Histone Deacetylase 6 Regulates Bladder Architecture and Host Susceptibility to Uropathogenic Escherichia coli

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
2.1. HDAC6−/− Murine Embryonic Fibroblasts Are Resistant to Bacterial Invasion
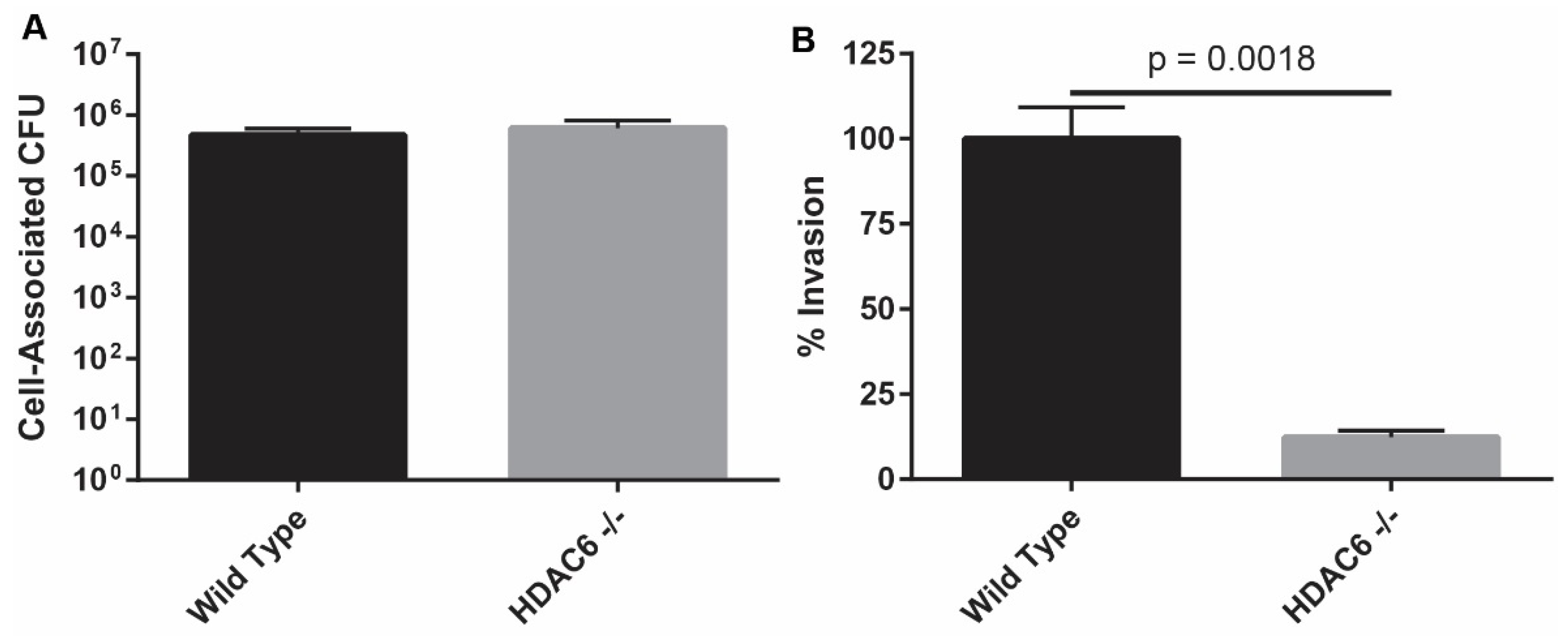
2.2. HDAC6−/−Mice are Transiently More Susceptible to Colonization by UPEC
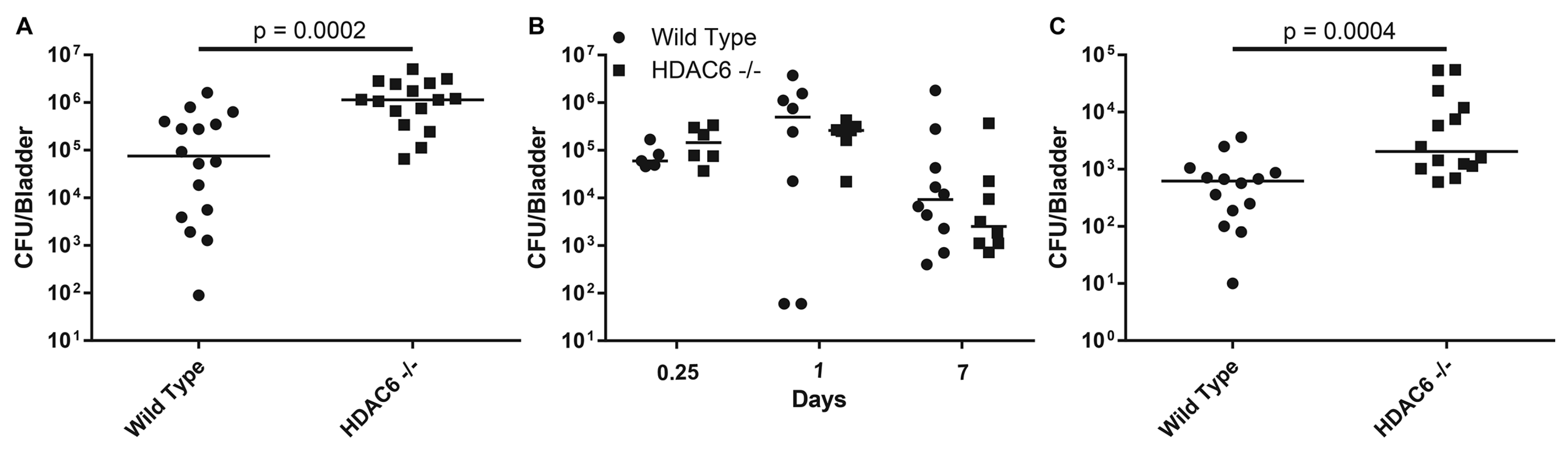
2.3. HDAC6−/− Mice Have Irregular Bladder Morphology and Larger Bladder Volume Capacity
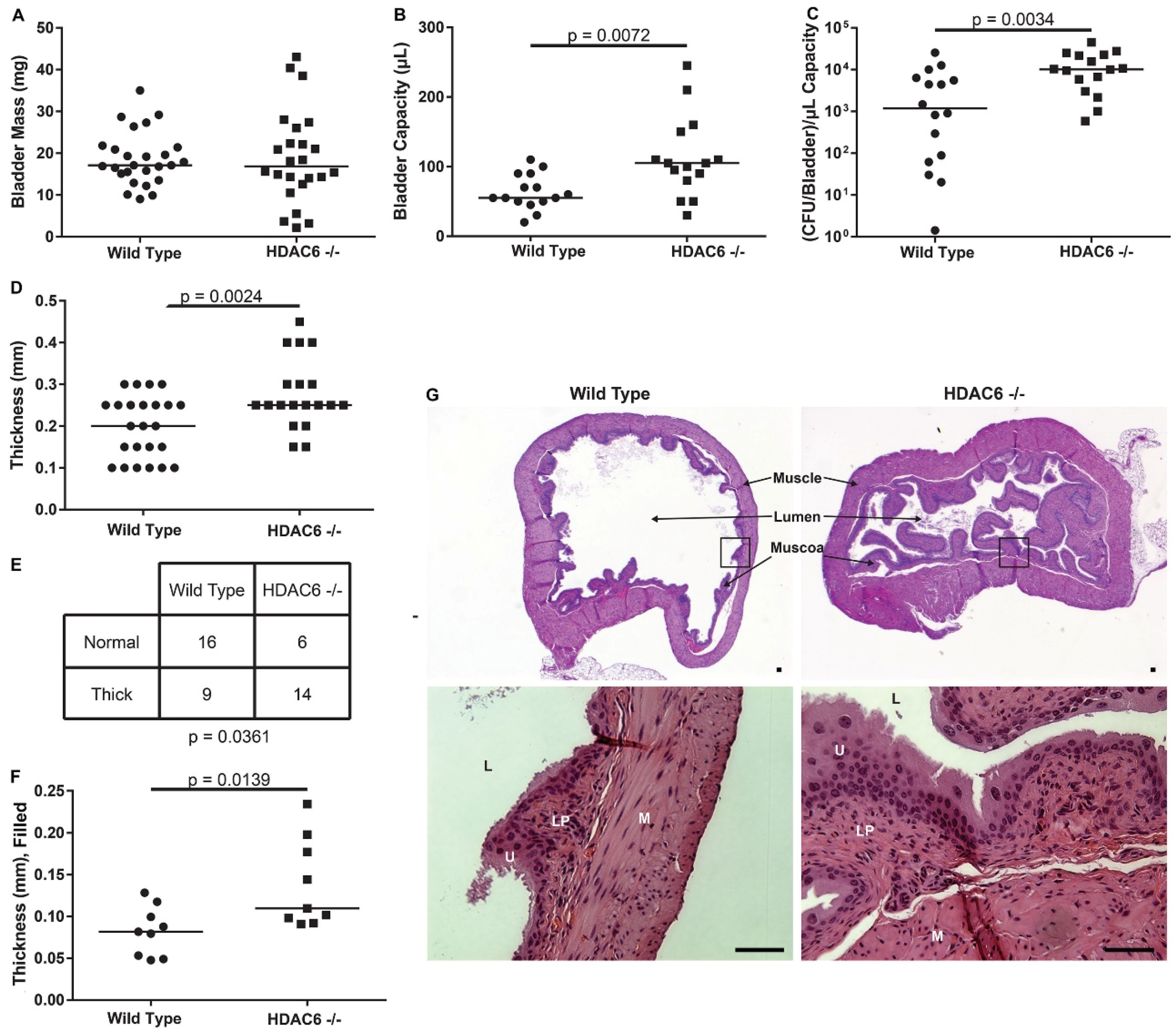
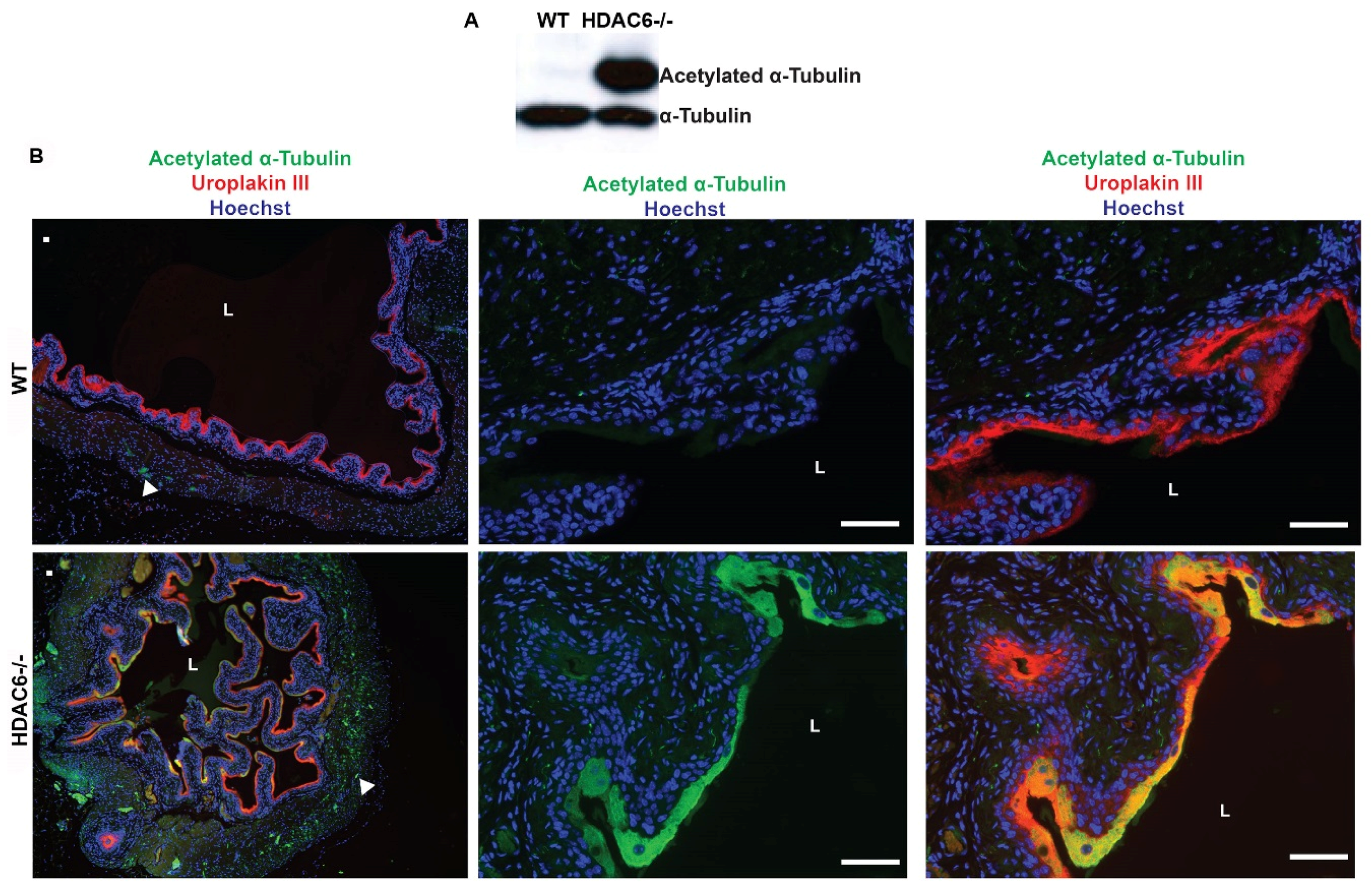
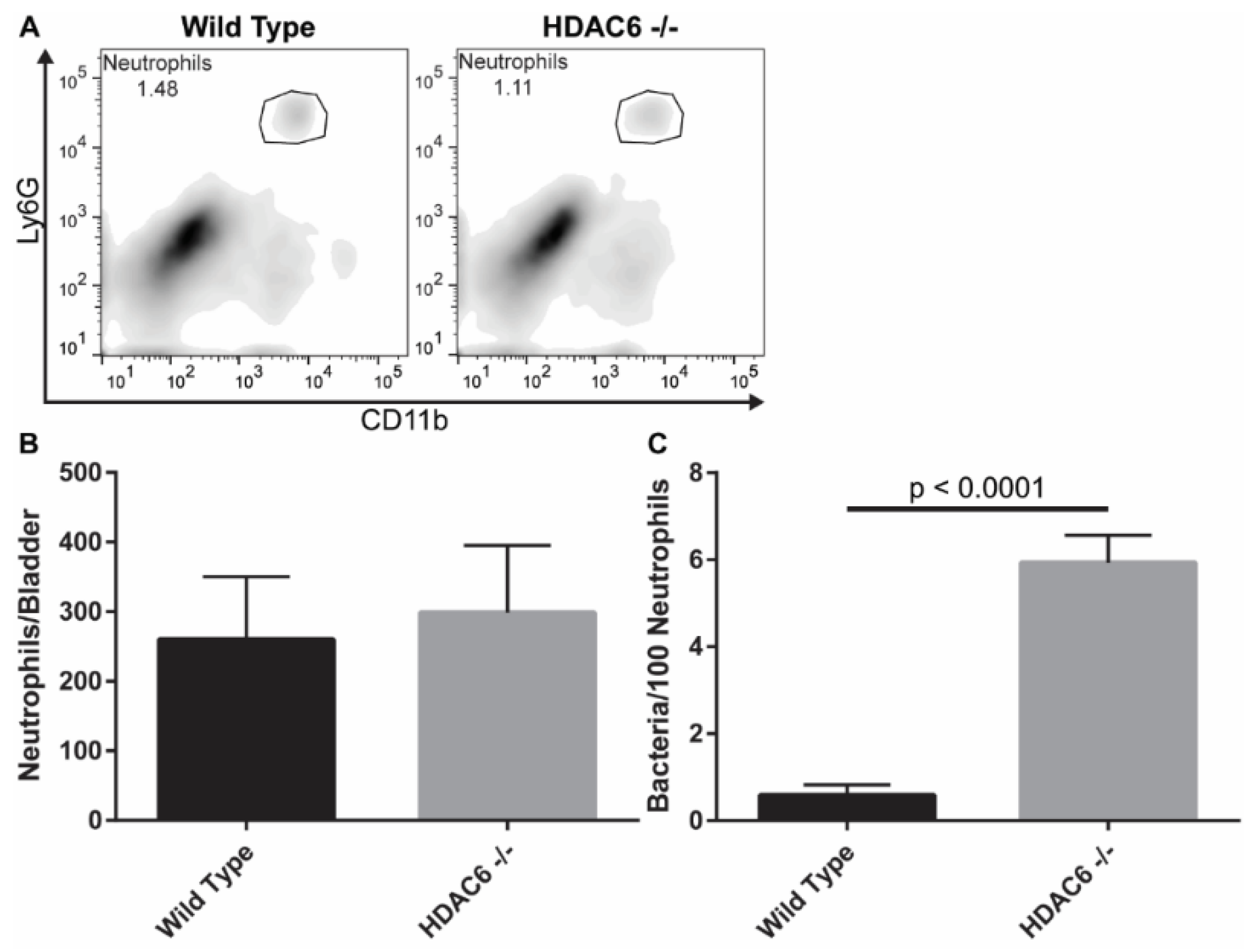
2.4. Neutrophil Recruitment Is Similar, But HDAC6−/− Neutrophils Have More Bacteria
3. Experimental Section
3.1. Chemicals, Reagents, and Antibodies
3.2. Bacterial and Cell Culture
3.3. Mouse Strains and Genotyping
3.4. Gentamicin Protection Assays
3.5. Bladder Catheterization
3.6. Bladder Capacity Measurements
3.7. H&E Staining and Immunofluorescence
3.8. Western Blotting
3.9. Bladder Dissociation and Flow Cytometry
3.10. Statistics
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Barber, A.E.; Norton, J.P.; Spivak, A.M.; Mulvey, M.A. Urinary tract infections: Current and emerging management strategies. Clin. Infect. Dis. 2013, 57, 719–724. [Google Scholar] [CrossRef] [PubMed]
- Dielubanza, E.J.; Schaeffer, A.J. Urinary tract infections in women. Med. Clin. N. Am. 2011, 95, 27–41. [Google Scholar] [CrossRef] [PubMed]
