
Pathophysiology of COVID-19: A Post Hoc Analysis of the ICAT-COVID Clinical Trial of the Bradykinin Antagonist Icatibant

 , , and
, , and
Abstract
1. Introduction
2. Materials and Methods
2.1. Study Design, Patients, Procedures, and Outcomes
2.2. Statistical Analysis
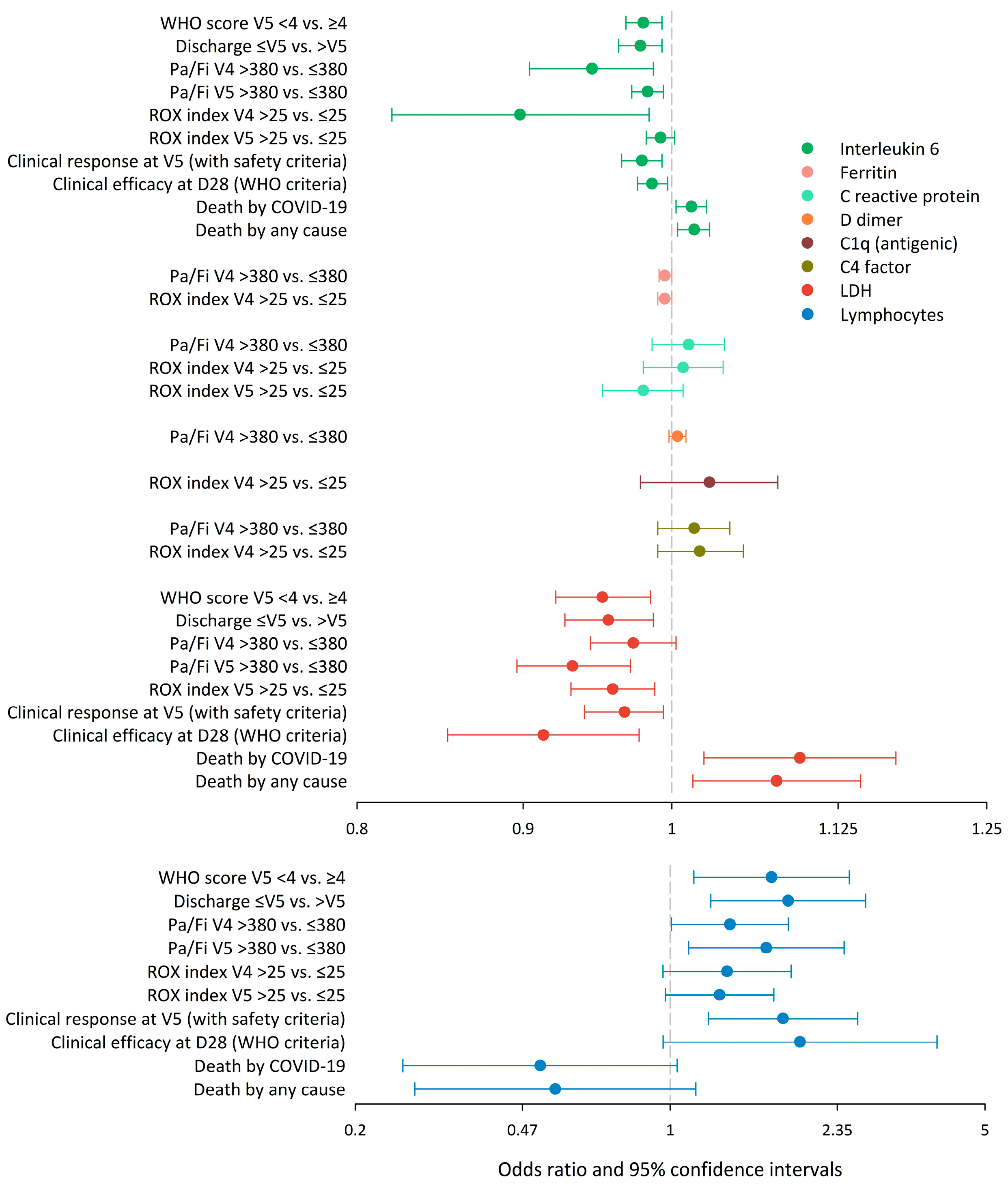
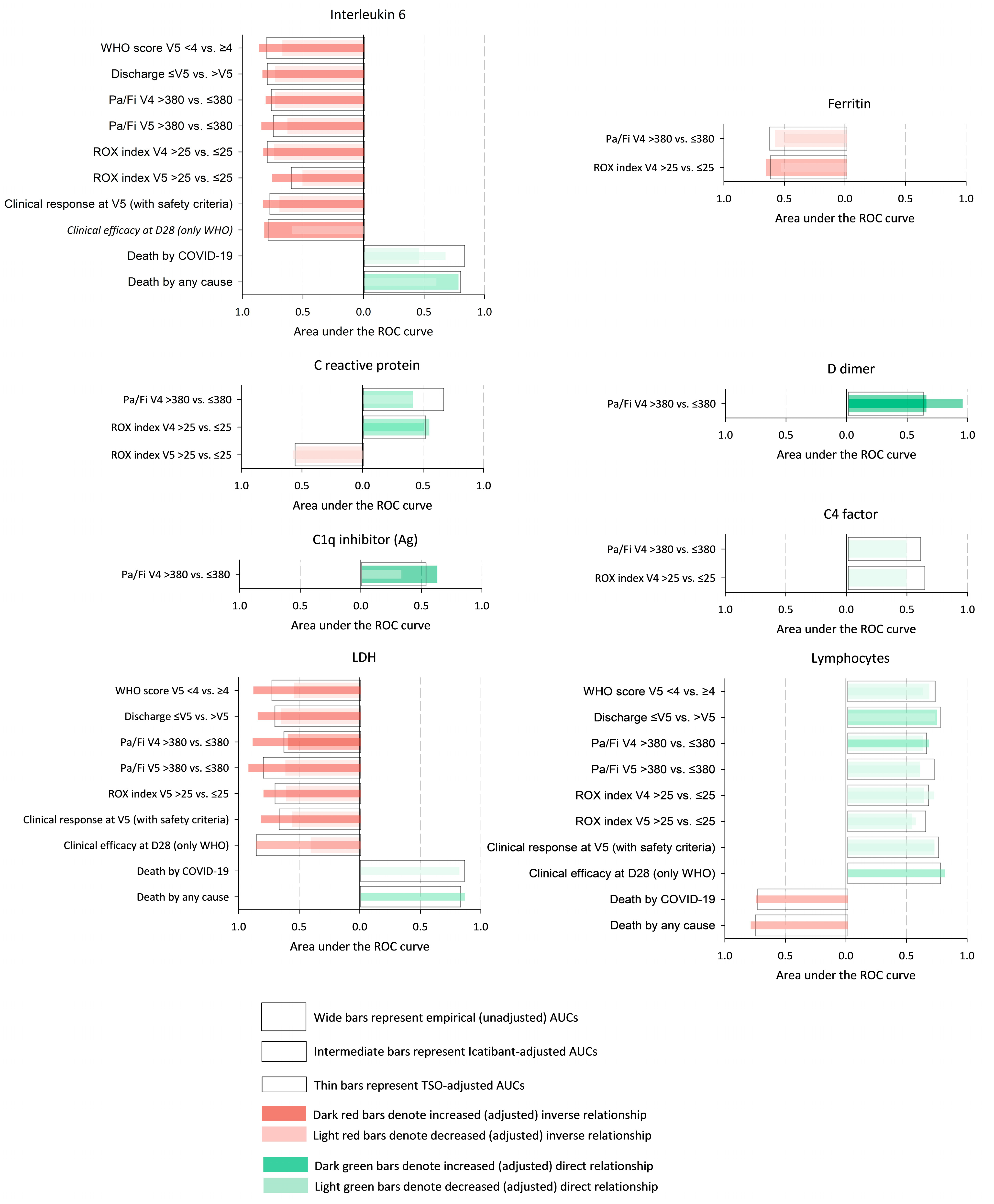
3. Results
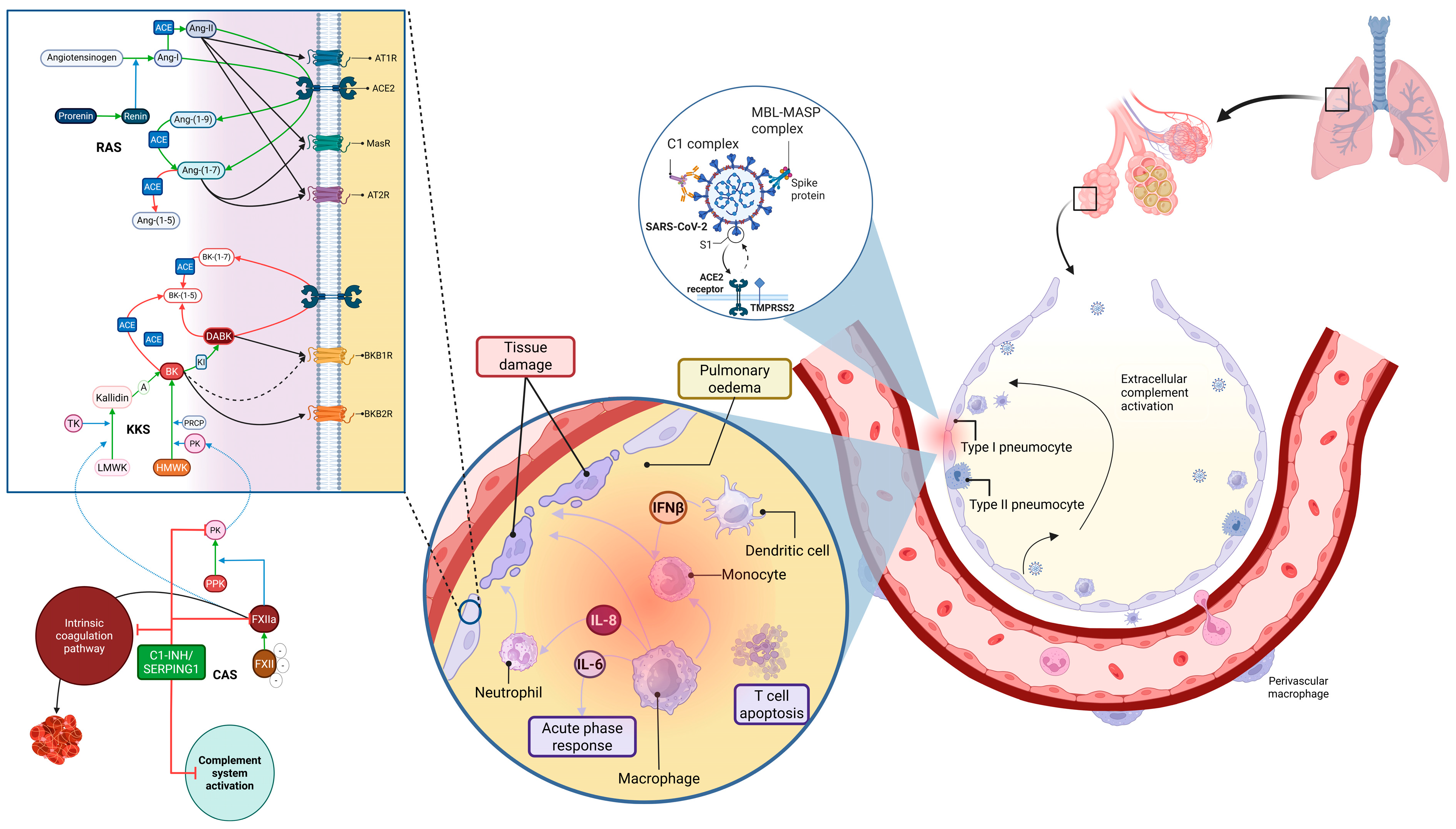
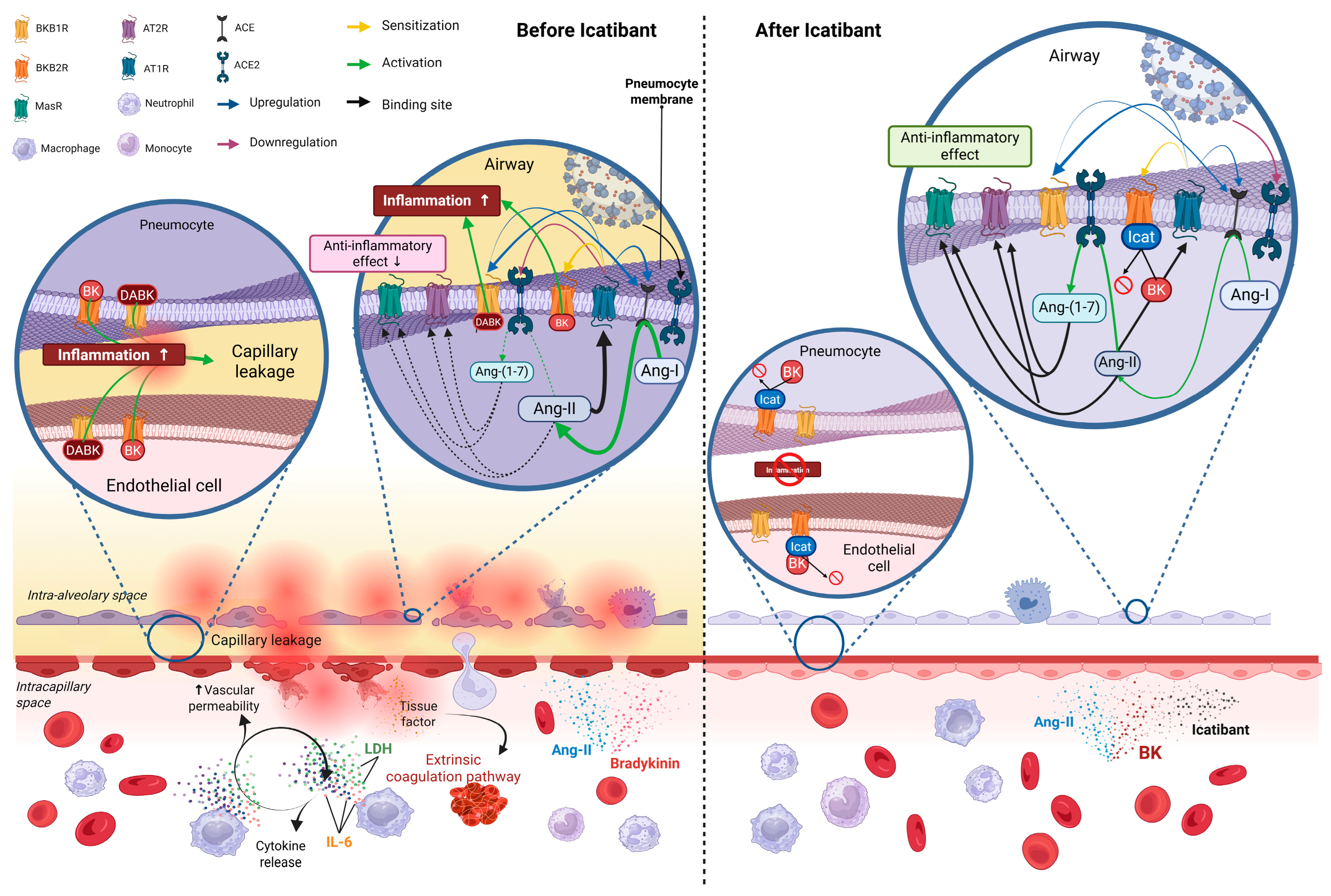
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ACE2 | Angiotensin converting enzyme 2 |
| Ang | Angiotensin |
| BK | Bradykinin |
| BKB1R | Bradykinin B1 receptor |
| BKB2R | Bradykinin B2 receptor |
| C | Complement system |
| CI | Confidence interval |
| COVID-19 | Coronavirus disease 2019 |
| DABK | Des-Arg9 bradykinin |
| Fi | Inspiratory oxygen fraction |
| IL | Interleukin |
| KKS | Kallikrein–kinin system |
| LDH | Lactate dehydrogenase |
| P | Probability |
| Pa | Partial (arterial) oxygen pressure |
| RAS | Renin angiotensin system |
| RD | Risk difference |
| ROC | Receiver operating characteristic |
| SARS-CoV-2 | Severe acute respiratory syndrome coronavirus 2 |
| SoC | Standard of care |
| V4 | Visit 4 |
| V5 | Visit 5 |
| VIP | Variable influence on projection |
| WHO | World Health Organization |
References
- Johns Hopkins University & Medicine. Coronavirus Resource Center, Baltimore, MD, USA, 2023. Available online: https://coronavirus.jhu.edu/map.html (accessed on 11 July 2023).
