Identifying Pathogenicity Islands in Bacterial Pathogenomics Using Computational Approaches

Abstract
:
1. Introduction
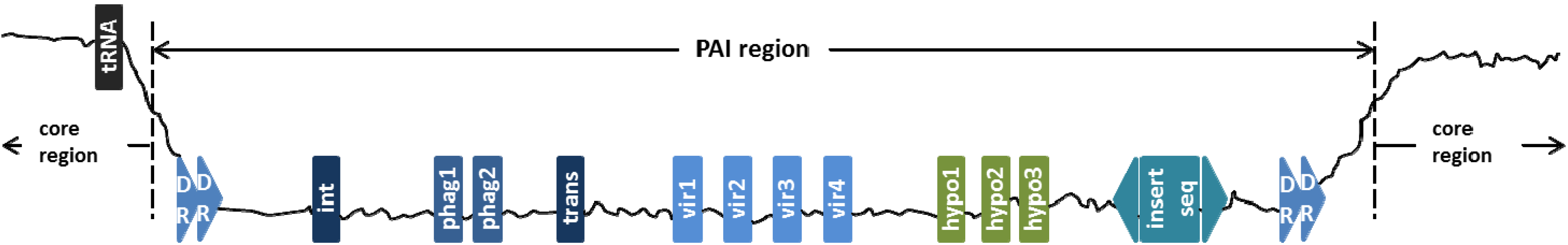
2. PAIs-Related Features
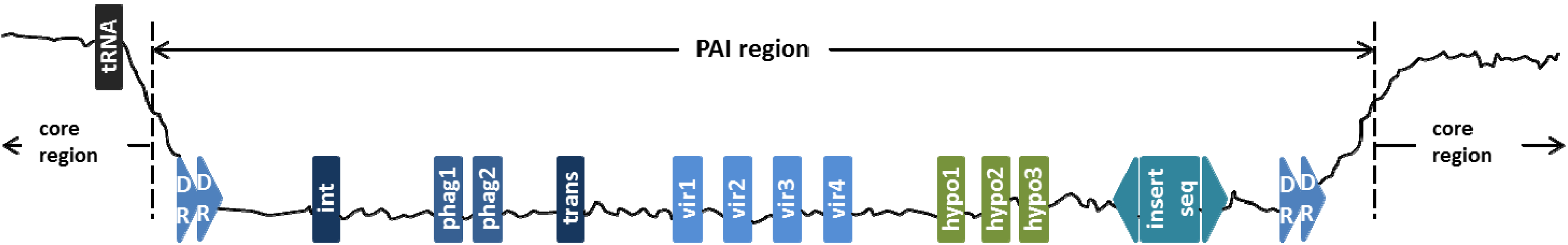

{kind=link}
{kind=link}
{kind=link}
| PAI-Associated Features | Feature Measurement Methods |
|---|---|
| Different genomic sequence signature | Compute G+C content, GC-skew, codon usage, or other sequence signature tools |
| Presence of virulence factors | Search through virulence factor database such as VFDB |
| Presence of mobility genes (integrases, transposes) | Search through NCBI-nr/nt, UniprotKB, Pfam or COG database |
| High percentage of phage-related genes | Search through NCBI-nr/nt, UniprotKB, Pfam or COG database |
| Presence of tRNA genes | Use tRNA gene search tool of tRNAscan-SE |
| High percentage of hypothetic protein genes | Search through NCBI-nr/nt, UniprotKB, Pfam or COG database |
| Presence of direct repeats | Use repeat finder software REPuter |
| Presence of insertion sequences | Search through ISfinder database |
2.1. Genomic Sequence Signature
2.1.1. G+C Content and GC-Skew
2.1.2. k-Mer Frequency
2.1.3. Codon Usage
2.1.4. Caveat
2.2. Virulence-Associated Genes
2.3. Mobility Genes
2.4. Phage-Related Genes
2.5. Transfer RNA
2.6. Hypothetical Protein Genes
2.7. Direct Repeat
2.8. Insertion Sequence (IS) Elements
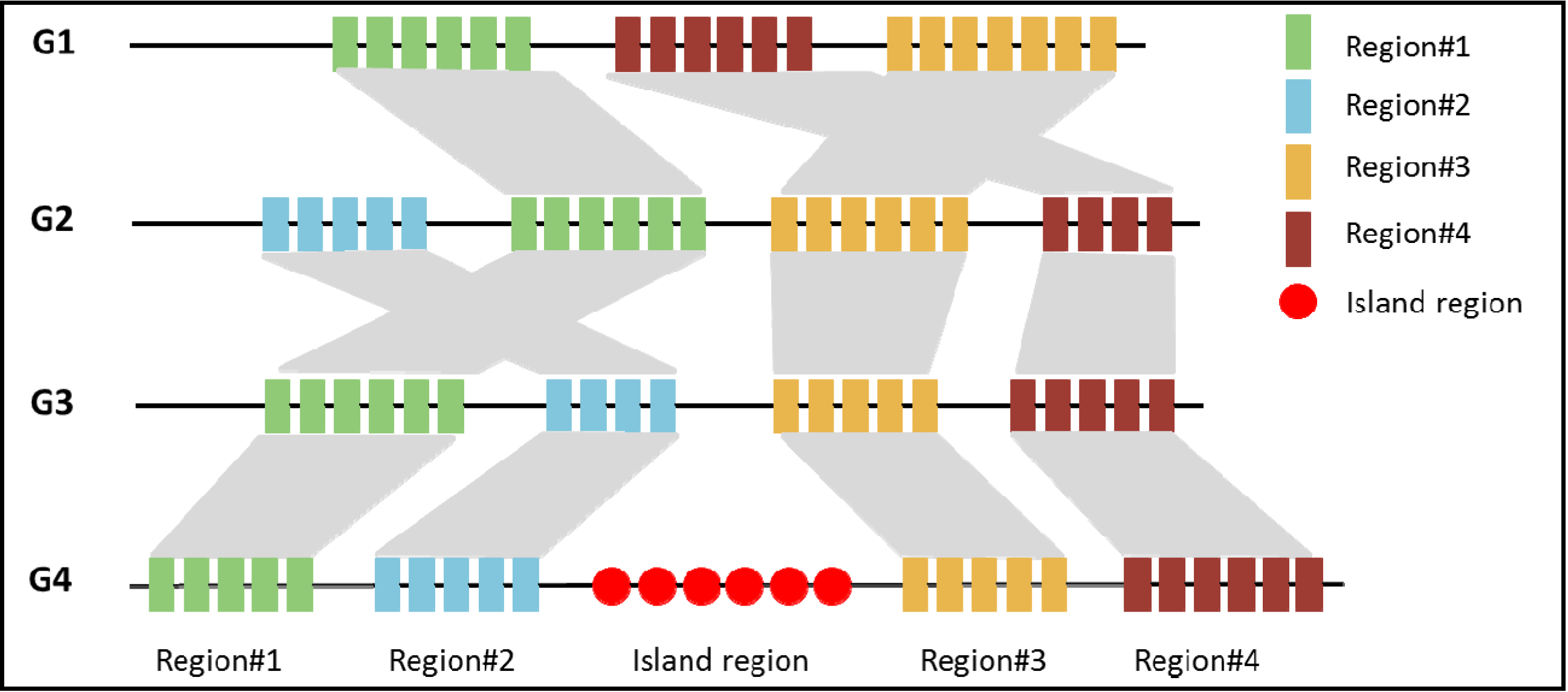
3. Computational Identification of PAIs
3.1. Comparative Genomics Based Approach
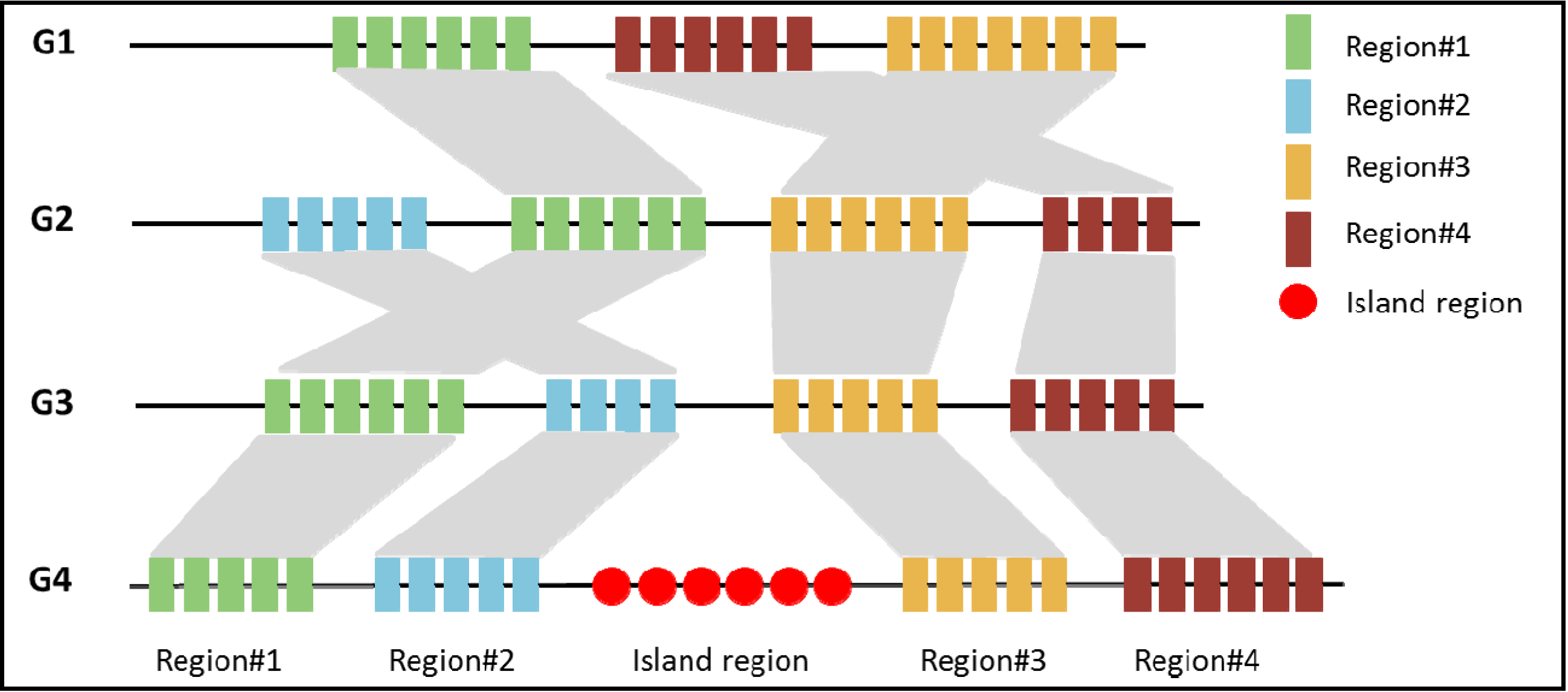
3.2. Sequence Composition Based Approach
| Software a | Main Principle | System Setup b | Website |
|---|---|---|---|
| AlienHunter | HMMs on various mer words | Unix/Linux OS, Java and Perl environment setup | http://www.sanger.ac.uk/resources/software/alien_hunter/ |
| Centroid | Centroid on k-mer word | Unix/Linux OS and C++ environment setup | Upon request |
| EGID | Ensembles the results of AlienHunter, IslandPath, SIGI-HMM, INDeGenIUS and PAI-IDA | Unix/Linux OS, Java, C++ and Perl environment setup | http://www5.esu.edu/cpsc/bioinfo/software/EGID |
| GIDetector | Decision-tree based bagging on IVOM score, insertion point, size, gene density, repeats, integrase, phage and non-coding RNA | Windows OS, C# with the support of Perl and Cygwin | http://www5.esu.edu/cpsc/bioinfo/software/GIDetector/ |
| GI-GPS | SVMs on sequence composition (including GC content, dinucleotide frequency, codon usage, and codon adaption usage), and with filtering steps including length of candidate segment, tRNA and repeat elements | Not available | http://gipop.life.nthu.edu.tw |
| GIHunter | Decision tree based bagging model using sequence composition, gene information and inter-genic distance, mobile genes, phage genes, tRNA, and gene density | Unix/Linux OS, Java, C++ and Perl environment setup | http://www5.esu.edu/cpsc/bioinfo/software/GIHunter |
| INDeGenIUS | Clustering/Centroid on k-mer word | Unix/Linux OS and C++ environment setup | Upon request |