- Barber, A.E.; Mulvey, M.A. Reply to Kaye and Sobel. Clin. Infect. Dis. 2014, 58, 444–445. [Google Scholar] [CrossRef] [PubMed]
- Martinez, J.J.; Mulvey, M.A.; Schilling, J.D.; Pinkner, J.S.; Hultgren, S.J. Type 1 pilus-mediated bacterial invasion of bladder epithelial cells. Embo J. 2000, 19, 2803–2812. [Google Scholar] [CrossRef] [PubMed]
- Dhakal, B.K.; Kulesus, R.R.; Mulvey, M.A. Mechanisms and consequences of bladder cell invasion by uropathogenic Escherichia coli. Eur. J. Clin. Investig. 2008, 38, 2–11. [Google Scholar] [CrossRef] [PubMed]
- Eto, D.S.; Sundsbak, J.L.; Mulvey, M.A. Actin-gated intracellular growth and resurgence of uropathogenic Escherichia coli. Cell Microbiol. 2006, 8, 704–717. [Google Scholar] [PubMed]
- Mysorekar, I.U.; Hultgren, S.J. Mechanisms of uropathogenic Escherichia coli persistence and eradication from the urinary tract. Proc. Natl. Acad. Sci. USA 2006, 103, 14170–14175. [Google Scholar] [CrossRef] [PubMed]
- Blango, M.G.; Mulvey, M.A. Persistence of uropathogenic Escherichia coli in the face of multiple antibiotics. Antimicrob. Agents Chemother. 2010, 54, 1855–1863. [Google Scholar] [CrossRef] [PubMed]
- Mulvey, M.A.; Lopez-Boado, Y.S.; Wilson, C.L.; Roth, R.; Parks, W.C.; Heuser, J.; Hultgren, S.J. Induction and evasion of host defenses by type 1-piliated uropathogenic Escherichia coli. Science 1998, 282, 1494–1497. [Google Scholar]
- Wang, H.; Liang, F.X.; Kong, X.P. Characteristics of the phagocytic cup induced by uropathogenic Escherichia coli. J. Histochem. Cytochem. 2008, 56, 597–604. [Google Scholar] [CrossRef] [PubMed]
