
Subversion of Host Innate Immunity by Uropathogenic Escherichia coli


Abstract
:
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Immune Evasion Strategies of UPEC
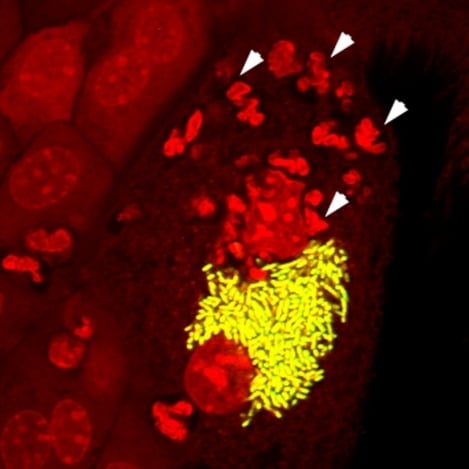
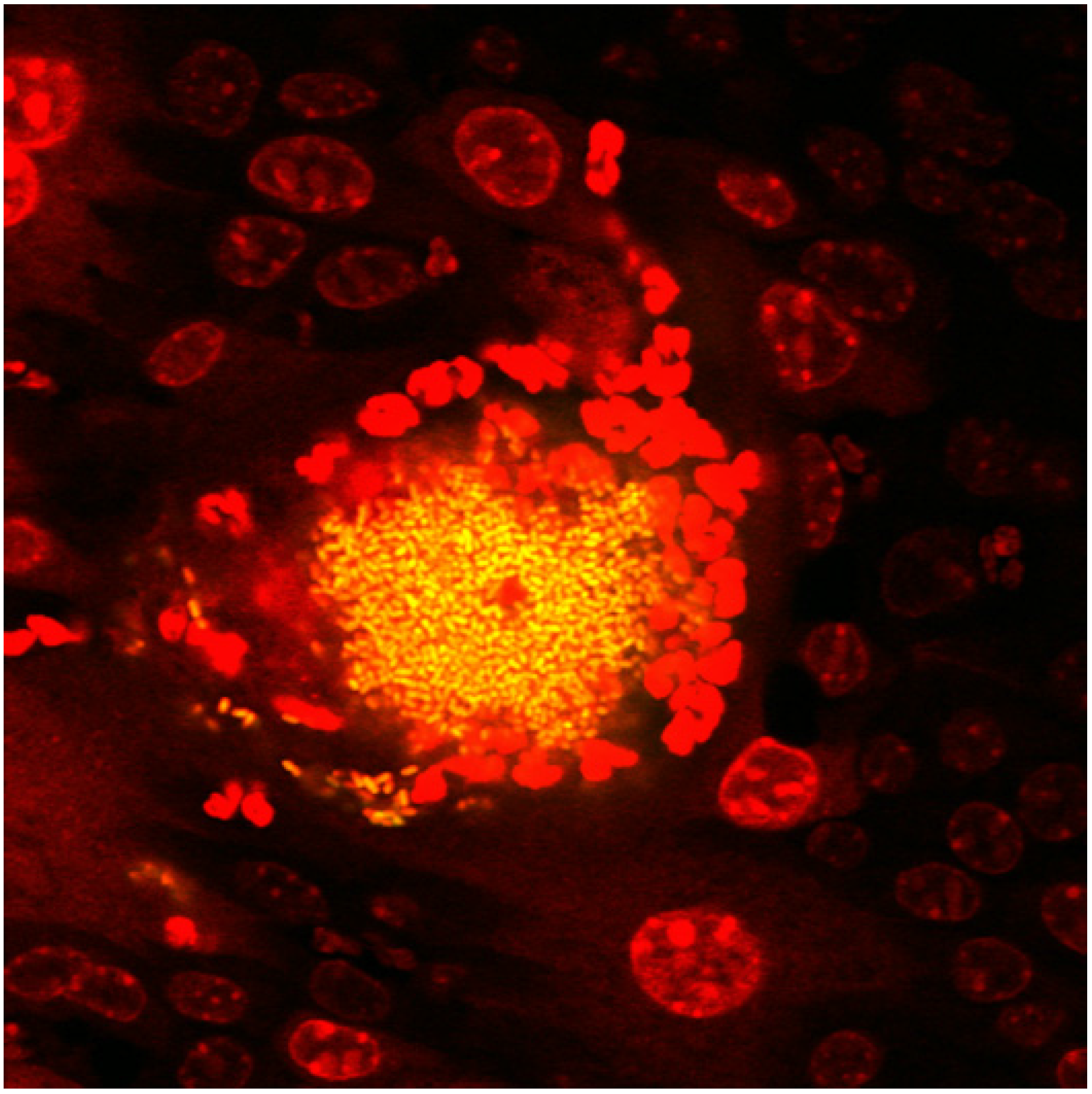
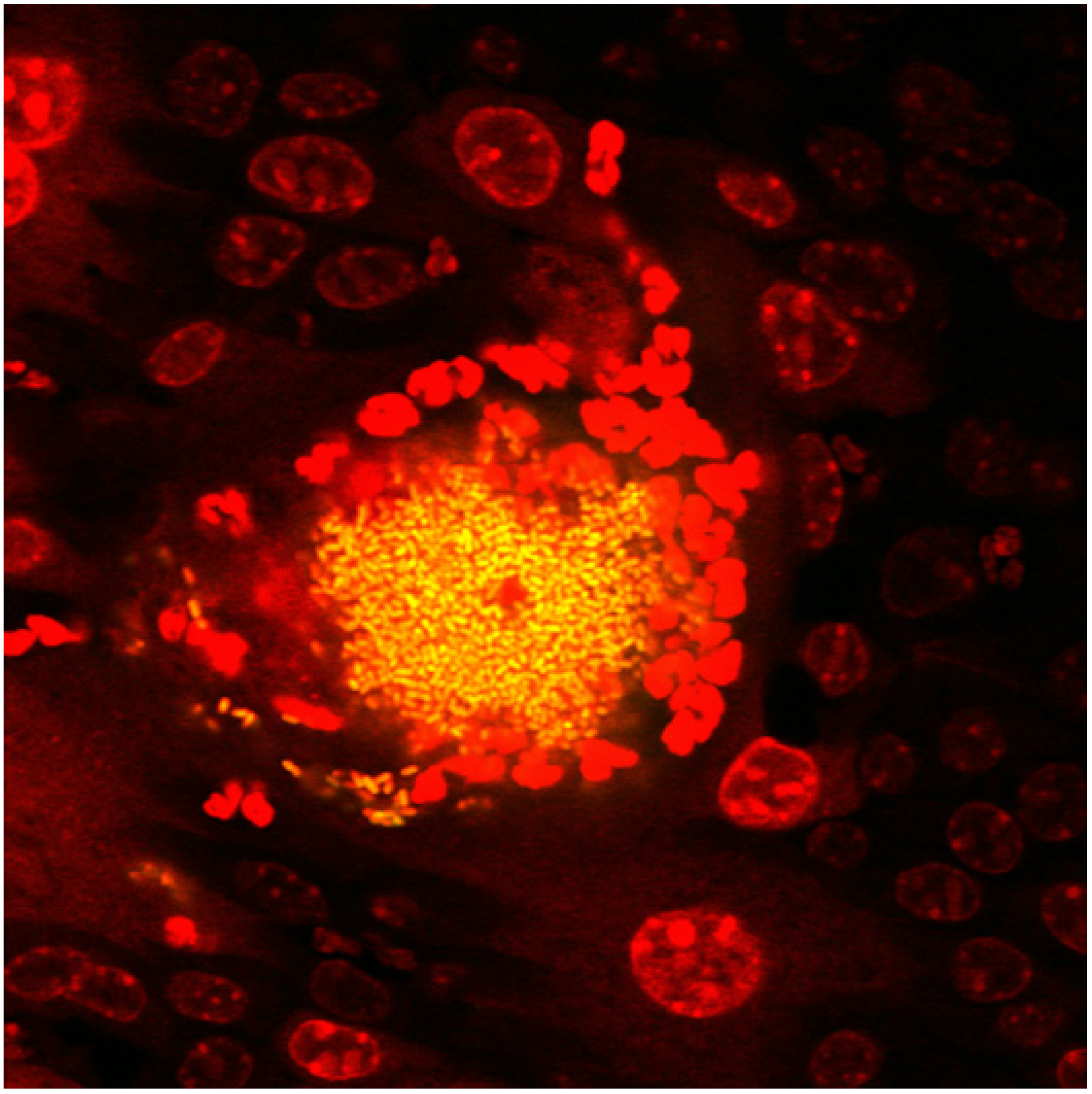
2.1. The UPEC Intracellular Bacterial Community
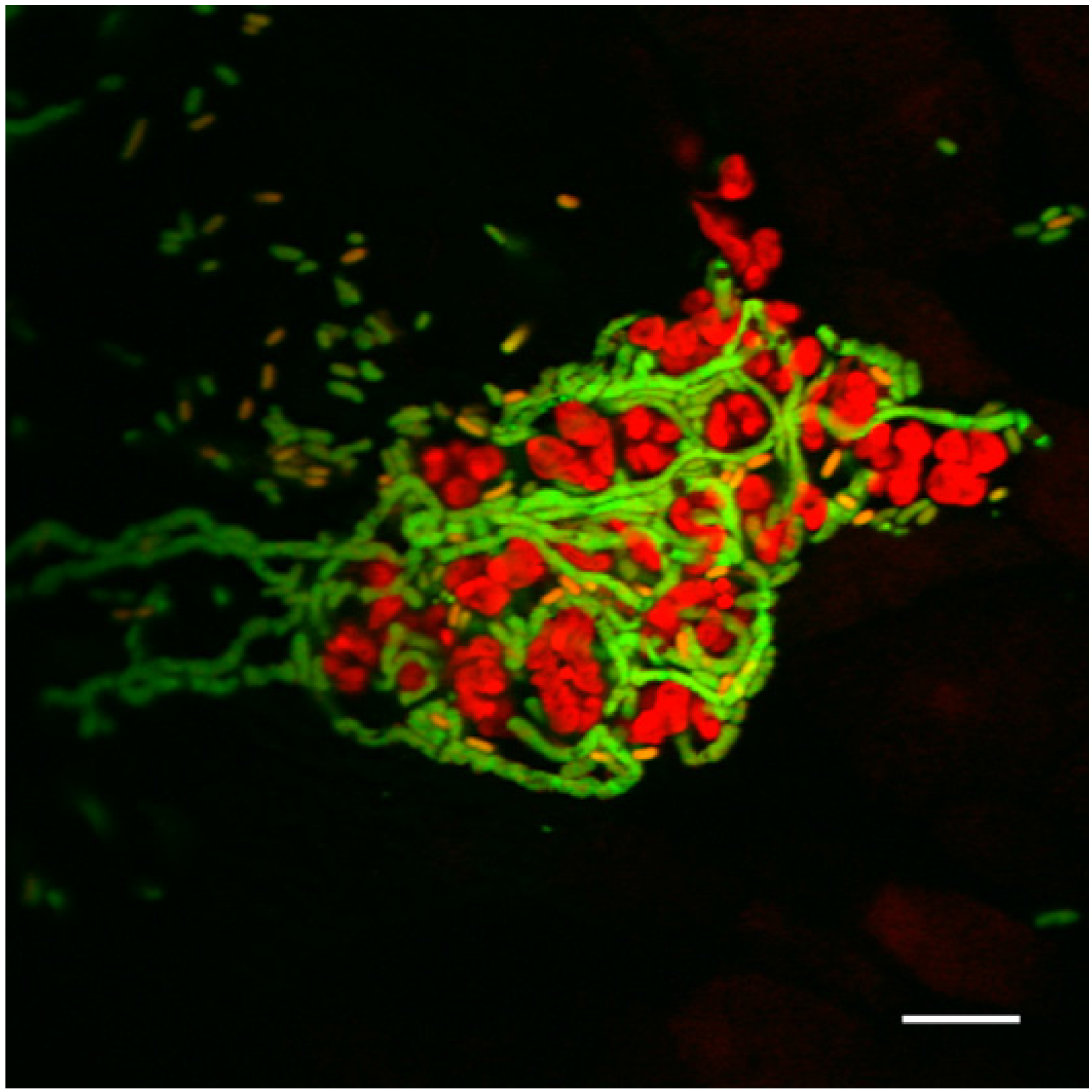
2.2. Filamentation
2.3. UPEC Attenuation of Early Uroepithelial Cytokine Production
2.4. UPEC Inhibition of Neutrophil Recruitment and Function
3. Does Inflammation Ultimately Promote UPEC Infection?
4. Conclusions and Future Directions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Alto, N.M.; Orth, K. Subversion of cell signaling by pathogens. Cold Spring Harb. Perspect. Biol. 2012, 4, a006114. [Google Scholar] [CrossRef] [PubMed]
- Giogha, C.; Lung, T.W.; Pearson, J.S.; Hartland, E.L. Inhibition of death receptor signaling by bacterial gut pathogens. Cytokine Growth Factor Rev. 2014, 25, 235–243. [Google Scholar] [CrossRef] [PubMed]
- Krachler, A.M.; Woolery, A.R.; Orth, K. Manipulation of kinase signaling by bacterial pathogens. J. Cell Biol. 2011, 195, 1083–1092. [Google Scholar] [CrossRef] [PubMed]
- Silke, J.; Hartland, E.L. Masters, marionettes and modulators: Intersection of pathogen virulence factors and mammalian death receptor signaling. Curr. Opin. Immunol. 2013, 25, 436–440. [Google Scholar] [CrossRef] [PubMed]
- Schappert, S.M.; Rechtsteiner, E.A. Ambulatory medical care utilization estimates for 2007. Vital Health Stat. Series 13, Data from the National Health Survey 2011, 169, 1–38. [Google Scholar]
- Litwin, M.S.; Saigal, C.S.; Yano, E.M.; Avila, C.; Geschwind, S.A.; Hanley, J.M.; Joyce, G.F.; Madison, R.; Pace, J.; Polich, S.M.; et al. Urologic diseases in America project: Analytical methods and principal findings. J. Urol. 2005, 173, 933–937. [Google Scholar] [CrossRef] [PubMed]
- Welch, R.A.; Burland, V.; Plunkett, G., 3rd; Redford, P.; Roesch, P.; Rasko, D.; Buckles, E.L.; Liou, S.R.; Boutin, A.; Hackett, J.; et al. Extensive mosaic structure revealed by the complete genome sequence of uropathogenic Escherichia coli. Proc. Natl. Acad. Sci. USA 2002, 99, 17020–17024. [Google Scholar] [CrossRef] [PubMed]
- Hedges, S.; Anderson, P.; Lidin-Janson, G.; de Man, P.; Svanborg, C. Interleukin-6 response to deliberate colonization of the human urinary tract with Gram-negative bacteria. Infect. Immun. 1991, 59, 421–427. [Google Scholar] [PubMed]