| IslandPath | G+C, dinucleotide, mobile genes, and codon usage | Unix/Linux OS and Perl environment setup | http://www.pathogenomics.sfu.ca/islandpath |
| PAI-IDA | Discriminant analysis on G+C, dinucleotide and codon usage | Unix/Linux OS, C++ and Perl environment setup | http://compbio.sibsnet.org/projects/pai-ida |
| PIPS | G+C content, codon usage deviation, virulence factors, hypothetical proteins, transposases, flanking tRNA and its absence in nonpathogenic organisms | Unix/Linux OS and Perl environment setup | http://www.genoma.ufpa.br/lgcm/pips |
| SIGI-HMM | HMM on codon usage | Unix/Linux OS and Java, environment setup | http://www.tcs.informatik.uni-goettingen.de/colombo |
4. Databases and Related Computational Resources
| Category | Description | Website |
|---|---|---|
| GI Databases/Servers | ||
| DGI | A database that contains genomic islands of more than 2,000 bacterial genomes, many of which are PAIs, and displays GIs in circular graphic images | http://www5.esu.edu/cpsc/bioinfo/dgi |
| GI-POP | A database that provides ongoing microbial gnome annotation, including ORF annotation, non-coding RNAs and GIs. GIs are predicted using GI-GPS | http://gipop.life.nthu.edu.tw |
| IGIPT | A web server that identifies islands based on standard deviation from sequence composition average | http://bioinf.iiit.ac.in/IGIPT/ |
| Islander | A database that contains a list of 89 islands in 106 bacterial genomes that harbor tRNA and tmRNA genes, and integrase genes | http://www.indiana.edu/~islander |
| IslandViewer | A database that contains predicted GI based on IslandPick, IslandPath-DIMOB and SIGI-HMM, and displays GIs in circular graphic images | http://www.pathogenomics.sfu.ca/islandviewer/query.php |
| MOSAIC | A database that contains conserved segments and various regions (i.e., GIs) in bacterial genomes, predicted by comparative genomic approach | http://genome.jouy.inra.fr/mosaic |
| PAI Databases/Servers | ||
| PAIDB | A database contains known PAIs, candidate PAIs which are homologous to known PAIs | http://www.gem.re.kr/paidb |
| PredictBias | A web server that calculates PAIs based on %G+C, dinucleotide, codon usage, virulence factor and absence of non-pathogenic species | http://www.davvbiotech.res.in/PredictBias |
| Virulence Factor Databases/Servers | ||
| MvirDB | A database that contains a collection of publicly available and organized sequences representing known toxins, virulence factors, and antibiotic resistance genes | http://mvirdb.llnl.gov/ |
| VFDB | A database that contains all known virulence factors, as well as homologous genes through similarity search | http://www.mgc.ac.cn/VFs/ |
| VirulentPred | A web server that predicts virulence factors based on input protein sequences | http://bioinfo.icgeb.res.in/virulent/ |
4.1. PAI Databases and Servers
4.2. Virulence Factor Databases and Servers
5. Concluding Remarks
Acknowledgments
Conflicts of Interest
References
- Pallen, M.J.; Wren, B.W. Bacterial pathogenomics. Nature 2007, 449, 835–842. [Google Scholar] [CrossRef]