- Dhakal, B.K.; Mulvey, M.A. Uropathogenic Escherichia coli invades host cells via an HDAC6-modulated microtubule-dependent pathway. J. Biol. Chem. 2009, 284, 446–454. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Shin, D.; Kwon, S.H. Histone deacetylase 6 plays a role as a distinct regulator of diverse cellular processes. FEBS J. 2013, 280, 775–793. [Google Scholar] [CrossRef] [PubMed]
- Simoes-Pires, C.; Zwick, V.; Nurisso, A.; Schenker, E.; Carrupt, P.A.; Cuendet, M. Hdac6 as a target for neurodegenerative diseases: What makes it different from the other hdacs? Mol. Neurodegener. 2013, 8, 7. [Google Scholar] [CrossRef] [PubMed]
- Yang, P.H.; Zhang, L.; Zhang, Y.J.; Zhang, J.; Xu, W.F. HDAC6: Physiological function and its selective inhibitors for cancer treatment. Drug Discov. Ther. 2013, 7, 233–242. [Google Scholar] [CrossRef] [PubMed]
- Yan, J. Interplay between HDAC6 and its interacting partners: Essential roles in the aggresome-autophagy pathway and neurodegenerative diseases. DNA Cell Biol. 2014, 33, 567–580. [Google Scholar] [CrossRef] [PubMed]
- Valenzuela-Fernandez, A.; Cabrero, J.R.; Serrador, J.M.; Sanchez-Madrid, F. HDAC6: A key regulator of cytoskeleton, cell migration and cell-cell interactions. Trends Cell Biol. 2008, 18, 291–297. [Google Scholar] [CrossRef] [PubMed]