- van de Veerdonk, F.L.; Giamarellos-Bourboulis, E.; Pickkers, P.; Derde, L.; Leavis, H.; van Crevel, R.; Engel, J.J.; Wiersinga, W.J.; Vlaar, A.P.J.; Shankar-Hari, M.; et al. A guide to immunotherapy for COVID-19. Nat. Med. 2022, 28, 39–50. [Google Scholar] [CrossRef] [PubMed]
- Yuki, K.; Fujiogi, M.; Koutsogiannaki, S. COVID-19 pathophysiology: A review. Clin. Immunol. 2020, 215, 108427. [Google Scholar] [CrossRef] [PubMed]
- Mehta, P.; McAuley, D.F.; Brown, M.; Sanchez, E.; Tattersall, R.S.; Manson, J.J.; Hlh Across Speciality Collaboration, U.K. COVID-19: Consider cytokine storm syndromes and immunosuppression. Lancet 2020, 395, 1033–1034. [Google Scholar] [CrossRef] [PubMed]
- Alfaro, E.; Diaz-Garcia, E.; Garcia-Tovar, S.; Zamarron, E.; Mangas, A.; Galera, R.; Nanwani-Nanwani, K.; Perez-de-Diego, R.; Lopez-Collazo, E.; Garcia-Rio, F.; et al. Impaired Kallikrein-Kinin System in COVID-19 Patients’ Severity. Front. Immunol. 2022, 13, 909342. [Google Scholar] [CrossRef]
- van de Veerdonk, F.L.; Netea, M.G.; van Deuren, M.; van der Meer, J.W.M.; de Mast, Q.; Brüggemann, R.J.; van der Hoeven, H. Kallikrein-kinin blockade in patients with COVID-19 to prevent acute respiratory distress syndrome. eLife 2020, 9, e57555. [Google Scholar] [CrossRef]