- Koskiniemi, S.; Sun, S.; Berg, O.G.; Andersson, D.I. Selection-driven gene loss in bacteria. PLoS Genet. 2012, 8, e1002787. [Google Scholar] [CrossRef]
- Maurelli, A.T. Black holes, antivirulence genes, and gene inactivation in the evolution of bacterial pathogens. FEMS Microbiol. Lett. 2007, 267, 1–8. [Google Scholar] [CrossRef]
- Penn, K.; Jenkins, C.; Nett, M.; Udwary, D.W.; Gontang, E.A.; McGlinchey, R.P.; Foster, B.; Lapidus, A.; Podell, S.; Allen, E.E.; et al. Genomic islands link secondary metabolism to functional adaptation in marine Actinobacteria. ISME J. 2009, 3, 1193–1203. [Google Scholar] [CrossRef]
- Hacker, J.; Bender, L.; Ott, M.; Wingender, J.; Lund, B.; Marre, R.; Goebel, W. Deletions of chromosomal regions coding for fimbriae and hemolysins occur in vitro and in vivo in various extraintestinal Escherichia coli isolates. Microb. Pathog. 1990, 8, 213–225. [Google Scholar] [CrossRef]
- Blum, G.; Falbo, V.; Caprioli, A.; Hacker, J. Gene clusters encoding the cytotoxic necrotizing factor type 1, Prs-fimbriae and alpha-hemolysin form the pathogenicity island II of the uropathogenic Escherichia coli strain J96. FEMS Microbiol. Lett. 1995, 126, 189–195. [Google Scholar]
- Blum, G.; Ott, M.; Lischewski, A.; Ritter, A.; Imrich, H.; Tschape, H.; Hacker, J. Excision of large DNA regions termed pathogenicity islands from tRNA-specific loci in the chromosome of an Escherichia coli wild-type pathogen. Infect. Immun. 1994, 62, 606–614. [Google Scholar]
- Swenson, D.L.; Bukanov, N.O.; Berg, D.E.; Welch, R.A. Two pathogenicity islands in uropathogenic Escherichia coli J96: Cosmid cloning and sample sequencing. Infect. Immun. 1996, 64, 3736–3743. [Google Scholar]
- McDaniel, T.K.; Jarvis, K.G.; Donnenberg, M.S.; Kaper, J.B. A genetic locus of enterocyte effacement conserved among diverse enterobacterial pathogens. Proc. Natl. Acad. Sci. USA 1995, 92, 1664–1668. [Google Scholar] [CrossRef]
- Billington, S.J.; Sinistaj, M.; Cheetham, B.F.; Ayres, A.; Moses, E.K.; Katz, M.E.; Rood, J.I. Identification of a native Dichelobacter nodosus plasmid and implications for the evolution of the vap regions. Gene 1996, 172, 111–116. [Google Scholar] [CrossRef]
- Censini, S.; Lange, C.; Xiang, Z.; Crabtree, J.E.; Ghiara, P.; Borodovsky, M.; Rappuoli, R.; Covacci, A. Cag, a pathogenicity island of Helicobacter pylori, encodes type I-specific and disease-associated virulence factors. Proc. Natl. Acad. Sci. USA 1996, 93, 14648–14653. [Google Scholar] [CrossRef]
- Fetherston, J.D.; Perry, R.D. The pigmentation locus of Yersinia pestis KIM6+ is flanked by an insertion sequence and includes the structural genes for pesticin sensitivity and HMWP2. Mol. Microbiol. 1994, 13, 697–708. [Google Scholar] [CrossRef]
- Du, P.; Yang, Y.; Wang, H.; Liu, D.; Gao, G.F.; Chen, C. A large scale comparative genomic analysis reveals insertion sites for newly acquired genomic islands in bacterial genomes. BMC Microbiol. 2011, 11, 135. [Google Scholar] [CrossRef]
- Karlin, S. Detecting anomalous gene clusters and pathogenicity islands in diverse bacterial genomes. Trends Microbiol. 2001, 9, 335–343. [Google Scholar] [CrossRef]
