A Novel flaB Gene-Based Profiling Approach for the Rapid and Accurate Detection of Borreliella and Borrelia Species in Ticks

 ,
,  ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. Tick Collection, DNA Extraction, and Pooling of Isolates
2.2. Designing PCR Primers for flaB Gene-Based Profiling
2.3. Screening for Borreliaceae DNA Using Nested PCR Test
2.4. Screening for Borreliaceae DNA Using V4 rrs Gene-Based Profiling
2.5. Screening for Borreliaceae DNA Using flaB Gene-Based Profiling
2.6. Library Preparation and Sequencing
2.7. Bioinformatic and Statistical Analyses
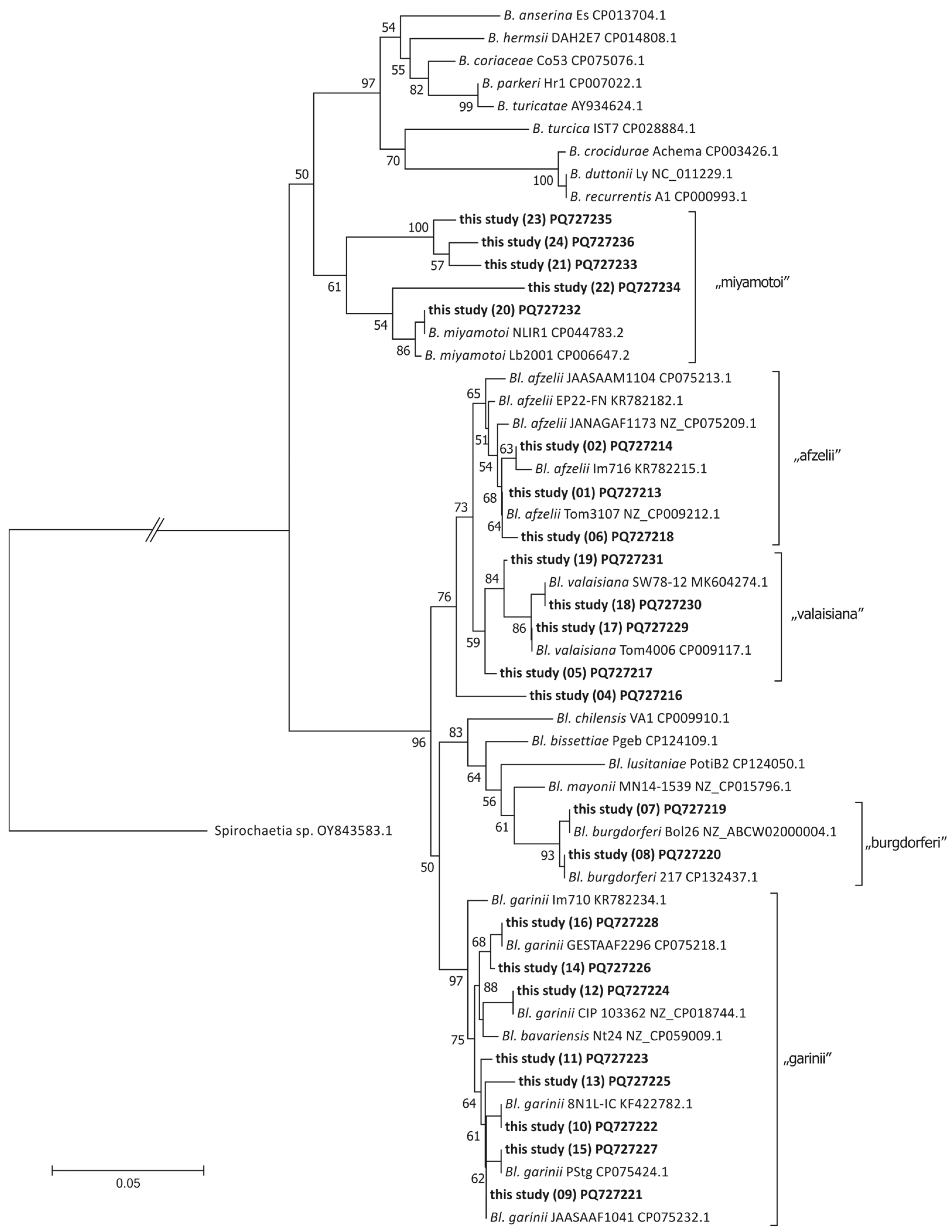
2.8. Phylogenetic Analysis
3. Results
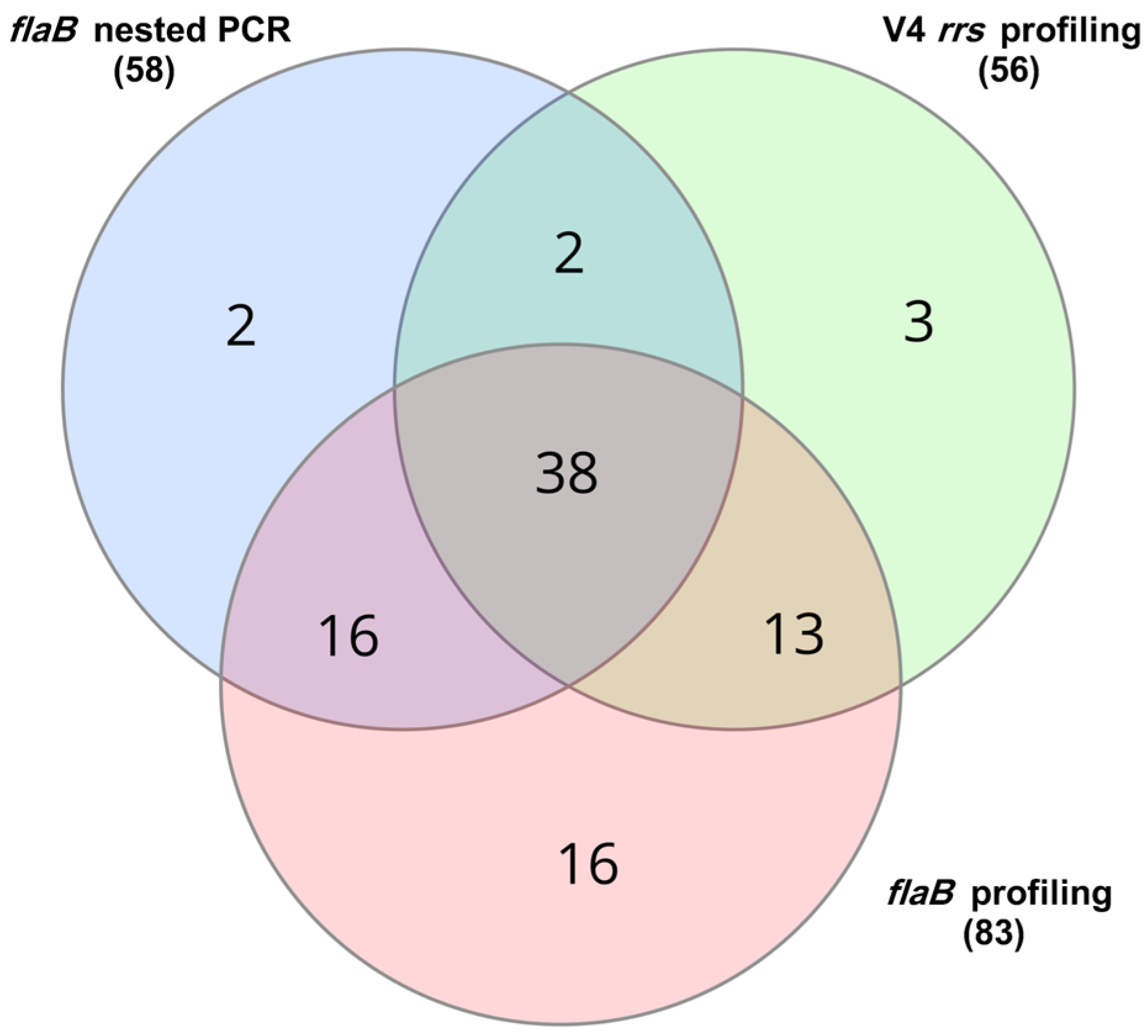
3.1. Screening by Conventional Nested PCR
3.2. Screening by V4 rrs Gene-Based Profiling
3.3. Screening by flaB Gene-Based Profiling
3.4. Flagellin Gene Sequence Variants
3.5. Borreliaceae Species Identified Using flaB Gene-Based Profiling
3.6. Microbial Profiling Using V4 rrs Marker
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kulisz, J.; Hoeks, S.; Kunc-Kozioł, R.; Woźniak, A.; Zając, Z.; Schipper, A.M.; Cabezas-Cruz, A.; Huijbregts, M.A.J. Spatiotemporal trends and covariates of Lyme borreliosis incidence in Poland, 2010–2019. Sci. Rep. 2024, 14, 10768. [Google Scholar] [CrossRef]
- Medlock, J.M.; Hansford, K.M.; Bormane, A.; Derdakova, M.; Estrada-Peña, A.; George, J.-C.; Golovljova, I.; Jaenson, T.G.T.; Jensen, J.-K.; Jensen, P.M.; et al. Driving forces for changes in geographical distribution of Ixodes ricinus ticks in Europe. Parasites Vectors 2013, 6, 1. [Google Scholar] [CrossRef] [PubMed]
- De La Fuente, J.; Estrada-Peña, A.; Rafael, M.; Almazán, C.; Bermúdez, S.; Abdelbaset, A.E.; Kasaija, P.D.; Kabi, F.; Akande, F.A.; Ajagbe, D.O.; et al. Perception of Ticks and Tick-Borne Diseases Worldwide. Pathogens 2023, 12, 1258. [Google Scholar] [CrossRef]
- Le Dortz, L.L.; Rouxel, C.; Polack, B.; Boulouis, H.-J.; Lagrée, A.-C.; Deshuillers, P.L.; Haddad, N. Tick-borne diseases in Europe: Current prevention, control tools and the promise of aptamers. Vet. Parasitol. 2024, 328, 110190. [Google Scholar] [CrossRef] [PubMed]