- Valenzuela-Fernandez, A.; Alvarez, S.; Gordon-Alonso, M.; Barrero, M.; Ursa, A.; Cabrero, J.R.; Fernandez, G.; Naranjo-Suarez, S.; Yanez-Mo, M.; Serrador, J.M.; et al. Histone deacetylase 6 regulates human immunodeficiency virus type 1 infection. Mol. Biol. Cell 2005, 16, 5445–5454. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, I.; Miyake, Y.; Nobs, S.P.; Schneider, C.; Horvath, P.; Kopf, M.; Matthias, P.; Helenius, A.; Yamauchi, Y. Influenza a virus uses the aggresome processing machinery for host cell entry. Science 2014, 346, 473–477. [Google Scholar] [CrossRef] [PubMed]
- Husain, M.; Cheung, C.Y. Histone deacetylase 6 inhibits influenza a virus release by downregulating the trafficking of viral components to the plasma membrane via its substrate, acetylated microtubules. J. Virol. 2014, 88, 11229–11239. [Google Scholar] [CrossRef] [PubMed]
- Shin, H.J.; DeCotiis, J.; Giron, M.; Palmeri, D.; Lukac, D.M. Histone deacetylase classes i and ii regulate kaposi's sarcoma-associated herpesvirus reactivation. J. Virol. 2014, 88, 1281–1292. [Google Scholar] [CrossRef] [PubMed]
- Legros, S.; Boxus, M.; Gatot, J.S.; Van Lint, C.; Kruys, V.; Kettmann, R.; Twizere, J.C.; Dequiedt, F. The HTLV-1 tax protein inhibits formation of stress granules by interacting with histone deacetylase 6. Oncogene 2011, 30, 4050–4062. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Coyne, C.B.; Sarkar, S.N. PKC alpha regulates Sendai virus-mediated interferon induction through HDAC6 and beta-catenin. EMBO J. 2011, 30, 4838–4849. [Google Scholar] [CrossRef] [PubMed]