- Samuelsson, P.; Hang, L.; Wullt, B.; Irjala, H.; Svanborg, C. Toll-like receptor 4 expression and cytokine responses in the human urinary tract mucosa. Infect. Immun. 2004, 72, 3179–3186. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Duncan, M.J.; Li, G.; Chan, C.; Grady, R.; Stapleton, A.; Abraham, S.N. A novel TLR4-mediated signaling pathway leading to IL-6 responses in human bladder epithelial cells. PLoS Pathog. 2007, 3, e60. [Google Scholar] [CrossRef] [PubMed]
- Andersen-Nissen, E.; Hawn, T.R.; Smith, K.D.; Nachman, A.; Lampano, A.E.; Uematsu, S.; Akira, S.; Aderem, A. Cutting edge: Tlr5−/− mice are more susceptible to Escherichia coli urinary tract infection. J. Immunol. 2007, 178, 4717–4720. [Google Scholar] [CrossRef] [PubMed]
- Smith, N.J.; Varley, C.L.; Eardley, I.; Feather, S.; Trejdosiewicz, L.K.; Southgate, J. Toll-like receptor responses of normal human urothelial cells to bacterial flagellin and lipopolysaccharide. J. Urol. 2011, 186, 1084–1092. [Google Scholar] [CrossRef] [PubMed]
- Peck, A.; Mellins, E.D. Precarious balance: Th17 cells in host defense. Infect. Immun. 2010, 78, 32–38. [Google Scholar] [CrossRef] [PubMed]
- Sivick, K.E.; Schaller, M.A.; Smith, S.N.; Mobley, H.L. The innate immune response to uropathogenic Escherichia coli involves IL-17A in a murine model of urinary tract infection. J. Immunol. 2010, 184, 2065–2075. [Google Scholar] [CrossRef] [PubMed]
- Chromek, M.; Slamova, Z.; Bergman, P.; Kovacs, L.; Podracka, L.; Ehren, I.; Hokfelt, T.; Gudmundsson, G.H.; Gallo, R.L.; Agerberth, B.; et al. The antimicrobial peptide cathelicidin protects the urinary tract against invasive bacterial infection. Nat. Med. 2006, 12, 636–641. [Google Scholar] [CrossRef] [PubMed]
- Danka, E.S.; Hunstad, D.A. Cathelicidin augments epithelial receptivity and pathogenesis in experimental Escherichia coli cystitis. J. Infect. Dis. 2015, 211, 1164–1173. [Google Scholar] [CrossRef] [PubMed]
- Becknell, B.; Spencer, J.D.; Carpenter, A.R.; Chen, X.; Singh, A.; Ploeger, S.; Kline, J.; Ellsworth, P.; Li, B.; Proksch, E.; et al. Expression and antimicrobial function of beta-defensin 1 in the lower urinary tract. PLoS One 2013, 8, e77714. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, K.L.; Dynesen, P.; Larsen, P.; Jakobsen, L.; Andersen, P.S.; Frimodt-Moller, N. Role of urinary cathelicidin LL-37 and human beta-defensin 1 in uncomplicated Escherichia coli urinary tract infections. Infect. Immun. 2014, 82, 1572–1578. [Google Scholar] [CrossRef] [PubMed]