- Pustijanac, E.; Buršić, M.; Millotti, G.; Paliaga, P.; Iveša, N.; Cvek, M. Tick-Borne Bacterial Diseases in Europe: Threats to public health. Eur. J. Clin. Microbiol. Infect. Dis. 2024, 43, 1261–1295. [Google Scholar] [CrossRef] [PubMed]
- Burn, L.; Tran, T.M.P.; Pilz, A.; Vyse, A.; Fletcher, M.A.; Angulo, F.J.; Gessner, B.D.; Moïsi, J.C.; Jodar, L.; Stark, J.H. Incidence of Lyme Borreliosis in Europe from National Surveillance Systems (2005–2020). Vector-Borne Zoonotic Dis. 2023, 23, 156–171. [Google Scholar] [CrossRef]
- Paradowska-Stankiewicz, I.; Zbrzeźniak, J.; Skufca, J.; Nagarajan, A.; Ochocka, P.; Pilz, A.; Vyse, A.; Begier, E.; Dzingina, M.; Blum, M.; et al. A Retrospective Database Study of Lyme Borreliosis Incidence in Poland from 2015 to 2019: A Public Health Concern. Vector-Borne Zoonotic Dis. 2023, 23, 247–255. [Google Scholar] [CrossRef]
- Barbour, A.G.; Gupta, R.S. The Family Borreliaceae (Spirochaetales), a Diverse Group in Two Genera of Tick-Borne Spirochetes of Mammals, Birds, and Reptiles. J. Med. Entomol. 2021, 58, 1513–1524. [Google Scholar] [CrossRef]
- Gupta, R.S. Distinction between Borrelia and Borreliella is more robustly supported by molecular and phenotypic characteristics than all other neighbouring prokaryotic genera: Response to Margos’ et al. “The genus Borrelia reloaded” (PLoS ONE 13(12): e0208432). PLoS ONE 2019, 14, e0221397. [Google Scholar] [CrossRef]
- Estrada-Peña, A.; Cutler, S.; Potkonjak, A.; Vassier-Tussaut, M.; Van Bortel, W.; Zeller, H.; Fernández-Ruiz, N.; Mihalca, A.D. An updated meta-analysis of the distribution and prevalence of Borrelia burgdorferi s.l. in ticks in Europe. Int. J. Health Geogr. 2018, 17, 41. [Google Scholar] [CrossRef]
- Dong, Y.; Zhou, G.; Cao, W.; Xu, X.; Zhang, Y.; Ji, Z.; Yang, J.; Chen, J.; Liu, M.; Fan, Y.; et al. Global seroprevalence and sociodemographic characteristics of Borrelia burgdorferi sensu lato in human populations: A systematic review and meta-analysis. BMJ Glob. Health 2022, 7, e007744. [Google Scholar] [CrossRef]