- Cooper, S.L.; Boyle, E.; Jefferson, S.R.; Heslop, C.R.A.; Mohan, P.; Mohanraj, G.G.J.; Sidow, H.A.; Tan, R.C.P.; Hill, S.J.; Woolard, J. Role of the Renin-Angiotensin-Aldosterone and Kinin-Kallikrein Systems in the Cardiovascular Complications of COVID-19 and Long COVID. Int. J. Mol. Sci. 2021, 22, 8255. [Google Scholar] [CrossRef]
- Bastolla, U.; Chambers, P.; Abia, D.; Garcia-Bermejo, M.-L.; Fresno, M. Is COVID-19 Severity Associated With ACE2 Degradation? Front. Drug Discov. 2022, 1, 789710. [Google Scholar] [CrossRef]
- Simoes e Silva, A.C.; Silveira, K.D.; Ferreira, A.J.; Teixeira, M.M. ACE2, angiotensin-(1–7) and Mas receptor axis in inflammation and fibrosis. Br. J. Pharmacol. 2013, 169, 477–492. [Google Scholar] [CrossRef]
- Mahmudpour, M.; Roozbeh, J.; Keshavarz, M.; Farrokhi, S.; Nabipour, I. COVID-19 cytokine storm: The anger of inflammation. Cytokine 2020, 133, 155151. [Google Scholar] [CrossRef]
- Domingo, P.; Mur, I.; Pomar, V.; Corominas, H.; Casademont, J.; de Benito, N. The four horsemen of a viral Apocalypse: The pathogenesis of SARS-CoV-2 infection (COVID-19). EBioMedicine 2020, 58, 102887. [Google Scholar] [CrossRef]
- Rasaeifar, B.; Gomez-Gutierrez, P.; Perez, J.J. Molecular Features of Non-Selective Small Molecule Antagonists of the Bradykinin Receptors. Pharmaceuticals 2020, 13, 259. [Google Scholar] [CrossRef] [PubMed]
- Mugisho, O.O.; Robilliard, L.D.; Nicholson, L.F.B.; Graham, E.S.; O’Carroll, S.J. Bradykinin receptor-1 activation induces inflammation and increases the permeability of human brain microvascular endothelial cells. Cell Biol. Int. 2020, 44, 343–351. [Google Scholar] [CrossRef] [PubMed]