- Chattopadhyay, S.; Fensterl, V.; Zhang, Y.; Veleeparambil, M.; Wetzel, J.L.; Sen, G.C. Inhibition of viral pathogenesis and promotion of the septic shock response to bacterial infection by IRF-3 are regulated by the acetylation and phosphorylation of its coactivators. Mbio 2013, 4, e00636-12. [Google Scholar] [CrossRef] [PubMed]
- Cheng, X.; Liu, Z.; Liu, B.; Zhao, T.; Li, Y.; Alam, H.B. Selective histone deacetylase 6 inhibition prolongs survival in a lethal two-hit model. J. Surg. Res. 2015, 197, 39–44. [Google Scholar] [CrossRef] [PubMed]
- Zhao, T.; Li, Y.; Bronson, R.T.; Liu, B.; Velmahos, G.C.; Alam, H.B. Selective histone deacetylase-6 inhibition attenuates stress responses and prevents immune organ atrophy in a lethal septic model. Surgery 2014, 156, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Thumbikat, P.; Berry, R.E.; Zhou, G.; Billips, B.K.; Yaggie, R.E.; Zaichuk, T.; Sun, T.T.; Schaeffer, A.J.; Klumpp, D.J. Bacteria-induced uroplakin signaling mediates bladder response to infection. PLoS Pathog. 2009. [Google Scholar] [CrossRef] [PubMed]
- Reed, N.A.; Cai, D.; Blasius, T.L.; Jih, G.T.; Meyhofer, E.; Gaertig, J.; Verhey, K.J. Microtubule acetylation promotes kinesin-1 binding and transport. Curr. Biol. 2006, 16, 2166–2172. [Google Scholar] [CrossRef] [PubMed]
- Dompierre, J.P.; Godin, J.D.; Charrin, B.C.; Cordelieres, F.P.; King, S.J.; Humbert, S.; Saudou, F. Histone deacetylase 6 inhibition compensates for the transport deficit in Huntington's disease by increasing tubulin acetylation. J. Neurosci. 2007, 27, 3571–3583. [Google Scholar] [CrossRef] [PubMed]