- Tokarz, R.; Lipkin, W.I. Discovery and Surveillance of Tick-Borne Pathogens. J. Med. Entomol. 2021, 58, 1525–1535. [Google Scholar] [CrossRef] [PubMed]
- Zinck, C.B.; Raveendram Thampy, P.; Uhlemann, E.M.E.; Adam, H.; Wachter, J.; Suchan, D.; Cameron, A.D.S.; Rego, R.O.M.; Brisson, D.; Bouchard, C.; et al. Variation among strains of Borrelia burgdorferi in host tissue abundance and lifetime transmission determine the population strain structure in nature. PLoS Pathog. 2023, 19, e1011572. [Google Scholar] [CrossRef]
- Carter, C.J.; Bergström, S.; Norris, S.J.; Barbour, A.G. A family of surface-exposed proteins of 20 kilodaltons in the genus Borrelia. Infect. Immun. 1994, 62, 2792–2799. [Google Scholar] [CrossRef] [PubMed]
- Stone, B.L.; Brissette, C.A. Host Immune Evasion by Lyme and Relapsing Fever Borreliae: Findings to Lead Future Studies for Borrelia miyamotoi. Front. Immunol. 2017, 8, 12. [Google Scholar] [CrossRef] [PubMed]
- Wodecka, B.; Leońska, A.; Skotarczak, B. A comparative analysis of molecular markers for the detection and identification of Borrelia spirochaetes in Ixodes ricinus. J. Med. Microbiol. 2010, 59, 309–314. [Google Scholar] [CrossRef]
- Graham, C.B.; Maes, S.E.; Hojgaard, A.; Fleshman, A.C.; Sheldon, S.W.; Eisen, R.J. A molecular algorithm to detect and differentiate human pathogens infecting Ixodes scapularis and Ixodes pacificus (Acari: Ixodidae). Ticks Tick-Borne Dis. 2018, 9, 390–403. [Google Scholar] [CrossRef]
- Wodecka, B. flaB Gene as a Molecular Marker for Distinct Identification of Borrelia Species in Environmental Samples by the PCR-Restriction Fragment Length Polymorphism Method. Appl. Environ. Microbiol. 2011, 77, 7088–7092. [Google Scholar] [CrossRef]
- Wodecka, B.; Kolomiiets, V. Genetic Diversity of Borreliaceae Species Detected in Natural Populations of Ixodes ricinus Ticks in Northern Poland. Life 2023, 13, 972. [Google Scholar] [CrossRef]