- Muus, C.; Luecken, M.D.; Eraslan, G.; Sikkema, L.; Waghray, A.; Heimberg, G.; Kobayashi, Y.; Vaishnav, E.D.; Subramanian, A.; Smillie, C.; et al. Single-cell meta-analysis of SARS-CoV-2 entry genes across tissues and demographics. Nat. Med. 2021, 27, 546–559. [Google Scholar] [CrossRef]
- Malchair, P.; Giol, J.; Garcia, V.; Rodriguez, O.; Ruibal, J.C.; Zarauza, A.; Llopis, F.; Matellan, L.; Bernal, T.; Solis, B.; et al. Three-Day Icatibant on Top of Standard Care in Patients With Coronavirus Disease 2019 Pneumonia: A Randomized, Open-Label, Phase 2, Proof-of-Concept Trial. Clin. Infect. Dis. 2023, 76, 1784–1792. [Google Scholar] [CrossRef]
- Pepe, M.S. Covariate effects on continuous and ordinal tests. In The Statistical Evaluation of Medical Tests for Classification and Prediction; Oxford University Press: New York, NY, USA, 2003; pp. 130–167. [Google Scholar]
- Pepe, M.S. The binormal ROC curve. In The Statistical Evaluation of Medical Tests for Classification and Prediction; Oxford University Press: New York, NY, USA, 2003; pp. 81–84. [Google Scholar]
- Arturi, F.; Melegari, G.; Giansante, A.; Giuliani, E.; Bertellini, E.; Barbieri, A. COVID-19 Biomarkers for Critically Ill Patients: A Compendium for the Physician. Neurol. Int. 2023, 15, 881–895. [Google Scholar] [CrossRef] [PubMed]
- Semiz, S. COVID19 biomarkers: What did we learn from systematic reviews? Front. Cell Infect. Microbiol. 2022, 12, 1038908. [Google Scholar] [CrossRef]
