Genomic Insights into the Pathogenicity of Hypervirulent Aeromonas hydrophila Strain D4 Isolated from Diseased Blunt Snout Bream with the Epidemic Sequence Type 251 Clones


Abstract
1. Introduction
2. Materials and Methods
2.1. Strains and Genomes
2.2. Phenotypic Identification of Strain D4
2.2.1. Swimming and Swarming Motility Assays
2.2.2. Hemolytic and Protease Activity Assays
2.2.3. Biofilm Formation Assay
2.2.4. Lethal Dose 50 Assay
2.3. Statistical Analyses
2.4. Comparative Genomics Analysis
2.4.1. ANI Analysis and General Features of Ten A. hydrophila Genomes
2.4.2. Genomic Collinearity and Rearrangement Analysis
2.4.3. Functional Annotation and Pathway Analysis
2.5. Virulence-Related Factors Analysis
2.5.1. Virulence Gene Conservation and Absence Analysis
2.5.2. Secondary Metabolite Gene Cluster Analysis
2.5.3. Prophage Content and Functional Analysis
2.5.4. Genomic Islands Analysis
2.5.5. Plasmid Content and Functional Analysis
3. Results
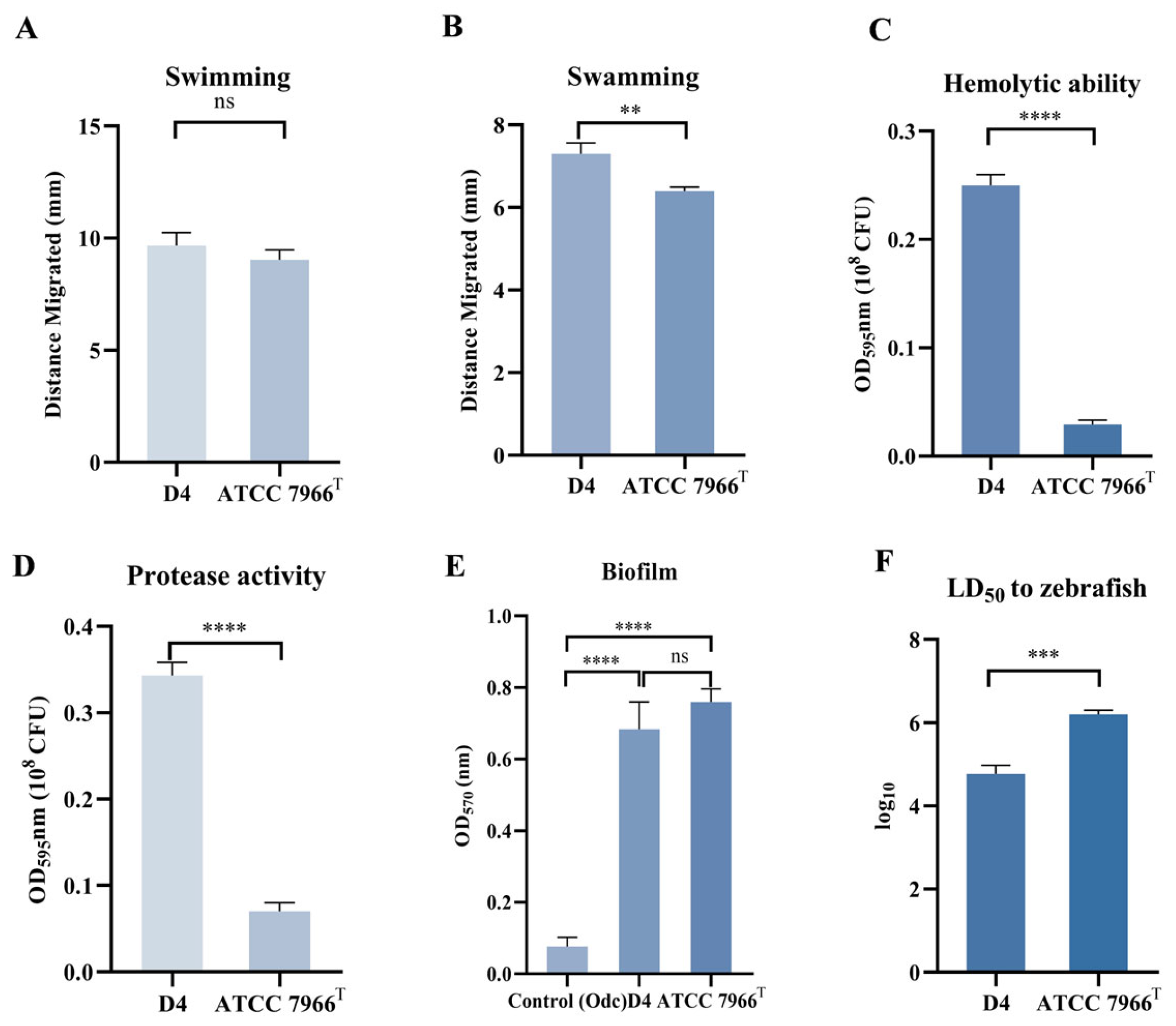
3.1. Phenotypic Characterization of Strain D4