- Vernikos, G.S.; Parkhill, J. Interpolated variable order motifs for identification of horizontally acquired DNA: Revisiting the Salmonella pathogenicity islands. Bioinformatics 2006, 22, 2196–2203. [Google Scholar] [CrossRef]
- Rajan, I.; Aravamuthan, S.; Mande, S.S. Identification of compositionally distinct regions in genomes using the centroid method. Bioinformatics 2007, 23, 2672–2677. [Google Scholar] [CrossRef]
- Waack, S.; Keller, O.; Asper, R.; Brodag, T.; Damm, C.; Fricke, W.F.; Surovcik, K.; Meinicke, P.; Merkl, R. Score-based prediction of genomic islands in prokaryotic genomes using hidden Markov models. BMC Bioinforma. 2006, 7, 142. [Google Scholar] [CrossRef]
- Karlin, S.; Mrazek, J. Predicted highly expressed genes of diverse prokaryotic genomes. J. Bacteriol. 2000, 182, 5238–5250. [Google Scholar] [CrossRef]
- Lawrence, J.G.; Ochman, H. Amelioration of bacterial genomes: Rates of change and exchange. J. Mol. Evol. 1997, 44, 383–397. [Google Scholar] [CrossRef]
- Gal-Mor, O.; Finlay, B.B. Pathogenicity islands: A molecular toolbox for bacterial virulence. Cell. Microbiol. 2006, 8, 1707–1719. [Google Scholar] [CrossRef]
- Hacker, J.; Kaper, J.B. Pathogenicity islands and the evolution of microbes. Annu. Rev. Microbiol. 2000, 54, 641–679. [Google Scholar] [CrossRef]
- Schmidt, H.; Hensel, M. Pathogenicity islands in bacterial pathogenesis. Clin. Microbiol. Rev. 2004, 17, 14–56. [Google Scholar] [CrossRef]
- Ho Sui, S.J.; Fedynak, A.; Hsiao, W.W.; Langille, M.G.; Brinkman, F.S. The association of virulence factors with genomic islands. PLoS One 2009, 4, e8094. [Google Scholar]
- Zhou, C.E.; Smith, J.; Lam, M.; Zemla, A.; Dyer, M.D.; Slezak, T. MvirDB—A microbial database of protein toxins, virulence factors and antibiotic resistance genes for bio-defence applications. Nucleic Acids Res. 2007, 35, D391–D394. [Google Scholar] [CrossRef]
- Garg, A.; Gupta, D. VirulentPred: A SVM based prediction method for virulent proteins in bacterial pathogens. BMC Bioinforma. 2008, 9, 62. [Google Scholar] [CrossRef]
- Dobrindt, U.; Hochhut, B.; Hentschel, U.; Hacker, J. Genomic islands in pathogenic and environmental microorganisms. Nat. Rev. Microbiol. 2004, 2, 414–424. [Google Scholar] [CrossRef]
- Finn, R.D.; Clements, J.; Eddy, S.R. HMMER web server: Interactive sequence similarity searching. Nucleic Acids Res. 2011, 39, W29–W37. [Google Scholar] [CrossRef]
- Finn, R.D.; Mistry, J.; Tate, J.; Coggill, P.; Heger, A.; Pollington, J.E.; Gavin, O.L.; Gunasekaran, P.; Ceric, G.; Forslund, K.; et al. The Pfam protein families database. Nucleic Acids Res. 2010, 38, D211–D222. [Google Scholar] [CrossRef]
- Vernikos, G.S.; Parkhill, J. Resolving the structural features of genomic islands: A machine learning approach. Genome Res. 2008, 18, 331–342. [Google Scholar] [CrossRef]
- Nakamura, Y.; Itoh, T.; Matsuda, H.; Gojobori, T. Biased biological functions of horizontally transferred genes in prokaryotic genomes. Nat. Genet. 2004, 36, 760–766. [Google Scholar] [CrossRef]