- Andrianto; Al-Farabi, M.J.; Nugraha, R.A.; Marsudi, B.A.; Azmi, Y. Biomarkers of endothelial dysfunction and outcomes in coronavirus disease 2019 (COVID-19) patients: A systematic review and meta-analysis. Microvasc. Res. 2021, 138, 104224. [Google Scholar] [CrossRef]
- Spy Covid Consortium. Report of the first seven agents in the I-SPY COVID trial: A phase 2, open label, adaptive platform randomised controlled trial. eClinicalMedicine 2023, 58, 101889. [Google Scholar] [CrossRef]
- Murakami, M.; Kamimura, D.; Hirano, T. Pleiotropy and Specificity: Insights from the Interleukin 6 Family of Cytokines. Immunity 2019, 50, 812–831. [Google Scholar] [CrossRef]
- Coomes, E.A.; Haghbayan, H. Interleukin-6 in COVID-19: A systematic review and meta-analysis. Rev. Med. Virol. 2020, 30, 1–9. [Google Scholar] [CrossRef]
- REMAP-CAP Investigators; Gordon, A.C.; Mouncey, P.R.; Al-Beidh, F.; Rowan, K.M.; Nichol, A.D.; Arabi, Y.M.; Annane, D.; Beane, A.; van Bentum-Puijk, W.; et al. Interleukin-6 Receptor Antagonists in Critically Ill Patients with COVID-19. N. Engl. J. Med. 2021, 384, 1491–1502. [Google Scholar]
- Venet, F.; Cour, M.; Rimmele, T.; Viel, S.; Yonis, H.; Coudereau, R.; Amaz, C.; Abraham, P.; Monard, C.; Casalegno, J.S.; et al. Longitudinal assessment of IFN-I activity and immune profile in critically ill COVID-19 patients with acute respiratory distress syndrome. Crit. Care 2021, 25, 140. [Google Scholar] [CrossRef] [PubMed]
