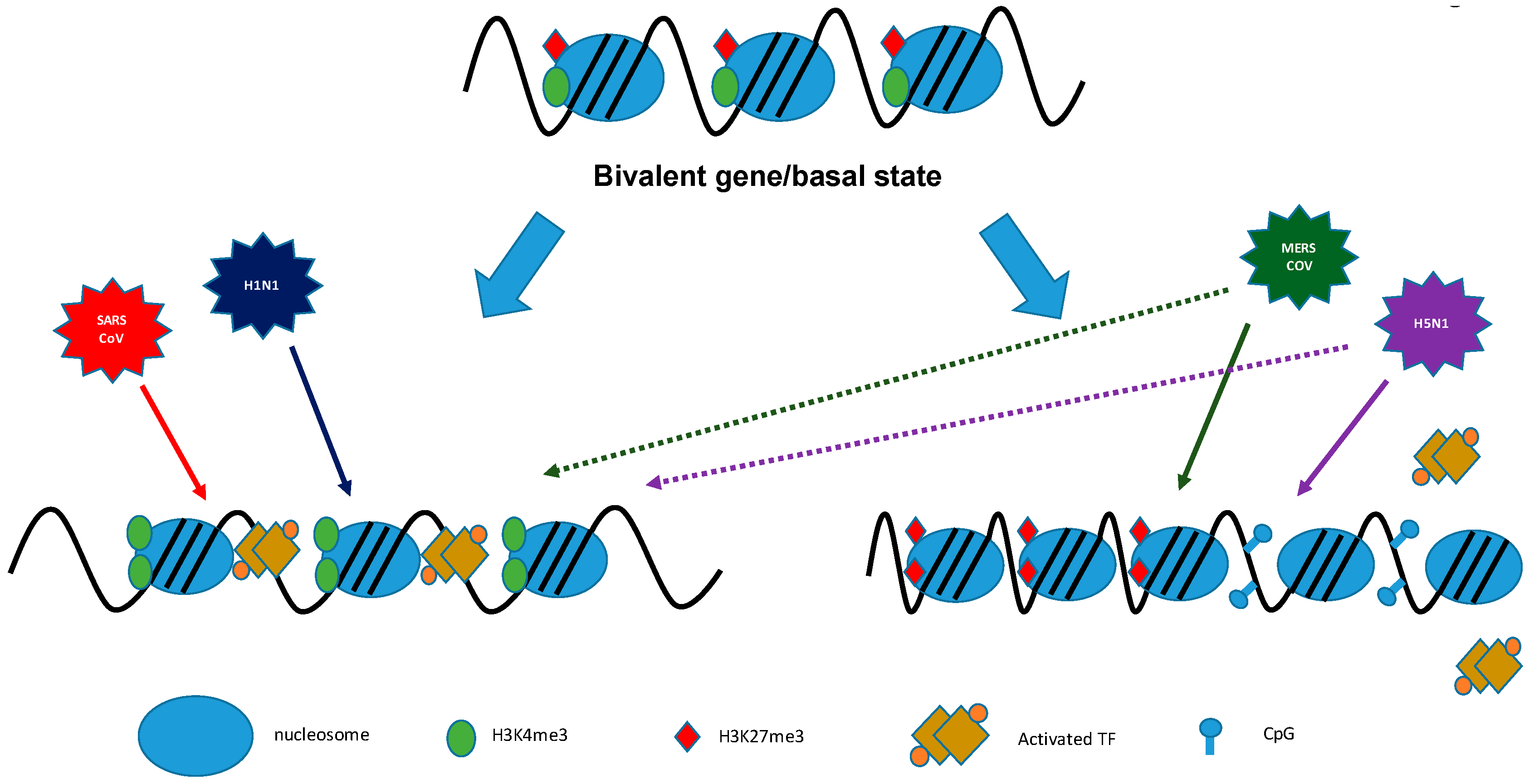
5.1. Histone Modifications and Transcription
5.2. DNA Methylation and De-Novo Methylation
5.3. Non-Coding RNAs and micro RNAs
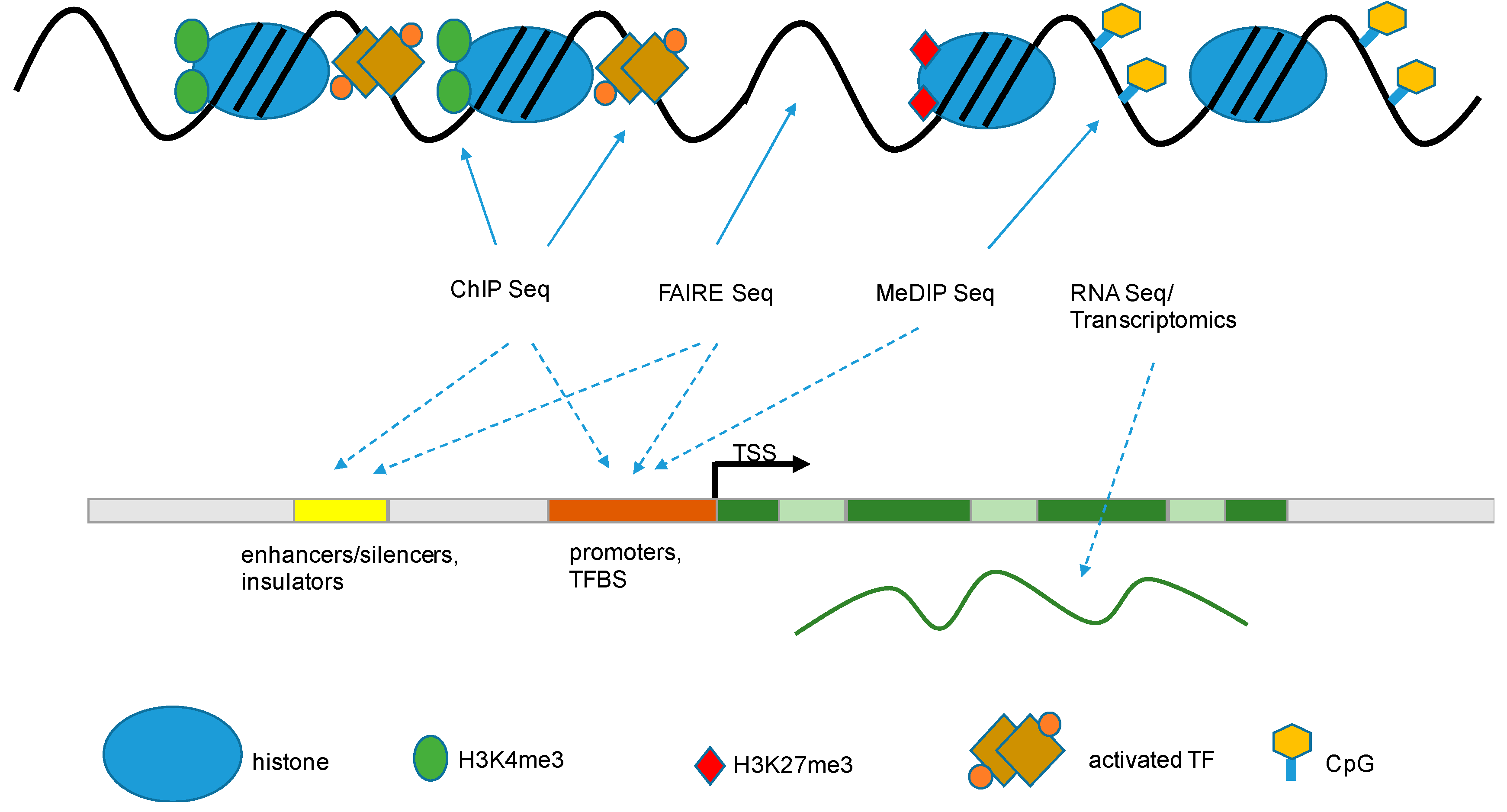
6. Methods to Study Epigenetics
6.1. Modifications of Histones and Localizations of Histone Marks within the Genome
6.1.1. Formaldehyde-Assisted Isolation of Regulatory Elements (FAIRE)
6.1.2. Chromatin Immuno-Precipitation (ChIP)
6.2. Whole-Genome Methylation Status
7. Immune System and Genetics
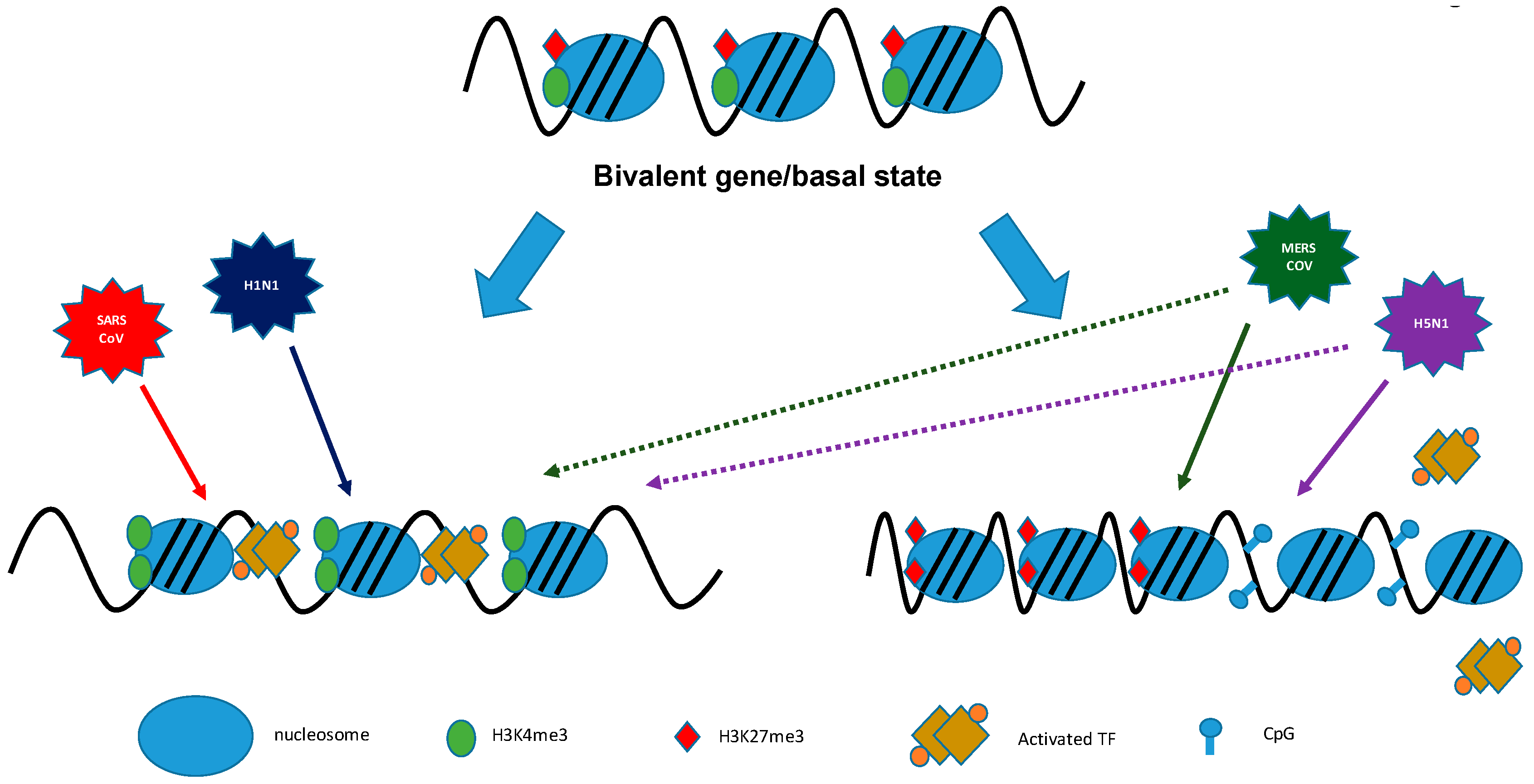
8. Epigenetic Regulation/Modulation of Host Response
- ➔
- Recruitment of transcription factors/machinery;
- ➔
- Prevention of unwanted expression of potent mediators; and
- ➔
9. Coronaviruses/Influenza Viruses: Viral Antagonism of Host Gene Expression by Altering Histone Modifications
10. Systems-Biology Approach
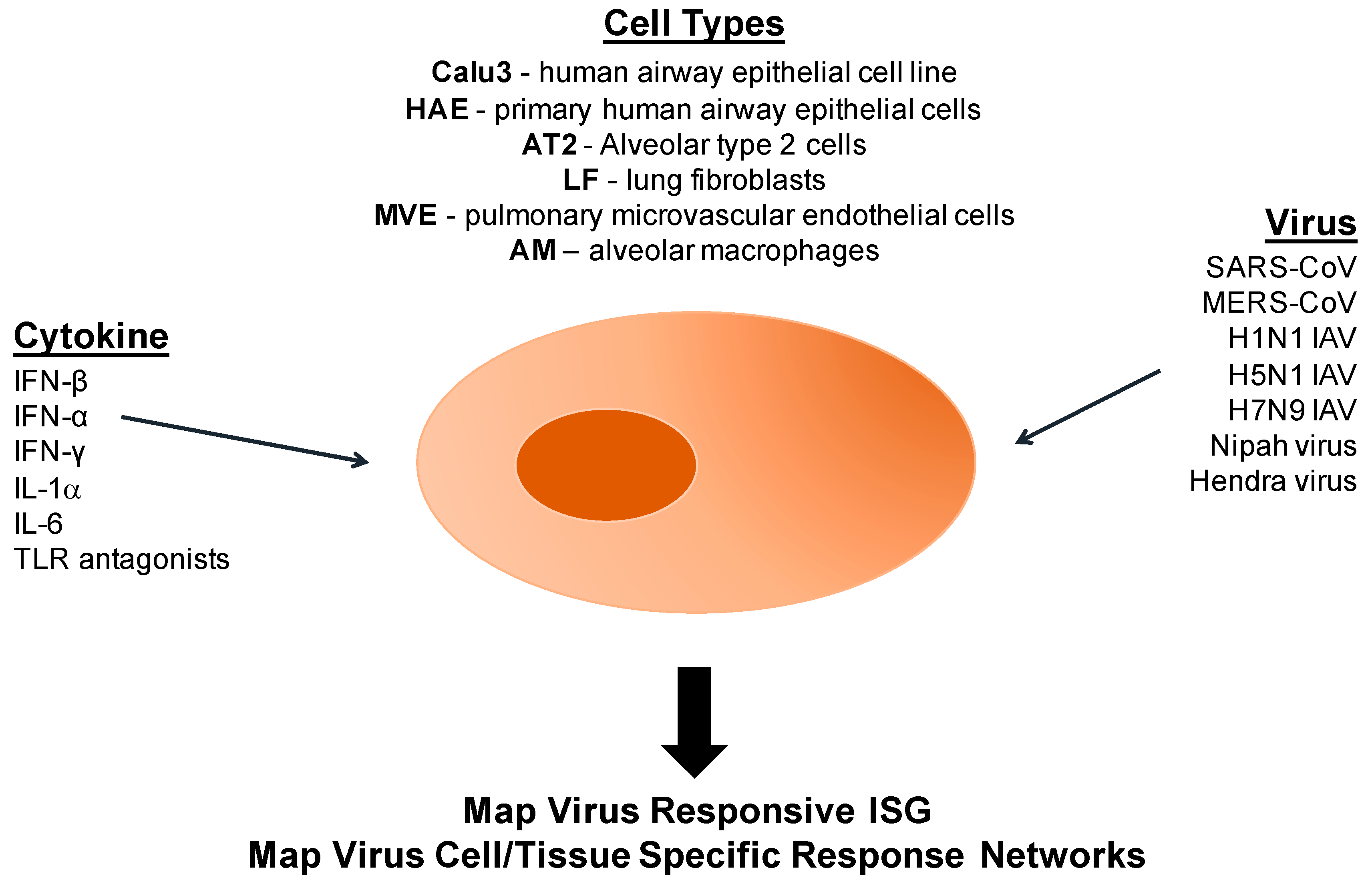
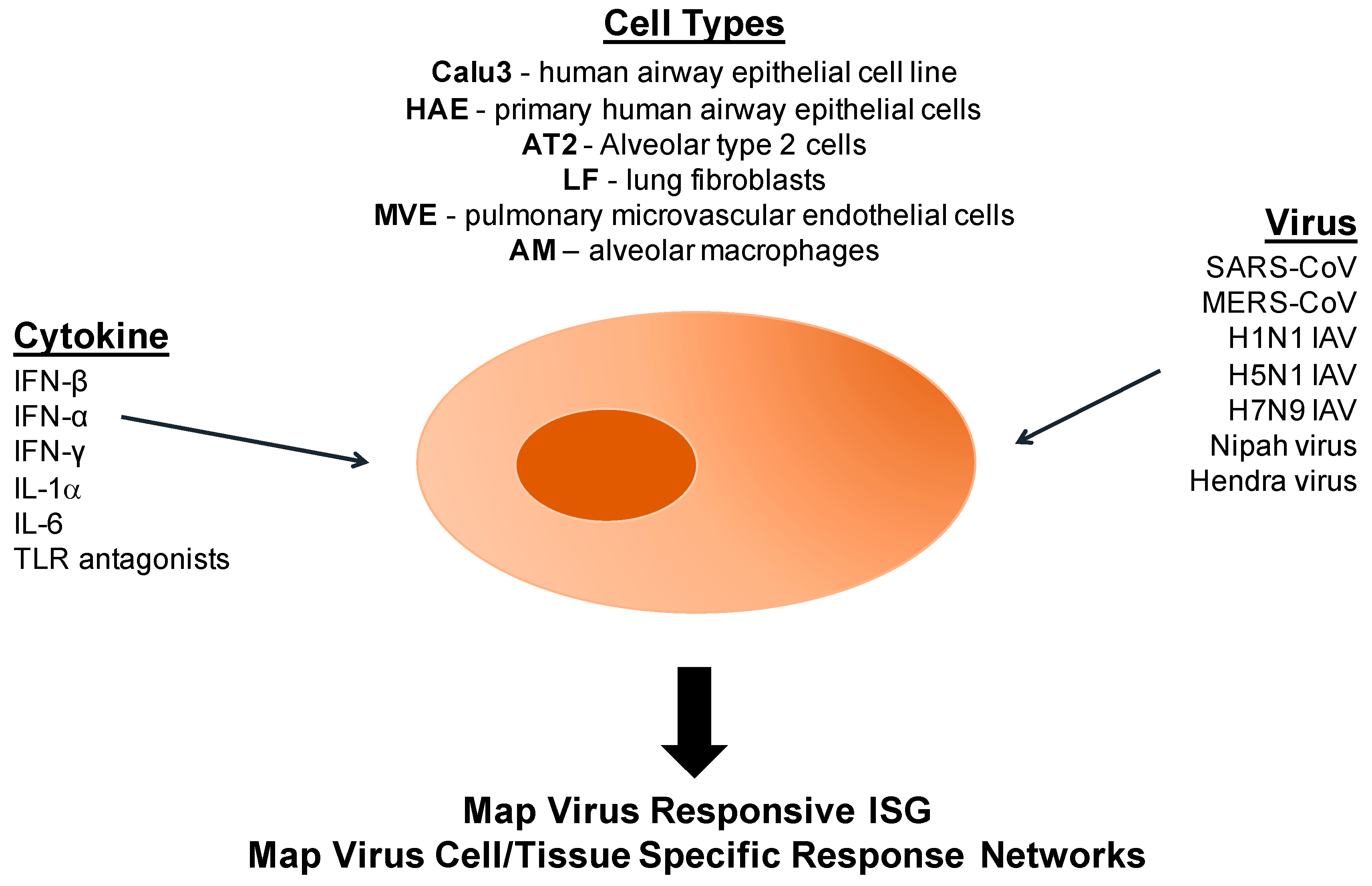
10.1. A Model Platform for Epigenetic Research Following Coronaviruses and Other Respiratory Virus Infections
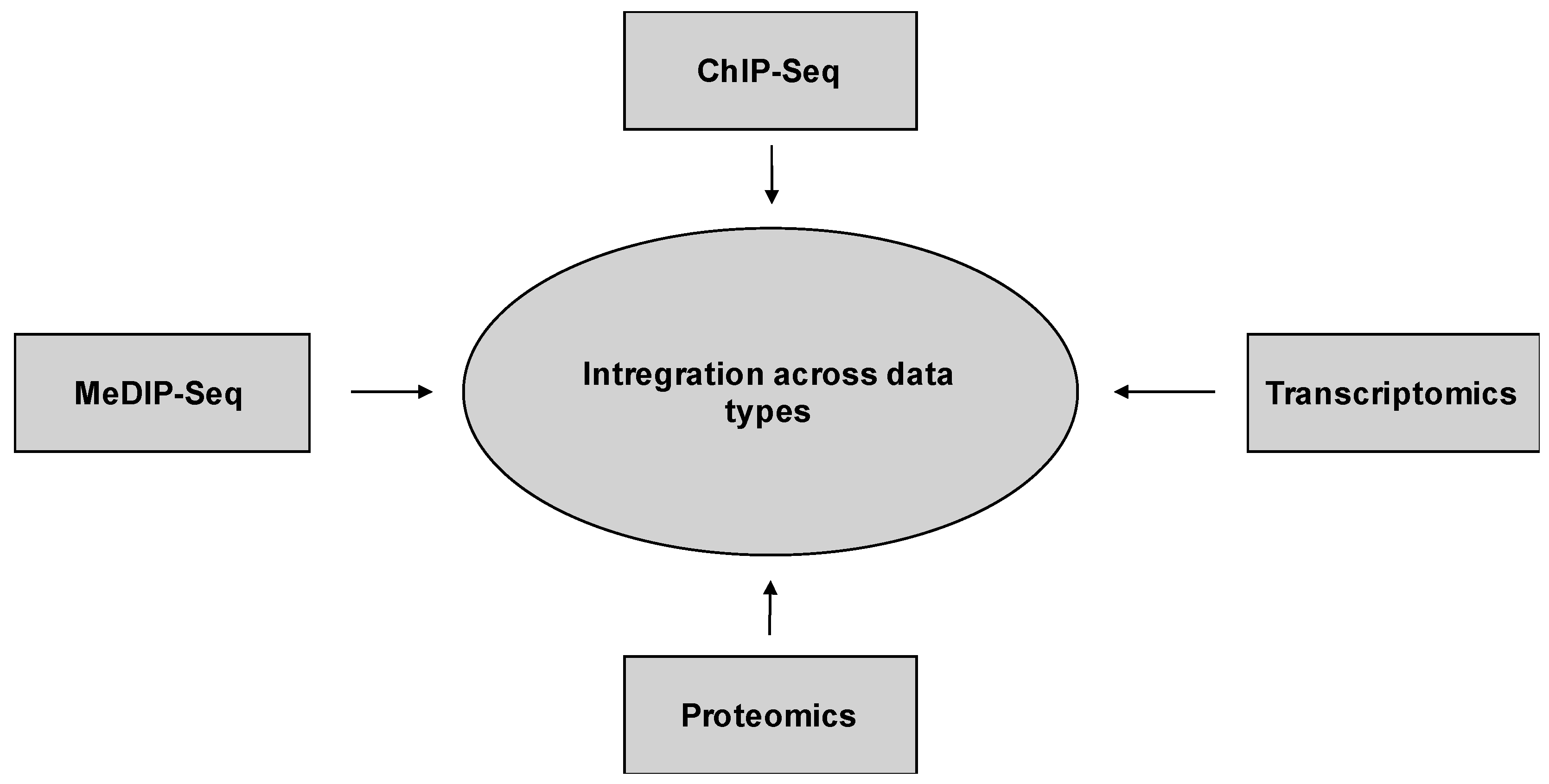
10.2. Integration across Data Types
11. Outlook
Acknowledgements
Conflicts of Interest
References
- Zhong, N. Management and prevention of SARS in China. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2004, 359, 1115–1116. [Google Scholar] [CrossRef] [PubMed]
- Peiris, S.J.; Guan, Y.; Yuen, K.Y. Severe acute respiratory syndrome. Nat. Med. 2004, 10 (Suppl. 12), S88–S97. [Google Scholar] [CrossRef] [PubMed]
- Cherry, J.D. The chronology of the 2002–2003 SARS mini pandemic. Paediatr. Respir. Rev. 2004, 5, 262–269. [Google Scholar] [CrossRef] [PubMed]
- Lau, S.K.; Woo, P.C.; Li, K.S.; Huang, Y.; Tsoi, H.W.; Wong, B.H.; Wong, S.S.; Leung, S.Y.; Chan, K.H.; Yuen, K.Y. Severe acute respiratory syndrome coronavirus-like virus in Chinese horseshoe bats. Proc. Natl. Acad. Sci. USA 2005, 102, 14040–14045. [Google Scholar] [CrossRef] [PubMed]
- Menachery, V.D.; Yount, B.L., Jr.; Sims, A.C.; Debbink, K.; Agnihothram, S.S.; Gralinski, L.E.; Graham, R.L.; Scobey, T.; Plante, J.A.; Royal, S.R.; et al. SARS-like WIV1-CoV poised for human emergence. Proc. Natl. Acad. Sci. USA 2016, 113, 3048–3053. [Google Scholar] [CrossRef] [PubMed]
- Menachery, V.D.; Yount, B.L., Jr.; Debbink, K.; Agnihothram, S.; Gralinski, L.E.; Plante, J.A.; Graham, R.L.; Scobey, T.; Ge, X.Y.; Donaldson, E.F.; et al. A SARS-like cluster of circulating bat coronaviruses shows potential for human emergence. Nat. Med. 2015, 21, 1508–1513. [Google Scholar] [CrossRef] [PubMed]
- Graham, R.L.; Donaldson, E.F.; Baric, R.S. A decade after SARS: Strategies for controlling emerging coronaviruses. Nat. Rev. Microbiol. 2013, 11, 836–848. [Google Scholar] [CrossRef] [PubMed]
- Lu, L.; Liu, Q.; Du, L.; Jiang, S. Middle East respiratory syndrome coronavirus (MERS-CoV): Challenges in identifying its source and controlling its spread. Microbes Infect. 2013, 15, 625–629. [Google Scholar] [CrossRef] [PubMed]
- De Wit, E.; van Doremalen, N.; Falzarano, D.; Munster, V.J. SARS and MERS: Recent insights into emerging coronaviruses. Nat. Rev. Microbiol. 2016, 14, 523–534. [Google Scholar] [CrossRef] [PubMed]
- Drosten, C.; Preiser, W.; Gunther, S.; Schmitz, H.; Doerr, H.W. Severe acute respiratory syndrome: Identification of the etiological agent. Trends Mol. Med. 2003, 9, 325–327. [Google Scholar] [CrossRef]
- Graham, R.L.; Becker, M.M.; Eckerle, L.D.; Bolles, M.; Denison, M.R.; Baric, R.S. A live, impaired-fidelity coronavirus vaccine protects in an aged, immunocompromised mouse model of lethal disease. Nat. Med. 2012, 18, 1820–1826. [Google Scholar] [CrossRef] [PubMed]
- Totura, A.L.; Baric, R.S. SARS coronavirus pathogenesis: Host innate immune responses and viral antagonism of interferon. Curr. Opin. Virol. 2012, 2, 264–275. [Google Scholar] [CrossRef] [PubMed]