3.1.1. Swimming and Swarming Motility
3.1.2. Hemolytic and Protease Activity
3.1.3. Biofilm
3.1.4. Virulence Assessment via LD50 Determination
3.2. Comparative Genomics Analysis
3.2.1. ANI Analysis and General Features of Ten A. hydrophila Genomes
3.2.2. Genomic Collinearity and Rearrangement Analysis
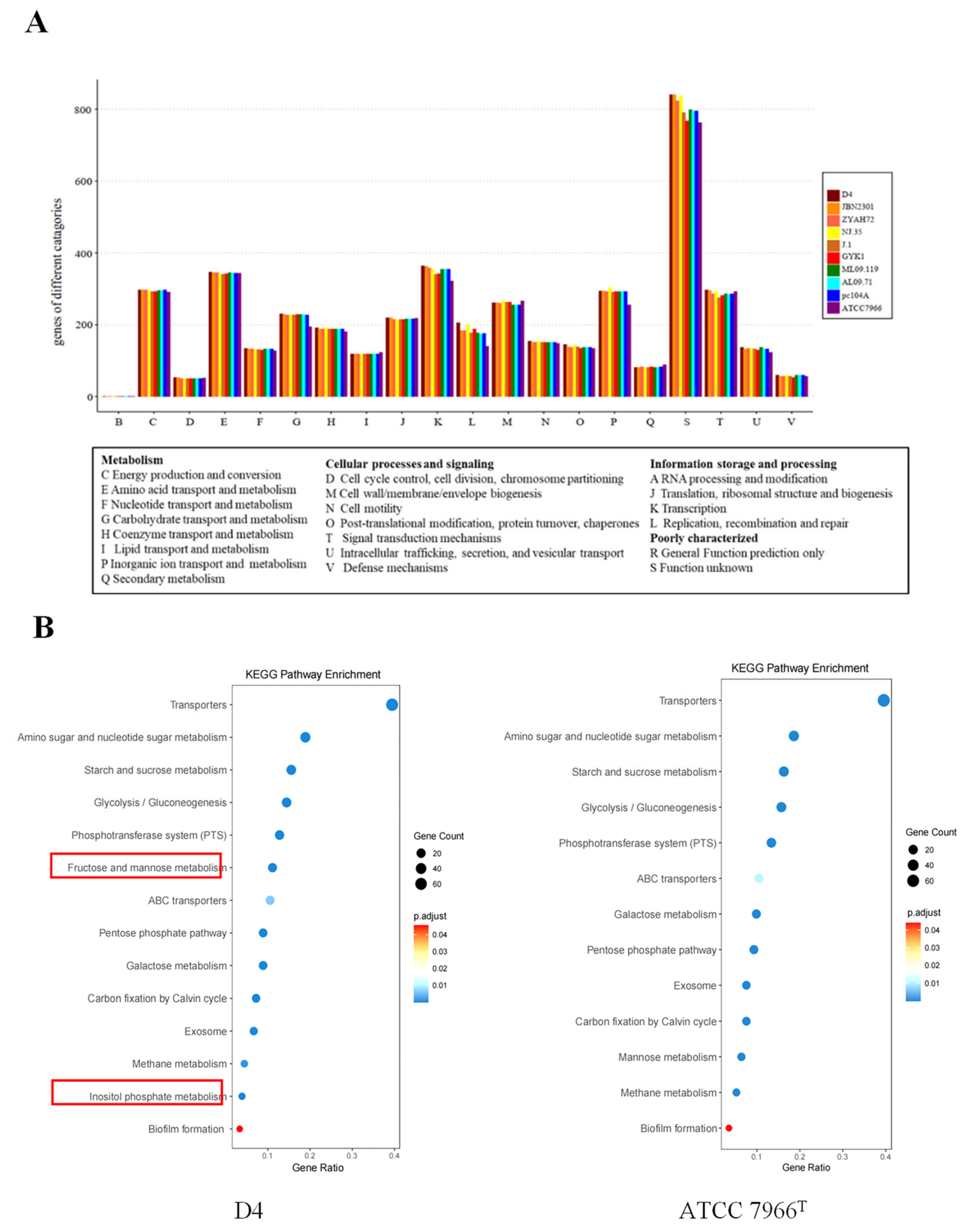
3.2.3. Functional Annotation and Pathway Analysis
3.3. Virulence-Related Factors Analysis
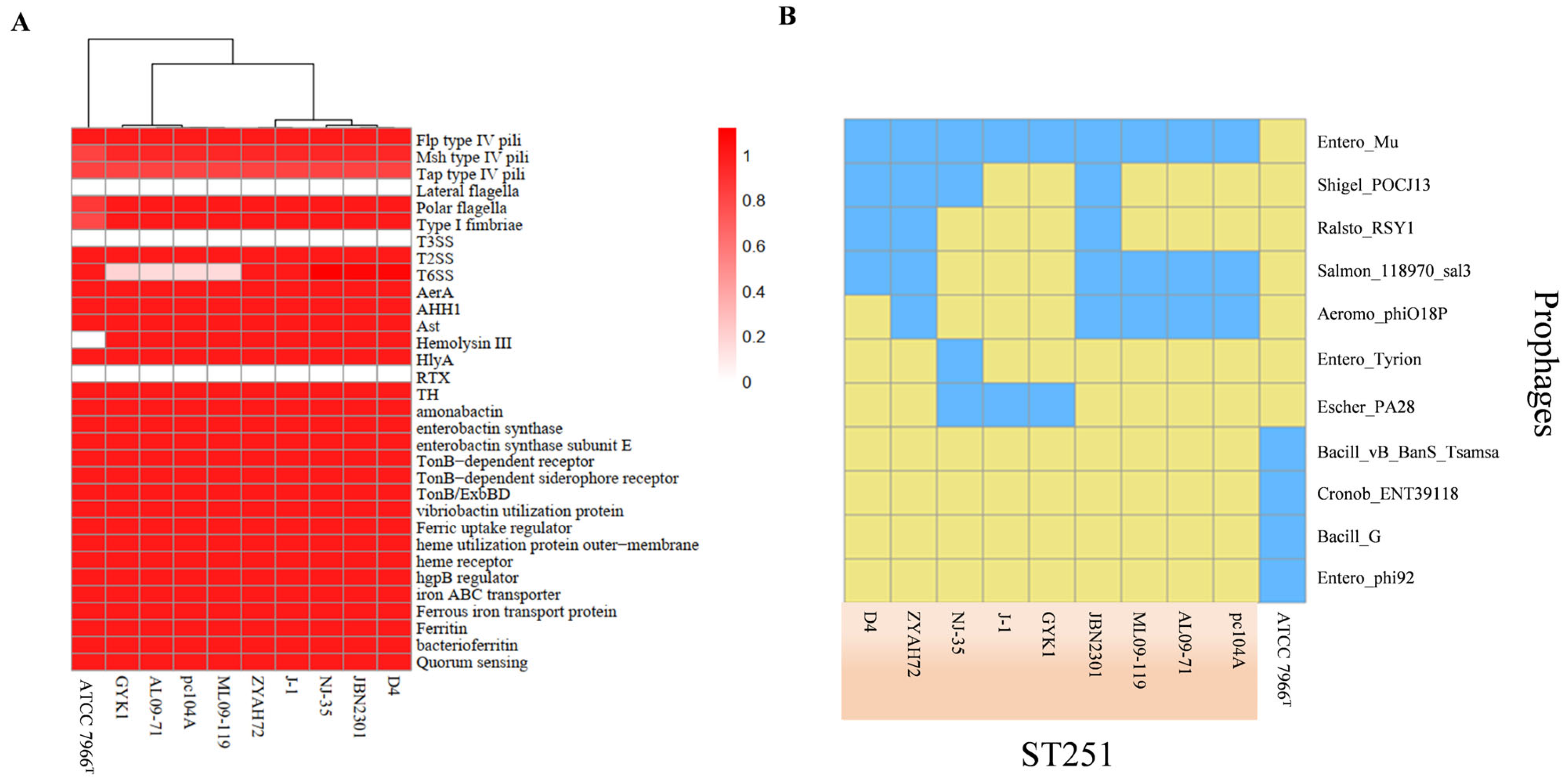
3.3.1. Virulence Gene Conservation and Absence Analysis
3.3.2. Secondary Metabolite Gene Cluster Analysis
3.3.3. Prophage Content and Functional Analysis
3.3.4. Genomic Island Analysis
3.3.5. Two-Component Regulatory System Profiling
3.3.6. Plasmid Content and Functional Analysis
4. Discussion
4.1. Phenotypic and Genomic Correlates of Virulence
4.2. Biofilm Formation and Functional Redundancy
4.3. Genomic Insights into ST251 Pathogenesis
4.4. Mobile Genetic Elements and Adaptive Evolution
4.5. Metabolic Adaptation as a Pathogenic Strategy
4.6. Limitations and Future Research Directions
- (1)
- Constructing deletion or overexpression mutants of key virulence or regulatory genes (e.g., pse genes, PtrR, IolR), combined with in vitro and in vivo infection models, will help clarify their roles in motility, biofilm formation, and virulence expression.
- (2)
- Transcriptomic analyses under various environmental stress conditions (e.g., high temperature and salinity and nutrient limitation) and natural host infection models (e.g., blunt snout bream) could provide mechanistic insights into transcriptional regulation and virulence adaptation in complex environments. Integrating histopathology and host immune gene expression will further improve ecological relevance.
- (3)
- Expanding the strain collection to include more ST251 isolates from diverse geographic regions and host sources, along with host response analyses (e.g., host transcriptomics, inflammatory markers, and immune cell activation), will facilitate a systems-level understanding of host–pathogen interactions and their impact on infection outcomes.