- Chan, F.K.; Moriwaki, K.; De Rosa, M.J. Detection of necrosis by release of lactate dehydrogenase activity. Methods Mol. Biol. 2013, 979, 65–70. [Google Scholar] [PubMed]
- Malik, P.; Patel, U.; Mehta, D.; Patel, N.; Kelkar, R.; Akrmah, M.; Gabrilove, J.L.; Sacks, H. Biomarkers and outcomes of COVID-19 hospitalisations: Systematic review and meta-analysis. BMJ Evid. Based Med. 2021, 26, 107–108. [Google Scholar] [CrossRef]
- Terpos, E.; Ntanasis-Stathopoulos, I.; Elalamy, I.; Kastritis, E.; Sergentanis, T.N.; Politou, M.; Psaltopoulou, T.; Gerotziafas, G.; Dimopoulos, M.A. Hematological findings and complications of COVID-19. Am. J. Hematol. 2020, 95, 834–847. [Google Scholar] [CrossRef]
- Levi, M.; Thachil, J.; Iba, T.; Levy, J.H. Coagulation abnormalities and thrombosis in patients with COVID-19. Lancet Haematol. 2020, 7, e438–e440. [Google Scholar] [CrossRef]
- Thachil, J. The protective rather than prothrombotic fibrinogen in COVID-19 and other inflammatory states. J. Thromb. Haemost. 2020, 18, 1849–1852. [Google Scholar] [CrossRef]
- Kaushal, K.; Kaur, H.; Sarma, P.; Bhattacharyya, A.; Sharma, D.J.; Prajapat, M.; Pathak, M.; Kothari, A.; Kumar, S.; Rana, S.; et al. Serum ferritin as a predictive biomarker in COVID-19. A systematic review, meta-analysis and meta-regression analysis. J. Crit. Care 2022, 67, 172–181. [Google Scholar] [CrossRef]