- Dobrindt, U.; Hentschel, U.; Kaper, J.B.; Hacker, J. Genome plasticity in pathogenic and nonpathogenic enterobacteria. Curr. Top. Microbiol. Immunol. 2002, 264, 157–175. [Google Scholar]
- Canchaya, C.; Proux, C.; Fournous, G.; Bruttin, A.; Brussow, H. Prophage genomics. Microbiol. Mol. Biol. Rev. 2003, 67, 238–276. [Google Scholar] [CrossRef]
- Ou, H.Y.; He, X.; Harrison, E.M.; Kulasekara, B.R.; Thani, A.B.; Kadioglu, A.; Lory, S.; Hinton, J.C.; Barer, M.R.; Deng, Z.; et al. MobilomeFINDER: Web-based tools for in silico and experimental discovery of bacterial genomic islands. Nucleic Acids Res. 2007, 35, W97–W104. [Google Scholar] [CrossRef]
- Fouts, D.E. Phage_Finder: Automated identification and classification of prophage regions in complete bacterial genome sequences. Nucleic Acids Res. 2006, 34, 5839–5851. [Google Scholar] [CrossRef]
- Boyd, E.F.; Almagro-Moreno, S.; Parent, M.A. Genomic islands are dynamic, ancient integrative elements in bacterial evolution. Trends Microbiol. 2009, 17, 47–53. [Google Scholar] [CrossRef]
- Lowe, T.M.; Eddy, S.R. tRNAscan-SE: A program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997, 25, 955–964. [Google Scholar]
- Hsiao, W.W.; Ung, K.; Aeschliman, D.; Bryan, J.; Finlay, B.B.; Brinkman, F.S. Evidence of a large novel gene pool associated with prokaryotic genomic islands. PLoS Genet. 2005, 1, e62. [Google Scholar] [CrossRef]
- Lukashin, A.V.; Borodovsky, M. GeneMark.hmm: New solutions for gene finding. Nucleic Acids Res. 1998, 26, 1107–1115. [Google Scholar] [CrossRef]
- Salzberg, S.L.; Delcher, A.L.; Kasif, S.; White, O. Microbial gene identification using interpolated Markov models. Nucleic Acids Res. 1998, 26, 544–548. [Google Scholar] [CrossRef]
- Altschul, S.F.; Madden, T.L.; Schaffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef]
- Bairoch, A.; Apweiler, R.; Wu, C.H.; Barker, W.C.; Boeckmann, B.; Ferro, S.; Gasteiger, E.; Huang, H.; Lopez, R.; Magrane, M.; et al. The universal protein resource (UniProt). Nucleic Acids Res. 2005, 33, D154–D159. [Google Scholar]
- Tatusov, R.L.; Natale, D.A.; Garkavtsev, I.V.; Tatusova, T.A.; Shankavaram, U.T.; Rao, B.S.; Kiryutin, B.; Galperin, M.Y.; Fedorova, N.D.; Koonin, E.V. The COG database: New developments in phylogenetic classification of proteins from complete genomes. Nucleic Acids Res. 2001, 29, 22–28. [Google Scholar] [CrossRef]
- Wang, H.; Fazekas, J.; Booth, M.; Liu, Q.; Che, D. An Integrative Approach for Genomic Island Prediction in Prokaryotic Genomes. In Bioinformatics Research and Applications; Chen, J., Wang, J., Zelikovsky, A., Eds.; Springer Berlin/Heidelberg: Berlin, Gremany, 2011; Volume 6674, pp. 404–415. [Google Scholar]
- Hacker, J.; Blum-Oehler, G.; Muhldorfer, I.; Tschape, H. Pathogenicity islands of virulent bacteria: Structure, function and impact on microbial evolution. Mol. Microbiol. 1997, 23, 1089–1097. [Google Scholar] [CrossRef]
- Kurtz, S.; Choudhuri, J.V.; Ohlebusch, E.; Schleiermacher, C.; Stoye, J.; Giegerich, R. REPuter: The manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res. 2001, 29, 4633–4642. [Google Scholar] [CrossRef]