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ANI | Average nucleotide identity |
| A. hydrophila | Aeromonas hydrophila |
| CAS | Chrome azurol-S |
| CDSs | Coding sequences |
| MAS | Motile Aeromonas Septicemia |
| PBS | Phosphate-buffered saline |
| T3SS | Type III secretion system |
| T6SS | Type VI secretion system |
| ST251 | Sequence type 251 |
References
- Little, D.C.; MacKenzie, S. Grand challenges for global aquaculture. Front. Aquac. 2023, 2, 1232936. [Google Scholar] [CrossRef]
- Michael Janda, J.; Sharon, L.A. The genus Aeromonas: Taxonomy, pathogenicity, and infection. Clin. Microbiol. Rev. 2010, 23, 35–73. [Google Scholar] [CrossRef] [PubMed]
- Bartie, K.L.; Ngô, T.P.H.; Bekaert, M.; Hoang Oanh, D.T.; Hoare, R.; Adams, A.; Desbois, A.P. Aeromonas hydrophila ST251 and Aeromonas dhakensis are major emerging pathogens of striped catfish in vietnam. Front. Microbiol. 2023, 13, 1067235. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Yang, W.; Wu, H.; Gong, X.; Li, A. Multilocus sequence typing revealed a clonal lineage of Aeromonas hydrophila caused Motile Aeromonas Septicemia outbreaks in pond-cultured cyprinid fish in an epidemic area in central china. Aquaculture 2014, 432, 1–6. [Google Scholar] [CrossRef]
- Rasmussen-Ivey, C.R.; Hossain, M.J.; Odom, S.E.; Terhune, J.S.; Hemstreet, W.G.; Shoemaker, C.A.; Zhang, D.; Xu, D.-H.; Griffin, M.J.; Liu, Y.-J.; et al. Classification of a hypervirulent Aeromonas hydrophila pathotype responsible for epidemic outbreaks in warm-water fishes. Front. Microbiol. 2016, 7, 1615. [Google Scholar] [CrossRef]
- Hossain, M.J.; Sun, D.; McGarey, D.J.; Wrenn, S.; Alexander, L.M.; Martino, M.E.; Xing, Y.; Terhune, J.S.; Liles, M.R. An Asian origin of virulent Aeromonas Hydrophila responsible for disease epidemics in United States-farmed catfish. mBio 2014, 5, e00848-14. [Google Scholar] [CrossRef]
- Rasmussen-Ivey, C.R.; Figueras, M.J.; McGarey, D.; Liles, M.R. Virulence factors of Aeromonas hydrophila: In the wake of reclassification. Front. Microbiol. 2016, 7, 1337. [Google Scholar] [CrossRef]
- Zhu, L.; Zheng, J.-S.; Wang, W.-M.; Luo, Y. Complete genome sequence of highly virulent Aeromonas hydrophila Strain D4, isolated from a diseased blunt-snout bream in China. Microbiol. Resour. Announc. 2019, 8, e01035-18. [Google Scholar] [CrossRef]
- Davey, M.E.; O’toole, G.A. Microbial biofilms: From ecology to molecular genetics. Microbiol. Mol. Biol. Rev. 2000, 64, 847–867. [Google Scholar] [CrossRef]
- Saganuwan, A. A modified arithmetical method of reed and muench for determination of a relatively ideal median lethal dose (LD50). Afr. J. Pharm. Pharmacol. 2011, 5, 1543–1546. [Google Scholar] [CrossRef]
- Medema, M.H.; Blin, K.; Cimermancic, P.; de Jager, V.; Zakrzewski, P.; Fischbach, M.A.; Weber, T.; Takano, E.; Breitling, R. antiSMASH: Rapid identification, annotation and analysis of secondary metabolite biosynthesis gene clusters in bacterial and fungal genome sequences. Nucleic Acids Res. 2011, 39, W339–W346. [Google Scholar] [CrossRef] [PubMed]
- McCarter, L.L. Dual flagellar systems enable motility under different circumstances. J. Mol. Microbiol. Biotechnol. 2004, 7, 18–29. [Google Scholar] [CrossRef] [PubMed]
- Chaban, B.; Hughes, H.V.; Beeby, M. The flagellum in bacterial pathogens: For motility and a whole lot more. Semin. Cell Dev. Biol. 2015, 46, 91–103. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Clark, C.G.; Liu, C.; Pucknell, C.; Munro, C.K.; Kruk, T.M.A.C.; Caldeira, R.; Woodward, D.L.; Rodgers, F.G. Detection and characterization of the hemolysin genes in Aeromonas hydrophila and Aeromonas sobria by multiplex PCR. J. Clin. Microbiol. 2003, 41, 1048–1054. [Google Scholar] [CrossRef]