- Smilowitz, N.R.; Kunichoff, D.; Garshick, M.; Shah, B.; Pillinger, M.; Hochman, J.S.; Berger, J.S. C-reactive protein and clinical outcomes in patients with COVID-19. Eur. Heart J. 2021, 42, 2270–2279. [Google Scholar] [CrossRef]
- Wang, G.; Ye, Y.; Zhang, X.; Song, J. Bradykinin stimulates IL-6 production and cell invasion in colorectal cancer cells. Oncol. Rep. 2014, 32, 1709–1714. [Google Scholar] [CrossRef]
- Tabassum, A.; Iqbal, M.S.; Sultan, S.; Alhuthali, R.A.; Alshubaili, D.I.; Sayyam, R.S.; Abyad, L.M.; Qasem, A.H.; Arbaeen, A.F. Dysregulated Bradykinin: Mystery in the Pathogenesis of COVID-19. Mediat. Inflamm. 2022, 2022, 7423537. [Google Scholar] [CrossRef] [PubMed]
- Ghosn, L.; Assi, R.; Evrenoglou, T.; Buckley, B.S.; Henschke, N.; Probyn, K.; Riveros, C.; Davidson, M.; Grana, C.; Bonnet, H.; et al. Interleukin-6 blocking agents for treating COVID-19: A living systematic review. Cochrane Database Syst. Rev. 2023, 6, CD013881. [Google Scholar] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Icatibant Group (n = 37) | SoC (Control) Group (n = 36) | |
|---|---|---|
| Median age (IQR)—years | 49.0 (41.0–59.0) | 56.5 (46.8–70.2) |
| Male sex—no. (%) | 27 (73.0) | 22 (61.1) |
| Median body mass index (IQR)—kg/m2 1 | 28.2 (25.7–36.3) | 30.3 (26.3–33.0) |
| Median respiratory rate (IQR)—breaths/minute | 21.0 (18.0–25.0) | 20.0 (18.0–22.0) |
| (IQR)—mmHg 2 | 71.0 (65.0–84.0) | 71.0 (63.8–79.2) |