- Osikowicz, L.M.; Rizzo, M.R.; Hojgaard, A.; Maes, S.E.; Eisen, R.J. Detection of Borrelia burgdorferi sensu lato species in host-seeking Ixodes species ticks in the United States. Ticks Tick-Borne Dis. 2024, 15, 102270. [Google Scholar] [CrossRef]
- Yang, J.; Liu, Z.; Guan, G.; Che, R.; Niu, Q.; Li, Y.; Liu, J.; Ma, M.; Ren, Q.; Liu, A.; et al. Evaluation of molecular methods for detection of Borrelia burgdorferi senso lato in ticks. Diagn. Microbiol. Infect. Dis. 2012, 73, 80–83. [Google Scholar] [CrossRef] [PubMed]
- Zhai, B.; Niu, Q.; Liu, Z.; Yang, J.; Pan, Y.; Li, Y.; Zhao, H.; Luo, J.; Yin, H. First detection and molecular identification of Borrelia species in Bactrian camel (Camelus bactrianus) from Northwest China. Infect. Genet. Evol. 2018, 64, 149–155. [Google Scholar] [CrossRef] [PubMed]
- Binetruy, F.; Garnier, S.; Boulanger, N.; Talagrand-Reboul, É.; Loire, E.; Faivre, B.; Noël, V.; Buysse, M.; Duron, O. A novel Borrelia species, intermediate between Lyme disease and relapsing fever groups, in neotropical passerine-associated ticks. Sci. Rep. 2020, 10, 10596. [Google Scholar] [CrossRef]
- Wodecka, B.; Michalik, J.; Grochowalska, R. Red Foxes (Vulpes vulpes) Are Exposed to High Diversity of Borrelia burgdorferi Sensu Lato Species Infecting Fox-Derived Ixodes Ticks in West-Central Poland. Pathogens 2022, 11, 696. [Google Scholar] [CrossRef]
- Trzebny, A.; Slodkowicz-Kowalska, A.; Becnel, J.J.; Sanscrainte, N.; Dabert, M. A new method of metabarcoding Microsporidia and their hosts reveals high levels of microsporidian infections in mosquitoes (Culicidae). Mol. Ecol. Resour. 2020, 20, 1486–1504. [Google Scholar] [CrossRef] [PubMed]
- Michelet, L.; Delannoy, S.; Devillers, E.; Umhang, G.; Aspan, A.; Juremalm, M.; Chirico, J.; van der Wal, F.J.; Sprong, H.; Boye Pihl, T.P.; et al. High-throughput screening of tick-borne pathogens in Europe. Front. Cell. Infect. Microbiol. 2014, 4, 103. [Google Scholar] [CrossRef]