- Shahin, R.D.; Engberg, I.; Hagberg, L.; Svanborg Eden, C. Neutrophil recruitment and bacterial clearance correlated with LPS responsiveness in local Gram-negative infection. J. Immunol. 1987, 138, 3475–3480. [Google Scholar] [PubMed]
- Haraoka, M.; Hang, L.; Frendeus, B.; Godaly, G.; Burdick, M.; Strieter, R.; Svanborg, C. Neutrophil recruitment and resistance to urinary tract infection. J. Infect. Dis. 1999, 180, 1220–1229. [Google Scholar] [CrossRef] [PubMed]
- Artifoni, L.; Negrisolo, S.; Montini, G.; Zucchetta, P.; Molinari, P.P.; Cassar, W.; Destro, R.; Anglani, F.; Rigamonti, W.; Zacchello, G.; et al. Interleukin-8 and CXCR1 receptor functional polymorphisms and susceptibility to acute pyelonephritis. J. Urol. 2007, 177, 1102–1106. [Google Scholar] [CrossRef] [PubMed]
- Ingersoll, M.A.; Kline, K.A.; Nielsen, H.V.; Hultgren, S.J. G-CSF induction early in uropathogenic Escherichia coli infection of the urinary tract modulates host immunity. Cell. Microbiol. 2008, 10, 2568–2578. [Google Scholar] [CrossRef] [PubMed]
- Svensson, M.; Irjala, H.; Svanborg, C.; Godaly, G. Effects of epithelial and neutrophil CXCR2 on innate immunity and resistance to kidney infection. Kidney Int. 2008, 74, 81–90. [Google Scholar] [CrossRef] [PubMed]
- Svensson, M.; Yadav, M.; Holmqvist, B.; Lutay, N.; Svanborg, C.; Godaly, G. Acute pyelonephritis and renal scarring are caused by dysfunctional innate immunity in mCXCR2 heterozygous mice. Kidney Int. 2011, 80, 1064–1072. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Bishop, B.L.; Li, G.; Duncan, M.J.; Abraham, S.N. TLR4-initiated and cAMP-mediated abrogation of bacterial invasion of the bladder. Cell Host Microbe 2007, 1, 287–298. [Google Scholar] [CrossRef] [PubMed]
- Wright, K.J.; Seed, P.C.; Hultgren, S.J. Development of intracellular bacterial communities of uropathogenic Escherichia coli depends on type 1 pili. Cell. Microbiol. 2007, 9, 2230–2241. [Google Scholar] [CrossRef] [PubMed]
- Eto, D.S.; Gordon, H.B.; Dhakal, B.K.; Jones, T.A.; Mulvey, M.A. Clathrin, AP-2, and the NPXY-binding subset of alternate endocytic adaptors facilitate FimH-mediated bacterial invasion of host cells. Cell. Microbiol. 2008, 10, 2553–2567. [Google Scholar] [CrossRef] [PubMed]