- Siguier, P.; Perochon, J.; Lestrade, L.; Mahillon, J.; Chandler, M. ISfinder: The reference centre for bacterial insertion sequences. Nucleic Acids Res. 2006, 34, D32–D36. [Google Scholar] [CrossRef]
- Langille, M.G.; Brinkman, F.S. Bioinformatic detection of horizontally transferred DNA in bacterial genomes. F1000 Biol. Rep. 2009, 1, 25. [Google Scholar]
- Avise, J.C. Gene trees and organismal histories: A phylogenetic approach to population biology. Evolution 1989, 43, 1192–1208. [Google Scholar] [CrossRef]
- Langille, M.G.; Hsiao, W.W.; Brinkman, F.S. Evaluation of genomic island predictors using a comparative genomics approach. BMC Bioinforma. 2008, 9, 329. [Google Scholar] [CrossRef]
- Qi, J.; Luo, H.; Hao, B. CVTree: A phylogenetic tree reconstruction tool based on whole genomes. Nucleic Acids Res. 2004, 32, W45–W47. [Google Scholar] [CrossRef]
- Darling, A.C.; Mau, B.; Blattner, F.R.; Perna, N.T. Mauve: Multiple alignment of conserved genomic sequence with rearrangements. Genome Res. 2004, 14, 1394–1403. [Google Scholar] [CrossRef]
- Chiapello, H.; Bourgait, I.; Sourivong, F.; Heuclin, G.; Gendrault-Jacquemard, A.; Petit, M.A.; El Karoui, M. Systematic determination of the mosaic structure of bacterial genomes: Species backbone versus strain-specific loops. BMC Bioinforma. 2005, 6, 171. [Google Scholar] [CrossRef]
- Chiapello, H.; Gendrault, A.; Caron, C.; Blum, J.; Petit, M.A.; El Karoui, M. MOSAIC: An online database dedicated to the comparative genomics of bacterial strains at the intra-species level. BMC Bioinforma. 2008, 9, 498. [Google Scholar] [CrossRef]
- Kurtz, S.; Phillippy, A.; Delcher, A.L.; Smoot, M.; Shumway, M.; Antonescu, C.; Salzberg, S.L. Versatile and open software for comparing large genomes. Genome Biol. 2004, 5, R12. [Google Scholar] [CrossRef]
- Hohl, M.; Kurtz, S.; Ohlebusch, E. Efficient multiple genome alignment. Bioinformatics 2002, 18, S312–S320. [Google Scholar] [CrossRef]
- Che, D.; Hasan, M.S.; Wang, H.; Fazekas, J.; Huang, J.; Liu, Q. EGID: An ensemble algorithm for improved genomic island detection in genomic sequences. Bioinformation 2011, 7, 311–314. [Google Scholar] [CrossRef]
- Hsiao, W.; Wan, I.; Jones, S.J.; Brinkman, F.S. IslandPath: Aiding detection of genomic islands in prokaryotes. Bioinformatics 2003, 19, 418–420. [Google Scholar] [CrossRef]
- Shrivastava, S.; Reddy Ch, V.; Mande, S.S. INDeGenIUS, a new method for high-throughput identification of specialized functional islands in completely sequenced organisms. J. Biosci. 2010, 35, 351–364. [Google Scholar] [CrossRef]
- Tu, Q.; Ding, D. Detecting pathogenicity islands and anomalous gene clusters by iterative discriminant analysis. FEMS Microbiol. Lett. 2003, 221, 269–275. [Google Scholar] [CrossRef]
- Hasan, M.S.; Liu, Q.; Wang, H.; Fazekas, J.; Chen, B.; Che, D. GIST: Genomic island suite of tools for predicting genomic islands in genomic sequences. Bioinformation 2012, 8, 203–205. [Google Scholar] [CrossRef]