- Hojgaard, A.; Osikowicz, L.M.; Eisen, L.; Eisen, R.J. Evaluation of a novel multiplex PCR amplicon sequencing assay for detection of human pathogens in Ixodes ticks. Ticks Tick. Borne Dis. 2020, 11, 101504. [Google Scholar] [CrossRef]
- Siuda, K. Kleszcze Polski (Acari: Ixodida).: Systematyka i Rozmieszczenie; Polskie Towarzystwo Parazytologiczne: Warsaw, Poland, 1993; Volume 2, ISSN 0540-6722. ISBN 839013490X, 9788390134901. [Google Scholar]
- Rijpkema, S.; Golubic, D.; Molkenboer, M.; Verbeek-De Kruif, N.; Schellekens, J. Identification of four genomic groups of Borrelia burgdorferi sensu lato in Ixodes ricinus ticks collected in a Lyme borreliosis endemic region in northern Croatia. Exp. Appl. Acarol. 1996, 20, 23–30. [Google Scholar] [CrossRef]
- Michalik, J.; Wodecka, B.; Liberska, J.; Dabert, M.; Postawa, T.; Piksa, K.; Stańczak, J. Diversity of Borrelia burgdorferi sensu lato species in Ixodes ticks (Acari: Ixodidae) associated with cave-dwelling bats from Poland and Romania. Ticks Tick-Borne Dis. 2020, 11, 101300. [Google Scholar] [CrossRef]
- Liberska, J.; Michalik, J.F.; Olechnowicz, J.; Dabert, M. Co-Occurrence of Borrelia burgdorferi Sensu Lato and Babesia spp. DNA in Ixodes ricinus Ticks Collected from Vegetation and Pets in the City of Poznań, Poland. Pathogens 2024, 13, 307. [Google Scholar] [CrossRef]
- Hall, T.A. BioEdit: A user-friendly biological sequence alignment editor and analysis program for Windows95/98/NT. Nucleic Acids Symp. Ser. 1999, 41, 9598. [Google Scholar]
- Makowska, N.; Philips, A.; Dabert, M.; Nowis, K.; Trzebny, A.; Koczura, R.; Mokracka, J. Metagenomic analysis of β-lactamase and carbapenemase genes in the wastewater resistome. Water Res. 2020, 170, 115277. [Google Scholar] [CrossRef]
- Therese, K.L.; Anand, A.R.; Madhavan, H.N. Polymerase chain reaction in the diagnosis of bacterial endophthalmitis. Br. J. Ophthalmol. 1998, 82, 1078–1082. [Google Scholar] [CrossRef]
- Hannon, G.J. FASTX-Toolkit. 2010. Available online: https://bio.tools/fastx-toolkit (accessed on 1 September 2024).