- Asthana, J.; Kapoor, S.; Mohan, R.; Panda, D. Inhibition of HDAC6 deacetylase activity increases its binding with microtubules and suppresses microtubule dynamic instability in mcf-7 cells. J. Biol. Chem. 2013, 288, 22516–22526. [Google Scholar] [CrossRef] [PubMed]
- Eto, D.S.; Gordon, H.B.; Dhakal, B.K.; Jones, T.A.; Mulvey, M.A. Clathrin, AP-2, and the NPXY-binding subset of alternate endocytic adaptors facilitate FimH-mediated bacterial invasion of host cells. Cell Microbiol. 2008. [Google Scholar] [CrossRef] [PubMed]
- Eto, D.S.; Jones, T.A.; Sundsbak, J.L.; Mulvey, M.A. Integrin-mediated host cell invasion by type 1-piliated uropathogenic Escherichia coli. PLoS Pathog. 2007. [Google Scholar] [CrossRef] [PubMed]
- Antao, E.M.; Ewers, C.; Gurlebeck, D.; Preisinger, R.; Homeier, T.; Li, G.; Wieler, L.H. Signature-tagged mutagenesis in a chicken infection model leads to the identification of a novel avian pathogenic Escherichia coli fimbrial adhesin. PLoS ONE 2009. [Google Scholar] [CrossRef] [PubMed]
- Mossman, K.L.; Mian, M.F.; Lauzon, N.M.; Gyles, C.L.; Lichty, B.; Mackenzie, R.; Gill, N.; Ashkar, A.A. Cutting edge: FimH adhesin of type 1 fimbriae is a novel TLR4 ligand. J. Immunol. 2008, 181, 6702–6706. [Google Scholar] [CrossRef] [PubMed]
- Ashkar, A.A.; Mossman, K.L.; Coombes, B.K.; Gyles, C.L.; Mackenzie, R. FimH adhesin of type 1 fimbriae is a potent inducer of innate antimicrobial responses which requires TLR4 and type 1 interferon signalling. PLoS Pathog. 2008. [Google Scholar] [CrossRef] [PubMed]