- Frieman, M.B.; Chen, J.; Morrison, T.E.; Whitmore, A.; Funkhouser, W.; Ward, J.M.; Lamirande, E.W.; Roberts, A.; Heise, M.; Subbarao, K.; et al. SARS-CoV pathogenesis is regulated by a STAT1 dependent but a type I, II and III interferon receptor independent mechanism. PLoS Pathog. 2010, 6, e1000849. [Google Scholar] [CrossRef] [PubMed]
- Sheahan, T.; Morrison, T.E.; Funkhouser, W.; Uematsu, S.; Akira, S.; Baric, R.S.; Heise, M.T. MyD88 is required for protection from lethal infection with a mouse-adapted SARS-CoV. PLoS Pathog. 2008, 4, e1000240. [Google Scholar] [CrossRef] [PubMed]
- Cervantes-Barragan, L.; Zust, R.; Weber, F.; Spiegel, M.; Lang, K.S.; Akira, S.; Thiel, V.; Ludewig, B. Control of coronavirus infection through plasmacytoid dendritic-cell-derived type I interferon. Blood 2007, 109, 1131–1137. [Google Scholar] [CrossRef] [PubMed]
- Totura, A.L.; Whitmore, A.; Agnihothram, S.; Schafer, A.; Katze, M.G.; Heise, M.T.; Baric, R.S. Toll-like receptor 3 signaling via TRIF contributes to a protective innate immune response to severe acute respiratory syndrome coronavirus infection. MBio 2015, 6, e00638-15. [Google Scholar] [CrossRef] [PubMed]
- Zalinger, Z.B.; Elliott, R.; Rose, K.M.; Weiss, S.R. MDA5 Is Critical to host defense during infection with murine coronavirus. J. Virol. 2015, 89, 12330–12340. [Google Scholar] [CrossRef] [PubMed]
- Menachery, V.D.; Eisfeld, A.J.; Schafer, A.; Josset, L.; Sims, A.C.; Proll, S.; Fan, S.; Li, C.; Neumann, G.; Tilton, S.C.; et al. Pathogenic influenza viruses and coronaviruses utilize similar and contrasting approaches to control interferon-stimulated gene responses. MBio 2014, 5, e01174-14. [Google Scholar] [CrossRef] [PubMed]
- Matthews, K.; Schafer, A.; Pham, A.; Frieman, M. The SARS coronavirus papain like protease can inhibit IRF3 at a post activation step that requires deubiquitination activity. Virol. J. 2014, 11, 209. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Huang, J.B.; Mi, J.; He, Y.F.; Wu, X.H.; Luo, C.L.; Liang, S.M.; Li, J.B.; Tang, Y.X.; Li, J. Characterization of acute renal allograft rejection by proteomic analysis of renal tissue in rat. Mol. Biol. Rep. 2012, 39, 1315–1322. [Google Scholar] [CrossRef] [PubMed]
- Sims, A.C.; Tilton, S.C.; Menachery, V.D.; Gralinski, L.E.; Schafer, A.; Matzke, M.M.; Webb-Robertson, B.J.; Chang, J.; Luna, M.L.; Long, C.E.; et al. Release of severe acute respiratory syndrome coronavirus nuclear import block enhances host transcription in human lung cells. J. Virol. 2013, 87, 3885–3902. [Google Scholar] [CrossRef] [PubMed]
- MMenachery, V.D.; Yount, B.L., Jr.; Josset, L.; Gralinski, L.E.; Scobey, T.; Agnihothram, S.; Katze, M.G.; Baric, R.S. Attenuation and restoration of severe acute respiratory syndrome coronavirus mutant lacking 2′-O-methyltransferase activity. J. Virol. 2014, 88, 4251–4264. [Google Scholar] [CrossRef] [PubMed]
- Daffis, S.; Szretter, K.J.; Schriewer, J.; Li, J.; Youn, S.; Errett, J.; Lin, T.Y.; Schneller, S.; Zust, R.; Dong, H.; et al. 2′-O-methylation of the viral mRNA cap evades host restriction by IFIT family members. Nature 2010, 468, 452–456. [Google Scholar] [CrossRef] [PubMed]
- Sperry, S.M.; Kazi, L.; Graham, R.L.; Baric, R.S.; Weiss, S.R.; Denison, M.R. Single-amino-acid substitutions in open reading frame (ORF) 1b-nsp14 and ORF 2a proteins of the coronavirus mouse hepatitis virus are attenuating in mice. J. Virol. 2005, 79, 3391–3400. [Google Scholar] [CrossRef] [PubMed]
- Becares, M.; Pascual-Iglesias, A.; Nogales, A.; Sola, I.; Enjuanes, L.; Zuniga, S. Mutagenesis of coronavirus nsp14 reveals its potential role in modulation of the innate immune response. J. Virol. 2016, 90, 5399–5414. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Bi, W.; Weiss, S.R.; Leibowitz, J.L. Mouse hepatitis virus gene 5b protein is a new virion envelope protein. Virology 1994, 202, 1018–1023. [Google Scholar] [CrossRef] [PubMed]