- Schilling, J.D.; Mulvey, M.A.; Vincent, C.D.; Lorenz, R.G.; Hultgren, S.J. Bacterial invasion augments epithelial cytokine responses to Escherichia coli through a lipopolysaccharide-dependent mechanism. J. Immunol. 2001, 166, 1148–1155. [Google Scholar] [CrossRef] [PubMed]
- Berry, R.E.; Klumpp, D.J.; Schaeffer, A.J. Urothelial cultures support intracellular bacterial community formation by uropathogenic Escherichia coli. Infect. Immun. 2009, 77, 2762–2772. [Google Scholar] [CrossRef] [PubMed]
- Anderson, G.G.; Palermo, J.J.; Schilling, J.D.; Roth, R.; Heuser, J.; Hultgren, S.J. Intracellular bacterial biofilm-like pods in urinary tract infections. Science 2003, 301, 105–107. [Google Scholar] [CrossRef] [PubMed]
- Martinez, J.J.; Mulvey, M.A.; Schilling, J.D.; Pinkner, J.S.; Hultgren, S.J. Type 1 pilus-mediated bacterial invasion of bladder epithelial cells. EMBO J. 2000, 19, 2803–2812. [Google Scholar] [CrossRef] [PubMed]
- Mulvey, M.A.; Schilling, J.D.; Hultgren, S.J. Establishment of a persistent Escherichia coli reservoir during the acute phase of a bladder infection. Infect. Immun. 2001, 69, 4572–4579. [Google Scholar] [CrossRef] [PubMed]
- Eto, D.S.; Jones, T.A.; Sundsbak, J.L.; Mulvey, M.A. Integrin-mediated host cell invasion by type 1-piliated uropathogenic Escherichia coli. PLoS Pathog. 2007, 3, e100. [Google Scholar] [CrossRef] [PubMed]
- Duncan, M.J.; Li, G.; Shin, J.S.; Carson, J.L.; Abraham, S.N. Bacterial penetration of bladder epithelium through lipid rafts. J. Biol. Chem. 2004, 279, 18944–18951. [Google Scholar] [CrossRef] [PubMed]
- Bishop, B.L.; Duncan, M.J.; Song, J.; Li, G.; Zaas, D.; Abraham, S.N. Cyclic AMP-regulated exocytosis of Escherichia coli from infected bladder epithelial cells. Nat. Med. 2007, 13, 625–630. [Google Scholar] [CrossRef] [PubMed]
- Eto, D.S.; Sundsbak, J.L.; Mulvey, M.A. Actin-gated intracellular growth and resurgence of uropathogenic Escherichia coli. Cell. Microbiol. 2006, 8, 704–717. [Google Scholar] [CrossRef] [PubMed]
- Justice, S.S.; Hung, C.; Theriot, J.A.; Fletcher, D.A.; Anderson, G.G.; Footer, M.J.; Hultgren, S.J. Differentiation and developmental pathways of uropathogenic Escherichia coli in urinary tract pathogenesis. Proc. Natl. Acad. Sci. USA 2004, 101, 1333–1338. [Google Scholar] [CrossRef] [PubMed]