- Che, D.; Hockenbury, C.; Marmelstein, R.; Rasheed, K. Classification of genomic islands using decision trees and their ensemble algorithms. BMC Genomics 2010, 11, S1. [Google Scholar]
- Breiman, L. Bagging predictors. Mach. Learn. 1996, 24, 123–140. [Google Scholar]
- Breiman, L. Random forests. Mach. Learn. 2001, 45, 5–32. [Google Scholar] [CrossRef]
- Lee, C.C.; Chen, Y.P.; Yao, T.J.; Ma, C.Y.; Lo, W.C.; Lyu, P.C.; Tang, C.Y. GI-POP: A combinational annotation and genomic island prediction pipeline for ongoing microbial genome projects. Gene 2013, 518, 114–123. [Google Scholar] [CrossRef]
- Soares, S.C.; Abreu, V.A.; Ramos, R.T.; Cerdeira, L.; Silva, A.; Baumbach, J.; Trost, E.; Tauch, A.; Hirata, R., Jr.; Mattos-Guaraldi, A.L.; et al. PIPS: Pathogenicity island prediction software. PLoS One 2012, 7, e30848. [Google Scholar] [CrossRef]
- Merkl, R. SIGI: Score-based identification of genomic islands. BMC Bioinforma. 2004, 5, 22. [Google Scholar] [CrossRef]
- Che, D.; Wang, H. GIV: A tool for genomic islands visualization. Bioinformation 2013, 9, 879–882. [Google Scholar] [CrossRef]
- Krzywinski, M.; Schein, J.; Birol, I.; Connors, J.; Gascoyne, R.; Horsman, D.; Jones, S.J.; Marra, M.A. Circos: An information aesthetic for comparative genomics. Genome Res. 2009, 19, 1639–1645. [Google Scholar] [CrossRef]
- Stewart, A.C.; Osborne, B.; Read, T.D. DIYA: A bacterial annotation pipeline for any genomics lab. Bioinformatics 2009, 25, 962–963. [Google Scholar] [CrossRef]
- Jain, R.; Ramineni, S.; Parekh, N. IGIPT—Integrated genomic island prediction tool. Bioinformation 2011, 7, 307–310. [Google Scholar] [CrossRef]
- Mantri, Y.; Williams, K.P. Islander: A database of integrative islands in prokaryotic genomes, the associated integrases and their DNA site specificities. Nucleic Acids Res. 2004, 32, D55–D58. [Google Scholar] [CrossRef]
- Laslett, D.; Canback, B.; Andersson, S. BRUCE: A program for the detection of transfer-messenger RNA genes in nucleotide sequences. Nucleic Acids Res. 2002, 30, 3449–3453. [Google Scholar] [CrossRef]
- Langille, M.G.; Brinkman, F.S. IslandViewer: An integrated interface for computational identification and visualization of genomic islands. Bioinformatics 2009, 25, 664–665. [Google Scholar] [CrossRef]
- Dhillon, B.K.; Chiu, T.A.; Laird, M.R.; Langille, M.G.; Brinkman, F.S. IslandViewer update: Improved genomic island discovery and visualization. Nucleic Acids Res. 2013, 41, W129–W132. [Google Scholar] [CrossRef]
- Yoon, S.H.; Park, Y.K.; Lee, S.; Choi, D.; Oh, T.K.; Hur, C.G.; Kim, J.F. Towards pathogenomics: A web-based resource for pathogenicity islands. Nucleic Acids Res. 2007, 35, D395–D400. [Google Scholar] [CrossRef]
- Yoon, S.H.; Hur, C.G.; Kang, H.Y.; Kim, Y.H.; Oh, T.K.; Kim, J.F. A computational approach for identifying pathogenicity islands in prokaryotic genomes. BMC Bioinforma. 2005, 6, 184. [Google Scholar] [CrossRef]
- Pundhir, S.; Vijayvargiya, H.; Kumar, A. PredictBias: A server for the identification of genomic and pathogenicity islands in prokaryotes. In Silico Biol. 2008, 8, 223–234. [Google Scholar]