- Matthews, K.L.; Coleman, C.M.; van der Meer, Y.; Snijder, E.J.; Frieman, M.B. The ORF4b-encoded accessory proteins of Middle East respiratory syndrome coronavirus and two related bat coronaviruses localize to the nucleus and inhibit innate immune signalling. J. Gen. Virol. 2014, 95 Pt 4, 874–882. [Google Scholar] [CrossRef] [PubMed]
- Scobey, T.; Yount, B.L.; Sims, A.C.; Donaldson, E.F.; Agnihothram, S.S.; Menachery, V.D.; Graham, R.L.; Swanstrom, J.; Bove, P.F.; Kim, J.D.; et al. Reverse genetics with a full-length infectious cDNA of the Middle East respiratory syndrome coronavirus. Proc. Natl. Acad. Sci. USA 2013, 110, 16157–16162. [Google Scholar] [CrossRef] [PubMed]
- Thornbrough, J.M.; Jha, B.K.; Yount, B.; Goldstein, S.A.; Li, Y.; Elliott, R.; Sims, A.C.; Baric, R.S.; Silverman, R.H.; Weiss, S.R. Middle East respiratory syndrome coronavirus NS4b protein inhibits host RNase L activation. MBio 2016, 7, e00258. [Google Scholar] [CrossRef] [PubMed]
- Rabouw, H.H.; Langereis, M.A.; Knaap, R.C.; Dalebout, T.J.; Canton, J.; Sola, I.; Enjuanes, L.; Bredenbeek, P.J.; Kikkert, M.; de Groot, R.J.; et al. Middle East respiratory coronavirus accessory protein 4a inhibits PKR-mediated antiviral stress responses. PLoS Pathog. 2016, 12, e1005982. [Google Scholar] [CrossRef] [PubMed]
- Marazzi, I.; Ho, J.S.; Kim, J.; Manicassamy, B.; Dewell, S.; Albrecht, R.A.; Seibert, C.W.; Schaefer, U.; Jeffrey, K.L.; Prinjha, R.K.; et al. Suppression of the antiviral response by an influenza histone mimic. Nature 2012, 483, 428–433. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, A.D.; Allis, C.D.; Bernstein, E. Epigenetics: A landscape takes shape. Cell 2007, 128, 635–638. [Google Scholar] [CrossRef] [PubMed]
- Bird, A. Perceptions of epigenetics. Nature 2007, 447, 396–398. [Google Scholar] [CrossRef] [PubMed]
- Esteller, M. Epigenetics in cancer. N. Engl. J. Med. 2008, 358, 1148–1159. [Google Scholar] [CrossRef] [PubMed]
- Obata, Y.; Furusawa, Y.; Hase, K. Epigenetic modifications of the immune system in health and disease. Immunol. Cell Biol. 2015, 93, 226–232. [Google Scholar] [CrossRef] [PubMed]
- Busslinger, M.; Tarakhovsky, A. Epigenetic control of immunity. Cold Spring Harb. Perspect. Biol. 2014, 6. [Google Scholar] [CrossRef]
- Collins, F.S.; Morgan, M.; Patrinos, A. The Human Genome Project: Lessons from large-scale biology. Science 2003, 300, 286–290. [Google Scholar] [CrossRef] [PubMed]
- Rivera, C.M.; Ren, B. Mapping human epigenomes. Cell 2013, 155, 39–55. [Google Scholar] [CrossRef] [PubMed]
- Schuster, S.C. Next-generation sequencing transforms today’s biology. Nat. Methods 2008, 5, 16–18. [Google Scholar] [CrossRef] [PubMed]
- Rakyan, V.K.; Down, T.A.; Balding, D.J.; Beck, S. Epigenome-wide association studies for common human diseases. Nat. Rev. Genet. 2011, 12, 529–541. [Google Scholar] [CrossRef] [PubMed]
- Birney, E.; Smith, G.D.; Greally, J.M. Epigenome-wide Association Studies and the Interpretation of Disease -Omics. PLoS Genet. 2016, 12, e1006105. [Google Scholar] [CrossRef] [PubMed]
- Consortium, E.P.; Birney, E.; Stamatoyannopoulos, J.A.; Dutta, A.; Guigo, R.; Gingeras, T.R.; Margulies, E.H.; Weng, Z.; Snyder, M.; Dermitzakis, E.T.; et al. Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature 2007, 447, 799–816. [Google Scholar]
- Consortium, E.P. A user’s guide to the encyclopedia of DNA elements (ENCODE). PLoS Biol. 2011, 9, e1001046. [Google Scholar]
- Derrien, T.; Johnson, R.; Bussotti, G.; Tanzer, A.; Djebali, S.; Tilgner, H.; Guernec, G.; Martin, D.; Merkel, A.; Knowles, D.G.; et al. The GENCODE v7 catalog of human long noncoding RNAs: Analysis of their gene structure, evolution, and expression. Genome Res. 2012, 22, 1775–1789. [Google Scholar] [CrossRef] [PubMed]
- Harrow, J.; Frankish, A.; Gonzalez, J.M.; Tapanari, E.; Diekhans, M.; Kokocinski, F.; Aken, B.L.; Barrell, D.; Zadissa, A.; Searle, S.; et al. GENCODE: The reference human genome annotation for The ENCODE Project. Genome Res. 2012, 22, 1760–1774. [Google Scholar] [CrossRef] [PubMed]