- Goswami, R.; Ghosh, D.; Saha, D.R.; Padhy, P.K.; Mazumder, S. Effect of acute and chronic arsenic exposure on growth, structure and virulence of Aeromonas hydrophila isolated from fish. Microb. Pathog. 2011, 50, 63–69. [Google Scholar] [CrossRef]
- Abreu, R.E.F.; Magalhães, T.C.; Souza, R.C.; Oliveira, S.T.; Ibelli, A.M.; Demarqui, F.N.; Gouveia, J.J.; Costa, M.M.; Gouveia, G.V. Environmental factors on virulence of Aeromonas hydrophila. Aquacult. Int. 2018, 26, 495–507. [Google Scholar] [CrossRef]
- Rodríguez, I.; Novoa, B.; Figueras, A. Immune response of zebrafish (Danio Rerio) against a newly isolated bacterial pathogen Aeromonas Hydrophila. Fish Shellfish Immunol. 2008, 25, 239–249. [Google Scholar] [CrossRef]
- Rodriguez-R, L.M.; Conrad, R.E.; Viver, T.; Feistel, D.J.; Lindner, B.G.; Venter, S.N.; Orellana, L.H.; Amann, R.; Rossello-Mora, R.; Konstantinidis, K.T. An ani gap within bacterial species that advances the definitions of intra-species units. mBio 2024, 15, e0269623. [Google Scholar] [CrossRef]
- Xie, Q.; Mei, W.; Ye, X.; Zhou, P.; Islam, M.S.; Elbassiony, K.R.A.; Yuan, C.; Li, J.; Zhou, Y. The two-component regulatory system CpxA/R is required for the pathogenesis of Aeromonas hydrophila. FEMS Microbiol. Lett. 2018, 365, fny218. [Google Scholar] [CrossRef]
- Pang, M.; Jiang, J.; Xie, X.; Wu, Y.; Dong, Y.; Kwok, A.H.Y.; Zhang, W.; Yao, H.; Lu, C.; Leung, F.C.; et al. Novel insights into the pathogenicity of epidemic Aeromonas hydrophila ST251 clones from comparative genomics. Sci. Rep. 2015, 5, 9833. [Google Scholar] [CrossRef]
- Bi, Z.X.; Liu, Y.J.; Lu, C.P. Contribution of AhyR to virulence of Aeromonas hydrophila J-1. Res. Vet Sci. 2007, 83, 150–156. [Google Scholar] [CrossRef] [PubMed]
- Hou-jun, P. Identification, virulence, hemolytic activity of GYK1, a Strain of pathogenic Aeromonas hydrophila isolated from mandarinfish. J. Shanghai Fish. Univ. 2004, 13, e29. [Google Scholar]
- Yang, W.; Li, N.; Li, M.; Zhang, D.; An, G. Complete genome sequence of fish pathogen Aeromonas hydrophila JBN2301. Genome Announc. 2016, 4, e01615-15. [Google Scholar] [CrossRef] [PubMed]
- Tekedar, H.C.; Waldbieser, G.C.; Karsi, A.; Liles, M.R.; Griffin, M.J.; Vamenta, S.; Sonstegard, T.; Hossain, M.; Schroeder, S.G.; Khoo, L.; et al. Complete genome sequence of a channel catfish epidemic isolate, Aeromonas hydrophila Strain ML09-119. Genome Announc. 2013, 1, e00755-15. [Google Scholar] [CrossRef] [PubMed]
- Pridgeon, J.W.; Zhang, D.; Zhang, L. Complete genome sequence of the highly virulent Aeromonas hydrophila AL09-71 isolated from diseased channel catfish in West Alabama. Genome Announc. 2014, 2, 10–1128. [Google Scholar] [CrossRef]
- Pridgeon, J.W.; Zhang, D.; Zhang, L. Complete genome sequence of a moderately virulent Aeromonas hydrophila Strain, pc104A, isolated from soil of a catfish pond in West Alabama. Genome Announc. 2014, 2, e00554-14. [Google Scholar] [CrossRef] [PubMed]
- Seshadri, R.; Joseph, S.W.; Chopra, A.K.; Sha, J.; Shaw, J.; Graf, J.; Haft, D.; Wu, M.; Ren, Q.; Rosovitz, M.J.; et al. Genome sequence of Aeromonas hydrophila ATCC 7966T: Jack of all trades. J. Bacteriol. 2006, 188, 8272–8282. [Google Scholar] [CrossRef]
- Fricke, W.F.; Mammel, M.K.; McDermott, P.F.; Tartera, C.; White, D.G.; Leclerc, J.E.; Ravel, J.; Cebula, T.A. Comparative genomics of 28 Salmonella Enterica isolates: Evidence for CRISPR-mediated adaptive sublineage evolution. J. Bacteriol. 2011, 193, 3556–3568. [Google Scholar] [CrossRef]
- Awan, F.; Dong, Y.; Liu, J.; Wang, N.; Mushtaq, M.H.; Lu, C.; Liu, Y. Comparative genome analysis provides deep insights into Aeromonas hydrophila taxonomy and virulence-related factors. BMC Genomics 2018, 19, 712. [Google Scholar] [CrossRef]