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef]
- Edgar, R.C. UNCROSS2: Identification of cross-talk in 16S rRNA OTU tables. BioRxiv 2018, 400762. [Google Scholar] [CrossRef]
- Glöckner, F.O.; Yilmaz, P.; Quast, C.; Gerken, J.; Beccati, A.; Ciuprina, A.; Bruns, G.; Yarza, P.; Peplies, J.; Westram, R.; et al. 25 years of serving the community with ribosomal RNA gene reference databases and tools. J. Biotechnol. 2017, 261, 169–176. [Google Scholar] [CrossRef]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2012, 41, D590–D596. [Google Scholar] [CrossRef] [PubMed]
- Yilmaz, P.; Parfrey, L.W.; Yarza, P.; Gerken, J.; Pruesse, E.; Quast, C.; Schweer, T.; Peplies, J.; Ludwig, W.; Glöckner, F.O.; et al. The SILVA and “All-species Living Tree Project (LTP)” taxonomic frameworks. Nucl. Acids Res. 2014, 42, D643–D648. [Google Scholar] [CrossRef]
- Zhang, Z.; Schwartz, S.; Wagner, L.; Miller, W. A Greedy Algorithm for Aligning DNA Sequences. J. Comput. Biol. 2000, 7, 203–214. [Google Scholar] [CrossRef]
- Morgulis, A.; Coulouris, G.; Raytselis, Y.; Madden, T.L.; Agarwala, R.; Schäffer, A.A. Database indexing for production MegaBLAST searches. Bioinformatics 2008, 24, 1757–1764. [Google Scholar] [CrossRef] [PubMed]
- Nicholas, K.B.; Nicholas, H.B. GeneDoc: A Tool for Editing and Annotating Multiple Sequence Alignments; 1997. Available online: https://api.semanticscholar.org/CorpusID:81058551 (accessed on 1 January 2024).
- McNemar, Q. Note on the sampling error of the difference between correlated proportions or percentages. Psychometrika 1947, 12, 153–157. [Google Scholar] [CrossRef]
- Shimodaira, H.; Hasegawa, M. Multiple Comparisons of Log-Likelihoods with Applications to Phylogenetic Inference. Mol. Biol. Evol. 1999, 16, 1114–1116. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef]
- Taberlet, P.; Griffin, S.; Goossens, B.; Questiau, S.; Manceau, V.; Escaravage, N.; Waits, L.P.; Bouvet, J. Reliable genotyping of samples with very low DNA quantities using PCR. Nucleic Acids Res. 1996, 24, 3189–3194. [Google Scholar] [CrossRef]
- Liberska, J.A.; Michalik, J.F.; Dabert, M. Exposure of dogs and cats to Borrelia miyamotoi infected Ixodes ricinus ticks in urban areas of the city of Poznań, west-central Poland. Ticks Tick-Borne Dis. 2023, 14, 102188. [Google Scholar] [CrossRef] [PubMed]
- Strnad, M.; Hönig, V.; Růžek, D.; Grubhoffer, L.; Rego, R.O.M. Europe-Wide Meta-Analysis of Borrelia burgdorferi Sensu Lato Prevalence in Questing Ixodes ricinus Ticks. Appl. Environ. Microbiol. 2017, 83, e00609-17. [Google Scholar] [CrossRef]
- Sawczyn-Domańska, A.; Zwoliński, J.; Kloc, A.; Wójcik-Fatla, A. Prevalence of Borrelia, Neoehrlichia mikurensis and Babesia in ticks collected from vegetation in eastern Poland. Exp. Appl. Acarol. 2023, 90, 409–428. [Google Scholar] [CrossRef] [PubMed]