- Gehlenborg, N.; O’Donoghue, S.I.; Baliga, N.S.; Goesmann, A.; Hibbs, M.A.; Kitano, H.; Kohlbacher, O.; Neuweger, H.; Schneider, R.; Tenenbaum, D.; et al. Visualization of Omics data for systems biology. Nat. Methods 2010, 7 (Suppl. 3), S56–S68. [Google Scholar] [CrossRef] [PubMed]
- Felsenfeld, G.; Groudine, M. Controlling the double helix. Nature 2003, 421, 448–453. [Google Scholar] [CrossRef] [PubMed]
- Ramakrishnan, V. Histone structure and the organization of the nucleosome. Annu. Rev. Biophys. Biomol. Struct. 1997, 26, 83–112. [Google Scholar] [CrossRef] [PubMed]
- Strahl, B.D.; Allis, C.D. The language of covalent histone modifications. Nature 2000, 403, 41–45. [Google Scholar] [CrossRef] [PubMed]
- Jenuwein, T.; Allis, C.D. Translating the histone code. Science 2001, 293, 1074–1080. [Google Scholar] [CrossRef] [PubMed]
- Zhou, V.W.; Goren, A.; Bernstein, B.E. Charting histone modifications and the functional organization of mammalian genomes. Nat. Rev. Genet. 2011, 12, 7–18. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, B.E.; Meissner, A.; Lander, E.S. The mammalian epigenome. Cell 2007, 128, 669–681. [Google Scholar] [CrossRef] [PubMed]
- Barski, A.; Cuddapah, S.; Cui, K.; Roh, T.Y.; Schones, D.E.; Wang, Z.; Wei, G.; Chepelev, I.; Zhao, K. High-resolution profiling of histone methylations in the human genome. Cell 2007, 129, 823–837. [Google Scholar] [CrossRef] [PubMed]
- Shlyueva, D.; Stampfel, G.; Stark, A. Transcriptional enhancers: From properties to genome-wide predictions. Nat. Rev. Genet. 2014, 15, 272–286. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, A. The promise of the health sciences in the 21st century. Nurs. Leadersh. (Tor Ont) 2005, 18, 41–43. [Google Scholar] [CrossRef] [PubMed]
- Guertin, M.J.; Lis, J.T. Mechanisms by which transcription factors gain access to target sequence elements in chromatin. Curr. Opin. Genet. Dev. 2013, 23, 116–123. [Google Scholar] [CrossRef] [PubMed]
- Lim, P.S.; Li, J.; Holloway, A.F.; Rao, S. Epigenetic regulation of inducible gene expression in the immune system. Immunology 2013, 139, 285–293. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, B.E.; Mikkelsen, T.S.; Xie, X.; Kamal, M.; Huebert, D.J.; Cuff, J.; Fry, B.; Meissner, A.; Wernig, M.; Plath, K.; et al. A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell 2006, 125, 315–326. [Google Scholar] [CrossRef] [PubMed]
- Tarakhovsky, A. Tools and landscapes of epigenetics. Nat. Immunol. 2010, 11, 565–568. [Google Scholar] [CrossRef] [PubMed]
- Rogatsky, I.; Adelman, K. Preparing the first responders: Building the inflammatory transcriptome from the ground up. Mol. Cell 2014, 54, 245–254. [Google Scholar] [CrossRef] [PubMed]
- Kouzarides, T. Chromatin modifications and their function. Cell 2007, 128, 693–705. [Google Scholar] [CrossRef] [PubMed]
- Bonev, B.; Cavalli, G. Organization and function of the 3D genome. Nat. Rev. Genet. 2016, 17, 661–678. [Google Scholar] [CrossRef] [PubMed]
- Torres, I.O.; Fujimori, D.G. Functional coupling between writers, erasers and readers of histone and DNA methylation. Curr. Opin. Struct. Biol. 2015, 35, 68–75. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.A. Functions of DNA methylation: Islands, start sites, gene bodies and beyond. Nat. Rev. Genet. 2012, 13, 484–492. [Google Scholar] [CrossRef] [PubMed]
- Jin, B.; Robertson, K.D. DNA methyltransferases, DNA damage repair, and cancer. Adv. Exp. Med. Biol. 2013, 754, 3–29. [Google Scholar] [PubMed]
- Jaenisch, R.; Bird, A. Epigenetic regulation of gene expression: How the genome integrates intrinsic and environmental signals. Nat. Genet. 2003, 33, 245–254. [Google Scholar] [CrossRef] [PubMed]
- Deaton, A.M.; Bird, A. CpG islands and the regulation of transcription. Genes Dev. 2011, 25, 1010–1022. [Google Scholar] [CrossRef] [PubMed]