- Robino, L.; Scavone, P.; Araujo, L.; Algorta, G.; Zunino, P.; Pirez, M.C.; Vignoli, R. Intracellular bacteria in the pathogenesis of Escherichia coli urinary tract infection in children. Clin. Infect. Dis. 2014, 59, e158–e164. [Google Scholar] [CrossRef] [PubMed]
- Robino, L.; Scavone, P.; Araujo, L.; Algorta, G.; Zunino, P.; Vignoli, R. Detection of intracellular bacterial communities in a child with Escherichia coli recurrent urinary tract infections. Pathog. Dis. 2013, 68, 78–81. [Google Scholar] [CrossRef] [PubMed]
- Rosen, D.A.; Hooton, T.M.; Stamm, W.E.; Humphrey, P.A.; Hultgren, S.J. Detection of intracellular bacterial communities in human urinary tract infection. PLoS Med. 2007, 4, e329. [Google Scholar] [CrossRef] [PubMed]
- Nicholson, T.F.; Watts, K.M.; Hunstad, D.A. OmpA of uropathogenic Escherichia coli promotes postinvasion pathogenesis of cystitis. Infect. Immun. 2009, 77, 5245–5251. [Google Scholar] [CrossRef] [PubMed]
- Justice, S.S.; Hunstad, D.A.; Seed, P.C.; Hultgren, S.J. Filamentation by Escherichia coli subverts innate defenses during urinary tract infection. Proc. Natl. Acad. Sci. USA 2006, 103, 19884–19889. [Google Scholar] [CrossRef] [PubMed]
- Horvath, D.J., Jr.; Li, B.; Casper, T.; Partida-Sanchez, S.; Hunstad, D.A.; Hultgren, S.J.; Justice, S.S. Morphological plasticity promotes resistance to phagocyte killing of uropathogenic Escherichia coli. Microbes Infect. 2011, 13, 426–437. [Google Scholar] [CrossRef] [PubMed]
- Justice, S.S.; Hunstad, D.A.; Cegelski, L.; Hultgren, S.J. Morphological plasticity as a bacterial survival strategy. Nat. Rev. Microbiol. 2008, 6, 162–168. [Google Scholar] [CrossRef] [PubMed]
- Champion, J.A.; Mitragotri, S. Role of target geometry in phagocytosis. Proc. Natl. Acad. Sci. USA 2006, 103, 4930–4934. [Google Scholar] [CrossRef] [PubMed]
- Klumpp, D.J.; Weiser, A.C.; Sengupta, S.; Forrestal, S.G.; Batler, R.A.; Schaeffer, A.J. Uropathogenic Escherichia coli potentiates type 1 pilus-induced apoptosis by suppressing NF-κB. Infect. Immun. 2001, 69, 6689–6695. [Google Scholar] [CrossRef] [PubMed]
- Billips, B.K.; Forrestal, S.G.; Rycyk, M.T.; Johnson, J.R.; Klumpp, D.J.; Schaeffer, A.J. Modulation of host innate immune response in the bladder by uropathogenic Escherichia coli. Infect. Immun. 2007, 75, 5353–5360. [Google Scholar] [CrossRef] [PubMed]