| (IQR)—unitless | 0.28 (0.21–0.32) | 0.28 (0.21–0.33) |
| (IQR)—unitless | 261.0 (212.0–317.0) | 263.0 (210.0–320.0) |
| (IQR)—% 3 | 96.0 (95.0–97.0) | 96.0 (95.0–97.0) |
| (IQR)—unitless 3 | 0.30 (0.28–0.35) | 0.31 (0.28–0.35) |
| (IQR)—unitless 3 | 326.0 (274.0–346.0) | 308.0 (275.0–343.0) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Malchair, P.; Giol, J.; Jacob, J.; Villoria, J.; Carnaval, T.; Videla, S. Pathophysiology of COVID-19: A Post Hoc Analysis of the ICAT-COVID Clinical Trial of the Bradykinin Antagonist Icatibant. Pathogens 2025, 14, 533. https://doi.org/10.3390/pathogens14060533
Malchair P, Giol J, Jacob J, Villoria J, Carnaval T, Videla S. Pathophysiology of COVID-19: A Post Hoc Analysis of the ICAT-COVID Clinical Trial of the Bradykinin Antagonist Icatibant. Pathogens. 2025; 14(6):533. https://doi.org/10.3390/pathogens14060533
Chicago/Turabian StyleMalchair, Pierre, Jordi Giol, Javier Jacob, Jesús Villoria, Thiago Carnaval, and Sebastián Videla. 2025. "Pathophysiology of COVID-19: A Post Hoc Analysis of the ICAT-COVID Clinical Trial of the Bradykinin Antagonist Icatibant" Pathogens 14, no. 6: 533. https://doi.org/10.3390/pathogens14060533
APA StyleMalchair, P., Giol, J., Jacob, J., Villoria, J., Carnaval, T., & Videla, S. (2025). Pathophysiology of COVID-19: A Post Hoc Analysis of the ICAT-COVID Clinical Trial of the Bradykinin Antagonist Icatibant. Pathogens, 14(6), 533. https://doi.org/10.3390/pathogens14060533