- He, Y.F.; Li, B.Z.; Li, Z.; Liu, P.; Wang, Y.; Tang, Q.; Ding, J.; Jia, Y.; Chen, Z.; Li, L.; et al. Tet-mediated formation of 5-carboxylcytosine and its excision by TDG in mammalian DNA. Science 2011, 333, 1303–1307. [Google Scholar] [CrossRef] [PubMed]
- Szulwach, K.E.; Li, X.; Li, Y.; Song, C.X.; Wu, H.; Dai, Q.; Irier, H.; Upadhyay, A.K.; Gearing, M.; Levey, A.I.; et al. 5-hmC-mediated epigenetic dynamics during postnatal neurodevelopment and aging. Nat. Neurosci. 2011, 14, 1607–1616. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, M.M.; Bird, A. DNA methylation landscapes: Provocative insights from epigenomics. Nat. Rev. Genet. 2008, 9, 465–476. [Google Scholar] [CrossRef] [PubMed]
- Palazzo, A.F.; Lee, E.S. Non-coding RNA: What is functional and what is junk? Front. Genet. 2015, 6, 2. [Google Scholar] [CrossRef] [PubMed]
- Geisler, S.; Coller, J. RNA in unexpected places: Long non-coding RNA functions in diverse cellular contexts. Nat. Rev. Mol. Cell Biol. 2013, 14, 699–712. [Google Scholar] [CrossRef] [PubMed]
- Carpenter, S.; Ricci, E.P.; Mercier, B.C.; Moore, M.J.; Fitzgerald, K.A. Post-transcriptional regulation of gene expression in innate immunity. Nat. Rev. Immunol. 2014, 14, 361–376. [Google Scholar] [CrossRef] [PubMed]
- Sato, F.; Tsuchiya, S.; Meltzer, S.J.; Shimizu, K. MicroRNAs and epigenetics. FEBS J. 2011, 278, 1598–1609. [Google Scholar] [CrossRef] [PubMed]
- Hilton, I.B.; Simon, J.M.; Lieb, J.D.; Davis, I.J.; Damania, B.; Dittmer, D.P. The open chromatin landscape of Kaposi’s sarcoma-associated herpesvirus. J. Virol. 2013, 87, 11831–11842. [Google Scholar] [CrossRef] [PubMed]
- Tsompana, M.; Buck, M.J. Chromatin accessibility: A window into the genome. Epigenet. Chromatin 2014, 7, 33. [Google Scholar] [CrossRef] [PubMed]
- Giresi, P.G.; Kim, J.; McDaniell, R.M.; Iyer, V.R.; Lieb, J.D. FAIRE (Formaldehyde-Assisted Isolation of Regulatory Elements) isolates active regulatory elements from human chromatin. Genome Res. 2007, 17, 877–885. [Google Scholar] [CrossRef] [PubMed]
- Giresi, P.G.; Lieb, J.D. Isolation of active regulatory elements from eukaryotic chromatin using FAIRE (Formaldehyde Assisted Isolation of Regulatory Elements). Methods 2009, 48, 233–239. [Google Scholar] [CrossRef] [PubMed]
- Robertson, G.; Hirst, M.; Bainbridge, M.; Bilenky, M.; Zhao, Y.; Zeng, T.; Euskirchen, G.; Bernier, B.; Varhol, R.; Delaney, A.; et al. Genome-wide profiles of STAT1 DNA association using chromatin immunoprecipitation and massively parallel sequencing. Nat. Methods 2007, 4, 651–657. [Google Scholar] [CrossRef] [PubMed]
- Park, P.J. ChIP-seq: Advantages and challenges of a maturing technology. Nat. Rev. Genet. 2009, 10, 669–680. [Google Scholar] [CrossRef] [PubMed]
- Landt, S.G.; Marinov, G.K.; Kundaje, A.; Kheradpour, P.; Pauli, F.; Batzoglou, S.; Bernstein, B.E.; Bickel, P.; Brown, J.B.; Cayting, P.; et al. ChIP-seq guidelines and practices of the ENCODE and modENCODE consortia. Genome Res. 2012, 22, 1813–1831. [Google Scholar] [CrossRef] [PubMed]
- Schones, D.E.; Zhao, K. Genome-wide approaches to studying chromatin modifications. Nat. Rev. Genet. 2008, 9, 179–191. [Google Scholar] [CrossRef] [PubMed]
- Weber, M.; Davies, J.J.; Wittig, D.; Oakeley, E.J.; Haase, M.; Lam, W.L.; Schubeler, D. Chromosome-wide and promoter-specific analyses identify sites of differential DNA methylation in normal and transformed human cells. Nat. Genet. 2005, 37, 853–862. [Google Scholar] [CrossRef] [PubMed]
- Nair, S.S.; Coolen, M.W.; Stirzaker, C.; Song, J.Z.; Statham, A.L.; Strbenac, D.; Robinson, M.D.; Clark, S.J. Comparison of methyl-DNA immunoprecipitation (MeDIP) and methyl-CpG binding domain (MBD) protein capture for genome-wide DNA methylation analysis reveal CpG sequence coverage bias. Epigenetics 2011, 6, 34–44. [Google Scholar] [CrossRef] [PubMed]