- Pietras, Z.; Hardwick, S.W.; Swiezewski, S.; Luisi, B.F. Potential regulatory interactions of Escherichia Coli RraA Protein with DEAD-Box helicases. J. Biol. Chem. 2013, 288, 31919–31929. [Google Scholar] [CrossRef]
- Xu, T.; Rasmussen-Ivey, C.R.; Moen, F.S.; Fernández-Bravo, A.; Lamy, B.; Beaz-Hidalgo, R.; Khan, C.D.; Castro Escarpulli, G.; Yasin, I.S.M.; Figueras, M.J.; et al. A global survey of hypervirulent Aeromonas hydrophila (vAh) identified vah strains in the lower Mekong River Basin and diverse opportunistic pathogens from farmed fish and other environmental sources. Microbiol. Spectr. 2023, 11, e0370522. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Wang, P.; Yuan, W.; Su, Z.; Bullard, S.H. Endocidal regulation of secondary metabolites in the producing organisms. Sci. Rep. 2016, 6, 29315. [Google Scholar] [CrossRef]
- Lian, L.; Sun, L.; Zhao, X.; Chen, L.; Zhang, B.; Liu, Y.; Lin, X. Analysis of pathogenicity factors in the highly virulent Aeromonas hydrophila strain LP-2. Aquaculture 2025, 598, 741982. [Google Scholar] [CrossRef]
- Huang, J.; Dai, X.; Wu, Z.; Hu, X.; Sun, J.; Tang, Y.; Zhang, W.; Han, P.; Zhao, J.; Liu, G.; et al. Conjugative transfer of streptococcal prophages harboring antibiotic resistance and virulence genes. ISME J. 2023, 17, 1467–1481. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; Shi, J.; Liu, C.; Jin, Y.; Li, K.; Chen, R.; Jin, S.; Wu, W. PrtR homeostasis contributes to Pseudomonas aeruginosa pathogenesis and resistance against ciprofloxacin. Infect. Immun. 2014, 82, 1638–1647. [Google Scholar] [CrossRef]
- Mehershahi, K.S.; Chen, S.L. DNA methylation by three Type I restriction modification systems of Escherichia Coli does not influence gene regulation of the host bacterium. Nucleic Acids Res. 2021, 49, 7375–7388. [Google Scholar] [CrossRef]
- Juhas, M.; van der Meer, J.R.; Gaillard, M.; Harding, R.M.; Hood, D.W.; Crook, D.W. Genomic islands: Tools of bacterial horizontal gene transfer and evolution. FEMS Microbiol. Rev. 2009, 33, 376–393. [Google Scholar] [CrossRef]
- Manske, C.; Schell, U.; Hilbi, H. Metabolism of myo-inositol by Legionella pneumophila promotes infection of amoebae and macrophages. Appl. Environ. Microbiol. 2016, 82, 5000–5014. [Google Scholar] [CrossRef]
- Krings, E.; Krumbach, K.; Bathe, B.; Kelle, R.; Wendisch, V.F.; Sahm, H.; Eggeling, L. Characterization of myo-inositol utilization by Corynebacterium glutamicum: The stimulon, identification of transporters, and influence on L-lysine formation. J. Bacteriol. 2006, 188, 8054–8061. [Google Scholar] [CrossRef]
- Dong, Y.; Li, S.; Zhao, D.; Liu, J.; Ma, S.; Geng, J.; Lu, C.; Liu, Y. IolR, a negative regulator of the myo-inositol metabolic pathway, inhibits cell autoaggregation and biofilm formation by downregulating rpma in Aeromonas hydrophila. NPJ Biofilms Microbiomes 2020, 6, 22. [Google Scholar] [CrossRef]
- da Silva Filho, A.C.; Marchaukoski, J.N.; Raittz, R.T.; De Pierri, C.R.; de Jesus Soares Machado, D.; Fadel-Picheth, C.M.T.; Picheth, G. Prediction and analysis in silico of genomic islands in Aeromonas hydrophila. Front. Microbiol. 2021, 12, 769380. [Google Scholar] [CrossRef] [PubMed]
- Salah Ud-Din, A.I.M.; Roujeinikova, A. Flagellin glycosylation with pseudaminic acid in Campylobacter and Helicobacter: Prospects for development of novel therapeutics. Cell. Mol. Life Sci. 2018, 75, 1163–1178. [Google Scholar] [CrossRef] [PubMed]
- Stahl, M.; Friis, L.M.; Nothaft, H.; Liu, X.; Li, J.; Szymanski, C.M.; Stintzi, A. L-fucose utilization provides campylobacter jejuni with a competitive advantage. Proc. Natl. Acad. Sci. USA 2011, 108, 7194–7199. [Google Scholar] [CrossRef] [PubMed]
- Zou, Y.; Li, C.; Brunzelle, J.S.; Nair, S.K. Molecular basis for substrate selectivity and specificity by an LPS biosynthetic enzyme. Biochemistry 2007, 46, 4294–4304. [Google Scholar] [CrossRef]