- Jungo, F.; Bairoch, A. Tox-Prot, the toxin protein annotation program of the Swiss-Prot protein knowledgebase. Toxicon 2005, 45, 293–301. [Google Scholar] [CrossRef]
- Srinivasan, K.N.; Gopalakrishnakone, P.; Tan, P.T.; Chew, K.C.; Cheng, B.; Kini, R.M.; Koh, J.L.; Seah, S.H.; Brusic, V. SCORPION, a molecular database of scorpion toxins. Toxicon 2002, 40, 23–31. [Google Scholar] [CrossRef]
- Paine, K.; Flower, D.R. Bacterial bioinformatics: Pathogenesis and the genome. J. Mol. Microbiol. Biotechnol. 2002, 4, 357–365. [Google Scholar]
- Chen, L.; Yang, J.; Yu, J.; Yao, Z.; Sun, L.; Shen, Y.; Jin, Q. VFDB: A reference database for bacterial virulence factors. Nucleic Acids Res. 2005, 33, D325–D328. [Google Scholar] [CrossRef]
- Scaria, J.; Chandramouli, U.; Verma, S.K. Antibiotic Resistance Genes Online (ARGO): A Database on vancomycin and beta-lactam resistance genes. Bioinformation 2005, 1, 5–7. [Google Scholar] [CrossRef]
- Alba, M.M.; Lee, D.; Pearl, F.M.; Shepherd, A.J.; Martin, N.; Orengo, C.A.; Kellam, P. VIDA: A virus database system for the organization of animal virus genome open reading frames. Nucleic Acids Res. 2001, 29, 133–136. [Google Scholar] [CrossRef]
- Yang, J.; Chen, L.; Sun, L.; Yu, J.; Jin, Q. VFDB 2008 release: An enhanced web-based resource for comparative pathogenomics. Nucleic Acids Res. 2008, 36, D539–D542. [Google Scholar] [CrossRef]
- Chen, L.; Xiong, Z.; Sun, L.; Yang, J.; Jin, Q. VFDB 2012 update: Toward the genetic diversity and molecular evolution of bacterial virulence factors. Nucleic Acids Res. 2012, 40, D641–D645. [Google Scholar] [CrossRef]
- Bairoch, A.; Apweiler, R. The SWISS-PROT protein sequence database and its supplement TrEMBL in 2000. Nucleic Acids Res. 2000, 28, 45–48. [Google Scholar] [CrossRef]
- Tompa, M.; Li, N.; Bailey, T.L.; Church, G.M.; de Moor, B.; Eskin, E.; Favorov, A.V.; Frith, M.C.; Fu, Y.; Kent, W.J.; et al. Assessing computational tools for the discovery of transcription factor binding sites. Nat. Biotechnol. 2005, 23, 137–144. [Google Scholar] [CrossRef]
- Brouwer, R.W.; Kuipers, O.P.; van Hijum, S.A. The relative value of operon predictions. Brief. Bioinforma. 2008, 9, 367–375. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Che, D.; Hasan, M.S.; Chen, B. Identifying Pathogenicity Islands in Bacterial Pathogenomics Using Computational Approaches. Pathogens 2014, 3, 36-56. https://doi.org/10.3390/pathogens3010036
Che D, Hasan MS, Chen B. Identifying Pathogenicity Islands in Bacterial Pathogenomics Using Computational Approaches. Pathogens. 2014; 3(1):36-56. https://doi.org/10.3390/pathogens3010036
Chicago/Turabian StyleChe, Dongsheng, Mohammad Shabbir Hasan, and Bernard Chen. 2014. "Identifying Pathogenicity Islands in Bacterial Pathogenomics Using Computational Approaches" Pathogens 3, no. 1: 36-56. https://doi.org/10.3390/pathogens3010036
APA StyleChe, D., Hasan, M. S., & Chen, B. (2014). Identifying Pathogenicity Islands in Bacterial Pathogenomics Using Computational Approaches. Pathogens, 3(1), 36-56. https://doi.org/10.3390/pathogens3010036