- Hunstad, D.A.; Justice, S.S.; Hung, C.S.; Lauer, S.R.; Hultgren, S.J. Suppression of bladder epithelial cytokine responses by uropathogenic Escherichia coli. Infect. Immun. 2005, 73, 3999–4006. [Google Scholar] [CrossRef] [PubMed]
- Billips, B.K.; Schaeffer, A.J.; Klumpp, D.J. Molecular basis of uropathogenic Escherichia coli evasion of the innate immune response in the bladder. Infect. Immun. 2008, 76, 3891–3900. [Google Scholar] [CrossRef] [PubMed]
- Vertommen, D.; Ruiz, N.; Leverrier, P.; Silhavy, T.J.; Collet, J.F. Characterization of the role of the Escherichia coli periplasmic chaperone SurA using differential proteomics. Proteomics 2009, 9, 2432–2443. [Google Scholar] [CrossRef] [PubMed]
- Hilbert, D.W.; Pascal, K.E.; Libby, E.K.; Mordechai, E.; Adelson, M.E.; Trama, J.P. Uropathogenic Escherichia coli dominantly suppress the innate immune response of bladder epithelial cells by a lipopolysaccharide- and Toll-like receptor 4-independent pathway. Microbes Infect. 2008, 10, 114–121. [Google Scholar] [CrossRef] [PubMed]
- Dhakal, B.K.; Mulvey, M.A. The UPEC pore-forming toxin α-hemolysin triggers proteolysis of host proteins to disrupt cell adhesion, inflammatory, and survival pathways. Cell Host Microbe 2012, 11, 58–69. [Google Scholar] [CrossRef] [PubMed]
- Hilbert, D.W.; Paulish-Miller, T.E.; Tan, C.K.; Carey, A.J.; Ulett, G.C.; Mordechai, E.; Adelson, M.E.; Gygax, S.E.; Trama, J.P. Clinical Escherichia coli isolates utilize α-hemolysin to inhibit in vitro epithelial cytokine production. Microbes Infect. 2012, 14, 628–638. [Google Scholar] [CrossRef] [PubMed]
- Cirl, C.; Wieser, A.; Yadav, M.; Duerr, S.; Schubert, S.; Fischer, H.; Stappert, D.; Wantia, N.; Rodriguez, N.; Wagner, H.; et al. Subversion of Toll-like receptor signaling by a unique family of bacterial Toll/interleukin-1 receptor domain-containing proteins. Nat. Med. 2008, 14, 399–406. [Google Scholar] [CrossRef] [PubMed]
- Yadav, M.; Zhang, J.; Fischer, H.; Huang, W.; Lutay, N.; Cirl, C.; Lum, J.; Miethke, T.; Svanborg, C. Inhibition of tir domain signaling by TcpC: MyD88-dependent and independent effects on Escherichia coli virulence. PLoS Pathog. 2010, 6, e1001120. [Google Scholar] [CrossRef] [PubMed]
- Lloyd, A.L.; Smith, S.N.; Eaton, K.A.; Mobley, H.L. Uropathogenic Escherichia coli suppresses the host inflammatory response via pathogenicity island genes SisA and SisB. Infect. Immun. 2009, 77, 5322–5333. [Google Scholar] [CrossRef] [PubMed]