- Hirakawa, H.; Kurushima, J.; Hashimoto, Y.; Tomita, H. Progress overview of bacterial two-component regulatory systems as potential targets for antimicrobial chemotherapy. Antibiotics 2020, 9, 635. [Google Scholar] [CrossRef] [PubMed]
- Beier, D.; Gross, R. Regulation of bacterial virulence by two-component systems. Curr. Opin. Microbiol. 2006, 9, 143–152. [Google Scholar] [CrossRef]
- Han, H.; Liu, C.; Wang, Q.; Xuan, C.; Zheng, B.; Tang, J.; Yan, J.; Zhang, J.; Li, M.; Cheng, H.; et al. The two-component system Ihk/Irr contributes to the virulence of streptococcus suis serotype 2 strain 05ZYH33 through alteration of the bacterial cell metabolism. Microbiology 2012, 158, 1852–1866. [Google Scholar] [CrossRef]
- Crosby, H.A.; Tiwari, N.; Kwiecinski, J.M.; Xu, Z.; Dykstra, A.; Jenul, C.; Fuentes, E.J.; Horswill, A.R. The Staphylococcus aureus ArlRS two-component system regulates virulence factor expression through MgrA. Mol. Microbiol. 2020, 113, 103–122. [Google Scholar] [CrossRef]
- Actis, L.A.; Tolmasky, M.E.; Crosa, J.H. Bacterial plasmids: Replication of extrachromosomal genetic elements encoding resistance to antimicrobial compounds. Front. Biosci. 1999, 4, D43–D62. [Google Scholar] [CrossRef]
- Zhao, Y.-L.; Zhou, Y.-H.; Chen, J.-Q.; Huang, Q.-Y.; Han, Q.; Liu, B.; Cheng, G.-D.; Li, Y.-H. Quantitative proteomic analysis of Sub-MIC erythromycin inhibiting biofilm formation of S. suis in vitro. J. Proteom. 2015, 116, 1–14. [Google Scholar] [CrossRef]
- Brüssow, H.; Canchaya, C.; Hardt, W.-D. Phages and the evolution of bacterial pathogens: From genomic rearrangements to lysogenic conversion. Microbiol. Mol. Biol. Rev. 2004, 68, 560–602. [Google Scholar] [CrossRef] [PubMed]
- Dougherty, P.E.; Nielsen, T.K.; Riber, L.; Lading, H.H.; Forero-Junco, L.M.; Kot, W.; Raaijmakers, J.M.; Hansen, L.H. Widespread and largely unknown prophage activity, diversity, and function in two genera of wheat phyllosphere bacteria. ISME J. 2023, 17, 2415–2425. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.-J.; Leung, P.M.; Wood, J.L.; Bay, S.K.; Hugenholtz, P.; Kessler, A.J.; Shelley, G.; Waite, D.W.; Franks, A.E.; Cook, P.L.M.; et al. Metabolic flexibility allows bacterial habitat generalists to become dominant in a frequently disturbed ecosystem. ISME J. 2021, 15, 2986–3004. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.-Z.; Zhao, L.-F.; Zhang, Q.; Fang, H.; Song, W.-L.; Li, W.-Z.; Ge, Y.-S.; Gao, P. Core fucosylation and its roles in gastrointestinal glycoimmunology. World J. Gastrointest. Oncol. 2023, 15, 1119–1134. [Google Scholar] [CrossRef]
- Corfield, A.P.; Carroll, D.; Myerscough, N.; Probert, C.S. Mucins in the gastrointestinal tract in health and disease. Front. Biosci. 2001, 6, D1321–D1357. [Google Scholar] [CrossRef]
- Kononova, S.; Litvinova, E.; Vakhitov, T.; Skalinskaya, M.; Sitkin, S. Acceptive immunity: The role of fucosylated glycans in human host–microbiome interactions. Int. J. Mol. Sci. 2021, 22, 3854. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| D4 | JBN2301 | ZYAH72 | NJ-35 | J-1 | GYK1 | ML09-119 | AL09-71 | pc104A | ATCC 7966T | |
|---|---|---|---|---|---|---|---|---|---|---|
| Accession No. | CP013965 | CP013178 | CP016989 | CP006870 | CP006883 | CP016392 | CP005966 | CP007566 | CP007576 | CP000462 |
| Date of isolation | 2012 | 2009 | 2015 | 2010 | 1989 | 2001 | 2009 | 2009 | 2010 | 1901 |
| Location | Wuhan, China | Wuhan, China | Wuhan, China | Nanjing, China | Nanjing, China | Guangzhou, China | Mississippi State USA | West Alabama USA | West Alabama USA | USA |
| Host/source | Diseased Fish (Megalobrama amblycephala) | Diseased Fish (Carassius auratus) | Diseased Fish (Carassius auratus) | Diseased Fish (Carassius auratus) | Diseased Fish (Carassius auratus) | Diseased Fish (Siniperca chuatsi) | Diseased Fish (Ictalurus punctatus) | Diseased Fish (Ictalurus punctatus) | Environment (Soil of a Catfish Pond) | Food (Fishy milk) |