- Kulisz, J.; Zając, Z.; Foucault-Simonin, A.; Woźniak, A.; Filipiuk, M.; Kloskowski, J.; Rudolf, R.; Corduneanu, A.; Bartosik, K.; Moutailler, S.; et al. Wide spectrum of tick-borne pathogens in juvenile Ixodes ricinus collected from autumn-migrating birds in the Vistula River Valley, Poland. BMC Vet. Res. 2024, 20, 556. [Google Scholar] [CrossRef]
- Zając, Z.; Kulisz, J.; Woźniak, A.; Bartosik, K.; Foucault-Simonin, A.; Moutailler, S.; Cabezas-Cruz, A. Tick Activity, Host Range, and Tick-Borne Pathogen Prevalence in Mountain Habitats of the Western Carpathians, Poland. Pathogens 2023, 12, 1186. [Google Scholar] [CrossRef]
- Dunaj, J.; Drewnowska, J.; Moniuszko-Malinowska, A.; Swiecicka, I.; Pancewicz, S. First metagenomic report of Borrelia americana and Borrelia carolinensis in Poland—A preliminary study. Ann. Agric. Environ. Med. 2021, 28, 49–55. [Google Scholar] [CrossRef] [PubMed]
- Lau, A.C.C.; Qiu, Y.; Moustafa, M.A.M.; Nakao, R.; Shimozuru, M.; Onuma, M.; Mohd-Azlan, J.; Tsubota, T. Detection of Borrelia burgdorferi sensu lato and relapsing fever borrelia in feeding Ixodes ticks and rodents in Sarawak, Malaysia: New geographical records of borrelia yangtzensis and borrelia miyamotoi. Pathogens 2020, 9, 846. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
| Compared Methods | McNemar Test Statistic 1 | p-Value | Significant Difference 2 |
|---|---|---|---|
| flaB profiling vs. nested PCR | 4 | 1.09 × 10−5 | Yes |
| flaB profiling vs. V4 rrs profiling | 5 | 7.43 × 10−6 | Yes |
| V4 rrs profiling vs. nested PCR | 16 | 0.8642 | No |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Taylor, A.D.; Trzebny, A.; Łośko, M.; Michalik, J.F.; Dabert, M. A Novel flaB Gene-Based Profiling Approach for the Rapid and Accurate Detection of Borreliella and Borrelia Species in Ticks. Pathogens 2025, 14, 506. https://doi.org/10.3390/pathogens14050506
Taylor AD, Trzebny A, Łośko M, Michalik JF, Dabert M. A Novel flaB Gene-Based Profiling Approach for the Rapid and Accurate Detection of Borreliella and Borrelia Species in Ticks. Pathogens. 2025; 14(5):506. https://doi.org/10.3390/pathogens14050506
Chicago/Turabian StyleTaylor, Abigail Dorothea, Artur Trzebny, Małgorzata Łośko, Jerzy Franciszek Michalik, and Miroslawa Dabert. 2025. "A Novel flaB Gene-Based Profiling Approach for the Rapid and Accurate Detection of Borreliella and Borrelia Species in Ticks" Pathogens 14, no. 5: 506. https://doi.org/10.3390/pathogens14050506
APA StyleTaylor, A. D., Trzebny, A., Łośko, M., Michalik, J. F., & Dabert, M. (2025). A Novel flaB Gene-Based Profiling Approach for the Rapid and Accurate Detection of Borreliella and Borrelia Species in Ticks. Pathogens, 14(5), 506. https://doi.org/10.3390/pathogens14050506