- Loughman, J.A.; Hunstad, D.A. Attenuation of human neutrophil migration and function by uropathogenic bacteria. Microbes Infect. 2011, 13, 555–565. [Google Scholar] [CrossRef] [PubMed]
- Lau, M.E.; Hunstad, D.A. Quantitative assessment of human neutrophil migration across a cultured bladder epithelium. J. Vis. Exp. 2013, e50919. [Google Scholar] [CrossRef] [PubMed]
- Lau, M.E.; Loughman, J.A.; Hunstad, D.A. YbcL of uropathogenic Escherichia coli suppresses transepithelial neutrophil migration. Infect. Immun. 2012, 80, 4123–4132. [Google Scholar] [CrossRef] [PubMed]
- Mellor, A.L.; Munn, D.H. IDO expression by dendritic cells: Tolerance and tryptophan catabolism. Nat. Rev. Immunol. 2004, 4, 762–774. [Google Scholar] [CrossRef] [PubMed]
- Loughman, J.A.; Hunstad, D.A. Induction of indoleamine 2,3-dioxygenase by uropathogenic bacteria attenuates innate responses to epithelial infection. J. Infect. Dis. 2012, 205, 1830–1839. [Google Scholar] [CrossRef] [PubMed]
- Cavalieri, S.J.; Snyder, I.S. Effect of Escherichia coli α-hemolysin on human peripheral leukocyte viability in vitro. Infect. Immun. 1982, 36, 455–461. [Google Scholar] [PubMed]
- Russo, T.A.; Davidson, B.A.; Genagon, S.A.; Warholic, N.M.; Macdonald, U.; Pawlicki, P.D.; Beanan, J.M.; Olson, R.; Holm, B.A.; Knight, P.R. E. coli virulence factor hemolysin induces neutrophil apoptosis and necrosis/lysis in vitro and necrosis/lysis and lung injury in a rat pneumonia model. Am. J. Physiol. Lung Cell. Mol. Biol. 2005, 289, L207–L216. [Google Scholar] [CrossRef] [PubMed]
- Davis, J.M.; Rasmussen, S.B.; O’Brien, A.D. Cytotoxic necrotizing factor type 1 production by uropathogenic Escherichia coli modulates polymorphonuclear leukocyte function. Infect. Immun. 2005, 73, 5301–5310. [Google Scholar] [CrossRef] [PubMed]
- Davis, J.M.; Carvalho, H.M.; Rasmussen, S.B.; O’Brien, A.D. Cytotoxic necrotizing factor type 1 delivered by outer membrane vesicles of uropathogenic Escherichia coli attenuates polymorphonuclear leukocyte antimicrobial activity and chemotaxis. Infect. Immun. 2006, 74, 4401–4408. [Google Scholar] [CrossRef] [PubMed]
- Lau, M.E.; Danka, E.S.; Tiemann, K.M.; Hunstad, D.A. Bacterial lysis liberates the neutrophil migration suppressor YbcL from the periplasm of uropathogenic Escherichia coli. Infect. Immun. 2014, 82, 4921–4930. [Google Scholar] [CrossRef] [PubMed]