- Jacinto, F.V.; Ballestar, E.; Esteller, M. Methyl-DNA immunoprecipitation (MeDIP): Hunting down the DNA methylome. Biotechniques 2008, 44, 35, 37, 39 passim. [Google Scholar] [CrossRef] [PubMed]
- Herman, J.G.; Graff, J.R.; Myohanen, S.; Nelkin, B.D.; Baylin, S.B. Methylation-specific PCR: A novel PCR assay for methylation status of CpG islands. Proc. Natl. Acad. Sci. USA 1996, 93, 9821–9826. [Google Scholar] [CrossRef] [PubMed]
- Beutler, B. Innate immunity: An overview. Mol. Immunol. 2004, 40, 845–859. [Google Scholar] [CrossRef] [PubMed]
- Medzhitov, R.; Janeway, C.A., Jr. Innate immunity: Impact on the adaptive immune response. Curr. Opin. Immunol. 1997, 9, 4–9. [Google Scholar] [CrossRef]
- Janeway, C.A., Jr.; Medzhitov, R. Innate immune recognition. Annu. Rev. Immunol. 2002, 20, 197–216. [Google Scholar] [CrossRef] [PubMed]
- Schoggins, J.W.; MacDuff, D.A.; Imanaka, N.; Gainey, M.D.; Shrestha, B.; Eitson, J.L.; Mar, K.B.; Richardson, R.B.; Ratushny, A.V.; Litvak, V.; et al. Pan-viral specificity of IFN-induced genes reveals new roles for cGAS in innate immunity. Nature 2014, 505, 691–695. [Google Scholar] [CrossRef] [PubMed]
- Ma, Z.; Damania, B. The cGAS-STING defense pathway and its counteraction by viruses. Cell Host Microbe 2016, 19, 150–158. [Google Scholar] [CrossRef] [PubMed]
- Akira, S.; Uematsu, S.; Takeuchi, O. Pathogen recognition and innate immunity. Cell 2006, 124, 783–801. [Google Scholar] [CrossRef] [PubMed]
- Medzhitov, R. Toll-like receptors and innate immunity. Nat. Rev. Immunol. 2001, 1, 135–145. [Google Scholar] [CrossRef] [PubMed]
- Medzhitov, R. CpG DNA: Security code for host defense. Nat. Immunol. 2001, 2, 15–16. [Google Scholar] [CrossRef] [PubMed]
- Paschos, K.; Allday, M.J. Epigenetic reprogramming of host genes in viral and microbial pathogenesis. Trends Microbiol. 2010, 18, 439–447. [Google Scholar] [CrossRef] [PubMed]
- Smale, S.T. Selective transcription in response to an inflammatory stimulus. Cell 2010, 140, 833–844. [Google Scholar] [CrossRef] [PubMed]
- Smale, S.T.; Tarakhovsky, A.; Natoli, G. Chromatin contributions to the regulation of innate immunity. Annu. Rev. Immunol. 2014, 32, 489–511. [Google Scholar] [CrossRef] [PubMed]
- Mehta, S.; Jeffrey, K.L. Beyond receptors and signaling: Epigenetic factors in the regulation of innate immunity. Immunol. Cell Biol. 2015, 93, 233–244. [Google Scholar] [CrossRef] [PubMed]
- Stender, J.D.; Glass, C.K. Epigenomic control of the innate immune response. Curr. Opin. Pharmacol. 2013, 13, 582–587. [Google Scholar] [CrossRef] [PubMed]
- Agalioti, T.; Lomvardas, S.; Parekh, B.; Yie, J.; Maniatis, T.; Thanos, D. Ordered recruitment of chromatin modifying and general transcription factors to the IFN-beta promoter. Cell 2000, 103, 667–678. [Google Scholar] [CrossRef]
- Fang, T.C.; Schaefer, U.; Mecklenbrauker, I.; Stienen, A.; Dewell, S.; Chen, M.S.; Rioja, I.; Parravicini, V.; Prinjha, R.K.; Chandwani, R.; et al. Histone H3 lysine 9 di-methylation as an epigenetic signature of the interferon response. J. Exp. Med. 2012, 209, 661–669. [Google Scholar] [CrossRef] [PubMed]
- Aevermann, B.D.; Pickett, B.E.; Kumar, S.; Klem, E.B.; Agnihothram, S.; Askovich, P.S.; Bankhead, A., 3rd; Bolles, M.; Carter, V.; Chang, J.; et al. A comprehensive collection of systems biology data characterizing the host response to viral infection. Sci. Data 2014, 1, 140033. [Google Scholar] [CrossRef] [PubMed]
- Kaikkonen, M.U.; Lam, M.T.; Glass, C.K. Non-coding RNAs as regulators of gene expression and epigenetics. Cardiovasc. Res. 2011, 90, 430–440. [Google Scholar] [CrossRef] [PubMed]
- Ivashkiv, L.B.; Donlin, L.T. Regulation of type I interferon responses. Nat. Rev. Immunol. 2014, 14, 36–49. [Google Scholar] [CrossRef] [PubMed]