- Visvikis, O.; Boyer, L.; Torrino, S.; Doye, A.; Lemonnier, M.; Lores, P.; Rolando, M.; Flatau, G.; Mettouchi, A.; Bouvard, D.; et al. Escherichia coli producing CNF1 toxin hijacks Tollip to trigger Rac1-dependent cell invasion. Traffic 2011, 12, 579–590. [Google Scholar] [CrossRef] [PubMed]
- Smith, Y.C.; Rasmussen, S.B.; Grande, K.K.; Conran, R.M.; O'Brien, A.D. Hemolysin of uropathogenic Escherichia coli evokes extensive shedding of the uroepithelium and hemorrhage in bladder tissue within the first 24 h after intraurethral inoculation of mice. Infect. Immun. 2008, 76, 2978–2990. [Google Scholar] [CrossRef] [PubMed]
- Schiwon, M.; Weisheit, C.; Franken, L.; Gutweiler, S.; Dixit, A.; Meyer-Schwesinger, C.; Pohl, J.M.; Maurice, N.J.; Thiebes, S.; Lorenz, K.; et al. Crosstalk between sentinel and helper macrophages permits neutrophil migration into infected uroepithelium. Cell 2014, 156, 456–468. [Google Scholar] [CrossRef] [PubMed]
- Haraoka, M.; Hang, L.; Frend#us, B.; Godaly, G.; Burdick, M.; Strieter, R.; Svanborg, C. Neutrophil recruitment and resistance to urinary tract infection. J. Infect. Dis. 1999, 180, 1220–1229. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Kwon, S.; Yamaguchi, T.; Cubizolles, F.; Rousseaux, S.; Kneissel, M.; Cao, C.; Li, N.; Cheng, H.L.; Chua, K.; et al. Mice lacking histone deacetylase 6 have hyperacetylated tubulin but are viable and develop normally. Mol. Cell Biol. 2008, 28, 1688–1701. [Google Scholar] [CrossRef] [PubMed]