- Hagberg, L.; Hull, R.; Hull, S.; McGhee, J.R.; Michalek, S.M.; Svanborg Eden, C. Difference in susceptibility to Gram-negative urinary tract infection between C3H/HeJ and C3H/HeN mice. Infect. Immun. 1984, 46, 839–844. [Google Scholar] [PubMed]
- Godaly, G.; Bergsten, G.; Hang, L.; Fischer, H.; Frendeus, B.; Lundstedt, A.C.; Samuelsson, M.; Samuelsson, P.; Svanborg, C. Neutrophil recruitment, chemokine receptors, and resistance to mucosal infection. J. Leukoc. Biol. 2001, 69, 899–906. [Google Scholar] [PubMed]
- Hannan, T.J.; Mysorekar, I.U.; Hung, C.S.; Isaacson-Schmid, M.L.; Hultgren, S.J. Early severe inflammatory responses to uropathogenic E. coli predispose to chronic and recurrent urinary tract infection. PLoS Pathog. 2010, 6, e1001042. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, D.J.; Conover, M.S.; Hannan, T.J.; Hultgren, S.J. Uropathogenic Escherichia coli superinfection enhances the severity of mouse bladder infection. PLoS Pathog. 2015, 11, e1004599. [Google Scholar] [CrossRef] [PubMed]
- Hannan, T.J.; Roberts, P.L.; Riehl, T.E.; van der Post, S.; Binkley, J.M.; Schwartz, D.J.; Miyoshi, H.; Mack, M.; Schwendener, R.A.; Hooton, T.M.; et al. Inhibition of cyclooxygenase-2 prevents chronic and recurrent cystitis. EBiomedicine 2014, 1, 46–57. [Google Scholar] [CrossRef] [PubMed]
- Serre, L.; Pereira de Jesus, K.; Zelwer, C.; Bureaud, N.; Schoentgen, F.; Benedetti, H. Crystal structures of YbhB and YbcL from Escherichia coli, two bacterial homologues to a Raf kinase inhibitor protein. J. Mol. Biol. 2001, 310, 617–634. [Google Scholar] [CrossRef] [PubMed]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Olson, P.D.; Hunstad, D.A. Subversion of Host Innate Immunity by Uropathogenic Escherichia coli. Pathogens 2016, 5, 2. https://doi.org/10.3390/pathogens5010002
Olson PD, Hunstad DA. Subversion of Host Innate Immunity by Uropathogenic Escherichia coli. Pathogens. 2016; 5(1):2. https://doi.org/10.3390/pathogens5010002
Chicago/Turabian StyleOlson, Patrick D., and David A. Hunstad. 2016. "Subversion of Host Innate Immunity by Uropathogenic Escherichia coli" Pathogens 5, no. 1: 2. https://doi.org/10.3390/pathogens5010002
APA StyleOlson, P. D., & Hunstad, D. A. (2016). Subversion of Host Innate Immunity by Uropathogenic Escherichia coli. Pathogens, 5(1), 2. https://doi.org/10.3390/pathogens5010002