- Schneider, W.M.; Chevillotte, M.D.; Rice, C.M. Interferon-stimulated genes: A complex web of host defenses. Annu. Rev. Immunol. 2014, 32, 513–545. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Sastre, A.; Biron, C.A. Type 1 interferons and the virus-host relationship: A lesson in detente. Science 2006, 312, 879–882. [Google Scholar] [CrossRef] [PubMed]
- Marban, C.; Suzanne, S.; Dequiedt, F.; de Walque, S.; Redel, L.; Van Lint, C.; Aunis, D.; Rohr, O.; et al. Recruitment of chromatin-modifying enzymes by CTIP2 promotes HIV-1 transcriptional silencing. EMBO J. 2007, 26, 412–423. [Google Scholar] [CrossRef] [PubMed]
- Taylor, T.J.; Knipe, D.M. Proteomics of herpes simplex virus replication compartments: Association of cellular DNA replication, repair, recombination, and chromatin remodeling proteins with ICP8. J. Virol. 2004, 78, 5856–5866. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Diaz, E.; Jorda, M.; Peinado, M.A.; Rivero, A. Epigenetics of host-pathogen interactions: The road ahead and the road behind. PLoS Pathog. 2012, 8, e1003007. [Google Scholar] [CrossRef] [PubMed]
- Hale, B.G.; Randall, R.E.; Ortin, J.; Jackson, D. The multifunctional NS1 protein of influenza A viruses. J. Gen. Virol. 2008, 89 Pt 10, 2359–2376. [Google Scholar] [CrossRef] [PubMed]
- Niemeyer, D.; Zillinger, T.; Muth, D.; Zielecki, F.; Horvath, G.; Suliman, T.; Barchet, W.; Weber, F.; Drosten, C.; Muller, M.A. Middle East respiratory syndrome coronavirus accessory protein 4a is a type I interferon antagonist. J. Virol. 2013, 87, 12489–12495. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Chidekel, A.; Shaffer, T.H. Cultured human airway epithelial cells (Calu-3): A model of human respiratory function, structure, and inflammatory responses. Crit. Care Res. Pract. 2010, 2010, 394578. [Google Scholar] [CrossRef] [PubMed]
- ims, A.C.; Baric, R.S.; Yount, B.; Burkett, S.E.; Collins, P.L.; Pickles, R.J. Severe acute respiratory syndrome coronavirus infection of human ciliated airway epithelia: Role of ciliated cells in viral spread in the conducting airways of the lungs. J. Virol. 2005, 79, 15511–15524. [Google Scholar]
- Marazzi, I.; Garcia-Sastre, A. Interference of viral effector proteins with chromatin, transcription, and the epigenome. Curr. Opin. Microbiol. 2015, 26, 123–129. [Google Scholar] [CrossRef] [PubMed]
- Silmon de Monerri, N.C.; Kim, K. Pathogens hijack the epigenome: A new twist on host-pathogen interactions. Am. J. Pathol. 2014, 184, 897–911. [Google Scholar] [CrossRef] [PubMed]
- Menachery, V.D.; Baric, R.S. Bugs in the system. Immunol. Rev. 2013, 255, 256–274. [Google Scholar] [CrossRef] [PubMed]
- Angelini, C.; Costa, V. Understanding gene regulatory mechanisms by integrating ChIP-seq and RNA-seq data: Statistical solutions to biological problems. Front. Cell Dev. Biol. 2014, 2, 51. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, M.D.; Holzinger, E.R.; Li, R.; Pendergrass, S.A.; Kim, D. Methods of integrating data to uncover genotype-phenotype interactions. Nat. Rev. Genet. 2015, 16, 85–97. [Google Scholar] [CrossRef] [PubMed]
- Van Lint, C.; Emiliani, S.; Ott, M.; Verdin, E. Transcriptional activation and chromatin remodeling of the HIV-1 promoter in response to histone acetylation. EMBO J. 1996, 15, 1112–1120. [Google Scholar] [PubMed]
- Liang, Y.; Vogel, J.L.; Narayanan, A.; Peng, H.; Kristie, T.M. Inhibition of the histone demethylase LSD1 blocks alpha-herpesvirus lytic replication and reactivation from latency. Nat. Med. 2009, 15, 1312–1317. [Google Scholar] [CrossRef] [PubMed]
 ) that are associated with functional gene components (dashed arrow
) that are associated with functional gene components (dashed arrow  ), like promoters, transcriptional start sites, enhancers, gene bodies, slicing sites, and transcriptional end sites [43]. TSS: transcription start site; TF: transcription factor; TFBS: transcription factor binding site; FAIRE: formaldehyde-assisted isolation of regulatory elements; ChIP: chromatin immuno-precipitation; MeDIP: methylated DNA immunoprecipitation.