- Mulvey, M.A.; Schilling, J.D.; Hultgren, S.J. Establishment of a persistent Escherichia coli reservoir during the acute phase of a bladder infection. Infect. Immun. 2001, 69, 4572–4579. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Mendonsa, G.R.; Symington, J.W.; Zhang, Q.; Cadwell, K.; Virgin, H.W.; Mysorekar, I.U. Atg16l1 deficiency confers protection from uropathogenic Escherichia coli infection in vivo. Proc. Natl. Acad. Sci. USA 2012, 109, 11008–11013. [Google Scholar] [CrossRef] [PubMed]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lewis, A.J.; Dhakal, B.K.; Liu, T.; Mulvey, M.A. Histone Deacetylase 6 Regulates Bladder Architecture and Host Susceptibility to Uropathogenic Escherichia coli. Pathogens 2016, 5, 20. https://doi.org/10.3390/pathogens5010020
Lewis AJ, Dhakal BK, Liu T, Mulvey MA. Histone Deacetylase 6 Regulates Bladder Architecture and Host Susceptibility to Uropathogenic Escherichia coli. Pathogens. 2016; 5(1):20. https://doi.org/10.3390/pathogens5010020
Chicago/Turabian StyleLewis, Adam J., Bijaya K. Dhakal, Ting Liu, and Matthew A. Mulvey. 2016. "Histone Deacetylase 6 Regulates Bladder Architecture and Host Susceptibility to Uropathogenic Escherichia coli" Pathogens 5, no. 1: 20. https://doi.org/10.3390/pathogens5010020
APA StyleLewis, A. J., Dhakal, B. K., Liu, T., & Mulvey, M. A. (2016). Histone Deacetylase 6 Regulates Bladder Architecture and Host Susceptibility to Uropathogenic Escherichia coli. Pathogens, 5(1), 20. https://doi.org/10.3390/pathogens5010020