| Genome size (bp) | 5,100,520 | 5,127,362 | 5,159,182 | 5,279,644 | 5,000,814 | 4,951,765 | 5,024,500 | 5,023,861 | 5,023,829 | 4,744,448 |
| G + C Content (%) | 60.80 | 60.78 | 60.70 | 60.50 | 60.90 | 60.80 | 60.80 | 60.80 | 60.80 | 61.51 |
| CDS | 4569 | 4438 | 4397 | 4526 | 4268 | 4219 | 4446 | 4297 | 4300 | 4151 |
| rRNAs | 31 | 31 | 31 | 31 | 31 | 31 | 31 | 31 | 31 | 31 |
| tRNAs | 117 | 129 | 123 | 102 | 110 | 114 | 112 | 111 | 111 | 126 |
| ncRNAs | 7 | 1 | 7 | 2 | 2 | 8 | 7 | 2 | 2 | 5 |
| Pseudo Genes | 50 | 47 | 59 | 55 | 51 | 43 | 102 | 51 | 49 | 31 |
| Plasmid | 4 | 3 | - | - | - | - | - | - | - | - |
| Cluster ID | Cluster Type | Most Similar Known Cluster | Similarity (%) | D4 | ZYAH72 | NJ-35 | J1 | GYK1 | JBN2301 | ML09-119 | AL09-71 | Pc104A | ATCC 7966T |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Cluster1 | Arylpolyene | Aryl polyenes, other | 61% | ||||||||||
| Cluster2 | Bacteriocin | ||||||||||||
| Cluster3 | NRPS | Amonabactin P 750, nrps | 100% | ||||||||||
| Cluster4 | Hserlactone | ||||||||||||
| Cluster5 | Bacteriocin |
| Strains | TCS Family/Number | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| OmpR Family | NarL Family | NtrC Family | Chemotaxis Family | Cellcycle Family | LuxR Family | Lux Family | LytTR Family | CitB Family | Sporulation Family | |
| D4 | 69 | 22 | 22 | 22 | 7 | 6 | 4 | 6 | 9 | 5 |
| ZYAH72 | 69 | 22 | 22 | 22 | 7 | 6 | 4 | 6 | 9 | 5 |
| NJ-35 | 69 | 22 | 22 | 22 | 7 | 6 | 4 | 6 | 9 | 5 |
| J-1 | 69 | 22 | 22 | 22 | 7 | 6 | 4 | 6 | 9 | 5 |
| GYK1 | 69 | 22 | 22 | 22 | 7 | 6 | 4 | 6 | 9 | 5 |
| JNB2301 | 69 | 22 | 22 | 22 | 7 | 6 | 4 | 6 | 9 | 5 |
| ML09-119 | 69 | 22 | 22 | 22 | 7 | 6 | 4 | 6 | 9 | 5 |
| AL09-71 | 69 | 22 | 22 | 22 | 7 | 6 | 4 | 6 | 9 | 5 |
| pc104A | 69 | 22 | 22 | 22 | 7 | 6 | 4 | 6 | 9 | 5 |
| ATCC 7966T | 68 | 22 | 21 | 22 | 7 | 6 | 4 | 6 | 10 | 5 |
| Plasmid | Plasmid Size (bp) | GC Content % | Numbers of CDSs | Most Similar Plasmid | Similarity % |
|---|---|---|---|---|---|
| pAhD4-1 | 156,086 | 50.12 | 164 | pHX3 | 81% |
| pAhD4-2 | 6318 | 56.25% | 11 | pAhJBN2301-1 | 100% |
| pAhD4-3 | 6163 | 54.28 | 6 | pAhJBN2301-2 | 99% |
| pAhD4-4 | 6045 | 51.50% | 9 | pAhJNB2301-3 | 100% |
| pAhJBN2301-1 | 6318 | 56.25% | 11 | pAhD4-2 | 100% |
| pAhJBN2301-2 | 6162 | 54.28% | 6 | pAhD4-3 | 99% |
| pAhJBN2301-3 | 6045 | 51.50% | 9 | pAhD4-4 | 100% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, L.; Kang, X.; Wang, Z.; Xiao, Z.; Luo, Y. Genomic Insights into the Pathogenicity of Hypervirulent Aeromonas hydrophila Strain D4 Isolated from Diseased Blunt Snout Bream with the Epidemic Sequence Type 251 Clones. Pathogens 2025, 14, 570. https://doi.org/10.3390/pathogens14060570
Xu L, Kang X, Wang Z, Xiao Z, Luo Y. Genomic Insights into the Pathogenicity of Hypervirulent Aeromonas hydrophila Strain D4 Isolated from Diseased Blunt Snout Bream with the Epidemic Sequence Type 251 Clones. Pathogens. 2025; 14(6):570. https://doi.org/10.3390/pathogens14060570
Chicago/Turabian StyleXu, Li, Xingyu Kang, Zhicheng Wang, Zuyuan Xiao, and Yi Luo. 2025. "Genomic Insights into the Pathogenicity of Hypervirulent Aeromonas hydrophila Strain D4 Isolated from Diseased Blunt Snout Bream with the Epidemic Sequence Type 251 Clones" Pathogens 14, no. 6: 570. https://doi.org/10.3390/pathogens14060570
APA StyleXu, L., Kang, X., Wang, Z., Xiao, Z., & Luo, Y. (2025). Genomic Insights into the Pathogenicity of Hypervirulent Aeromonas hydrophila Strain D4 Isolated from Diseased Blunt Snout Bream with the Epidemic Sequence Type 251 Clones. Pathogens, 14(6), 570. https://doi.org/10.3390/pathogens14060570