) that are associated with functional gene components (dashed arrow ), like promoters, transcriptional start sites, enhancers, gene bodies, slicing sites, and transcriptional end sites [43]. TSS: transcription start site; TF: transcription factor; TFBS: transcription factor binding site; FAIRE: formaldehyde-assisted isolation of regulatory elements; ChIP: chromatin immuno-precipitation; MeDIP: methylated DNA immunoprecipitation.
), like promoters, transcriptional start sites, enhancers, gene bodies, slicing sites, and transcriptional end sites [43]. TSS: transcription start site; TF: transcription factor; TFBS: transcription factor binding site; FAIRE: formaldehyde-assisted isolation of regulatory elements; ChIP: chromatin immuno-precipitation; MeDIP: methylated DNA immunoprecipitation.
) that are associated with functional gene components (dashed arrow ), like promoters, transcriptional start sites, enhancers, gene bodies, slicing sites, and transcriptional end sites [43]. TSS: transcription start site; TF: transcription factor; TFBS: transcription factor binding site; FAIRE: formaldehyde-assisted isolation of regulatory elements; ChIP: chromatin immuno-precipitation; MeDIP: methylated DNA immunoprecipitation.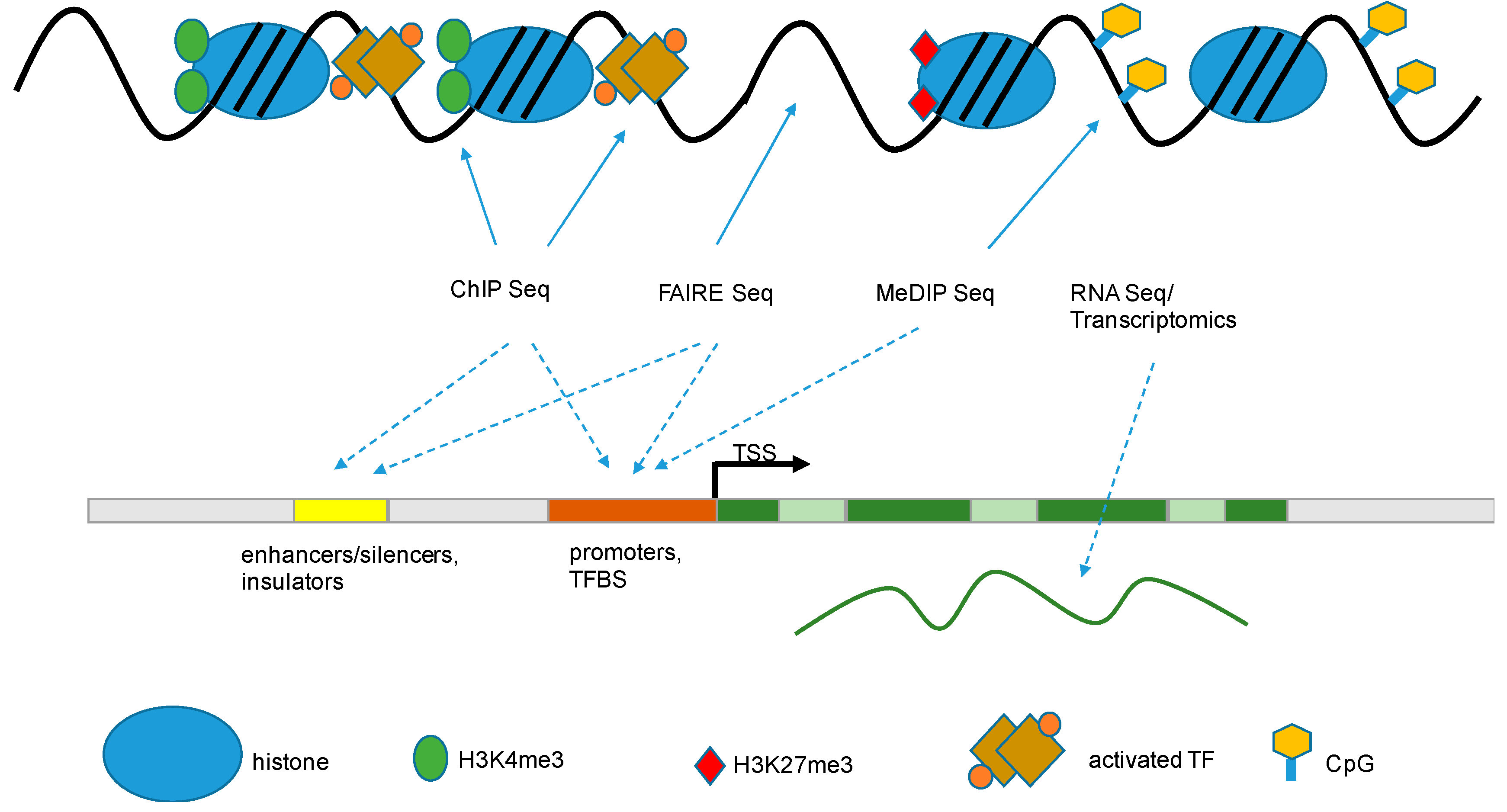


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Modification | Role in Transcription | Modification Site |
|---|---|---|
| Acetylation | Activation | H3ac, H3K9ac, H3K14ac, H3K27ac |
| Methylation | Activation | H3K4me1, H3K4me2, H3K4me3, H3K36me3, H3K79me2 |
| Methylation | Repression | H3K9me3, H3K27me3 |
| Phosphorylation | Activation | H3S10 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schäfer, A.; Baric, R.S. Epigenetic Landscape during Coronavirus Infection. Pathogens 2017, 6, 8. https://doi.org/10.3390/pathogens6010008
Schäfer A, Baric RS. Epigenetic Landscape during Coronavirus Infection. Pathogens. 2017; 6(1):8. https://doi.org/10.3390/pathogens6010008
Chicago/Turabian StyleSchäfer, Alexandra, and Ralph S. Baric. 2017. "Epigenetic Landscape during Coronavirus Infection" Pathogens 6, no. 1: 8. https://doi.org/10.3390/pathogens6010008
APA StyleSchäfer, A., & Baric, R. S. (2017). Epigenetic Landscape during Coronavirus Infection. Pathogens, 6(1), 8. https://doi.org/10.3390/pathogens6010008