
Orientia tsutsugamushi Modulates RIPK3 Cellular Levels but Does Not Inhibit Necroptosis

 , , , and
, , , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Cultivation and Bacterial Infections
2.2. Plasmid Constructs
2.3. Transfection
2.4. Co-Immunoprecipitation and Western Blot
2.5. Immunofluorescence Microscopy
2.6. Necroptosis Induction
2.7. Flow Cytometry
2.8. Statistical Analysis
3. Results
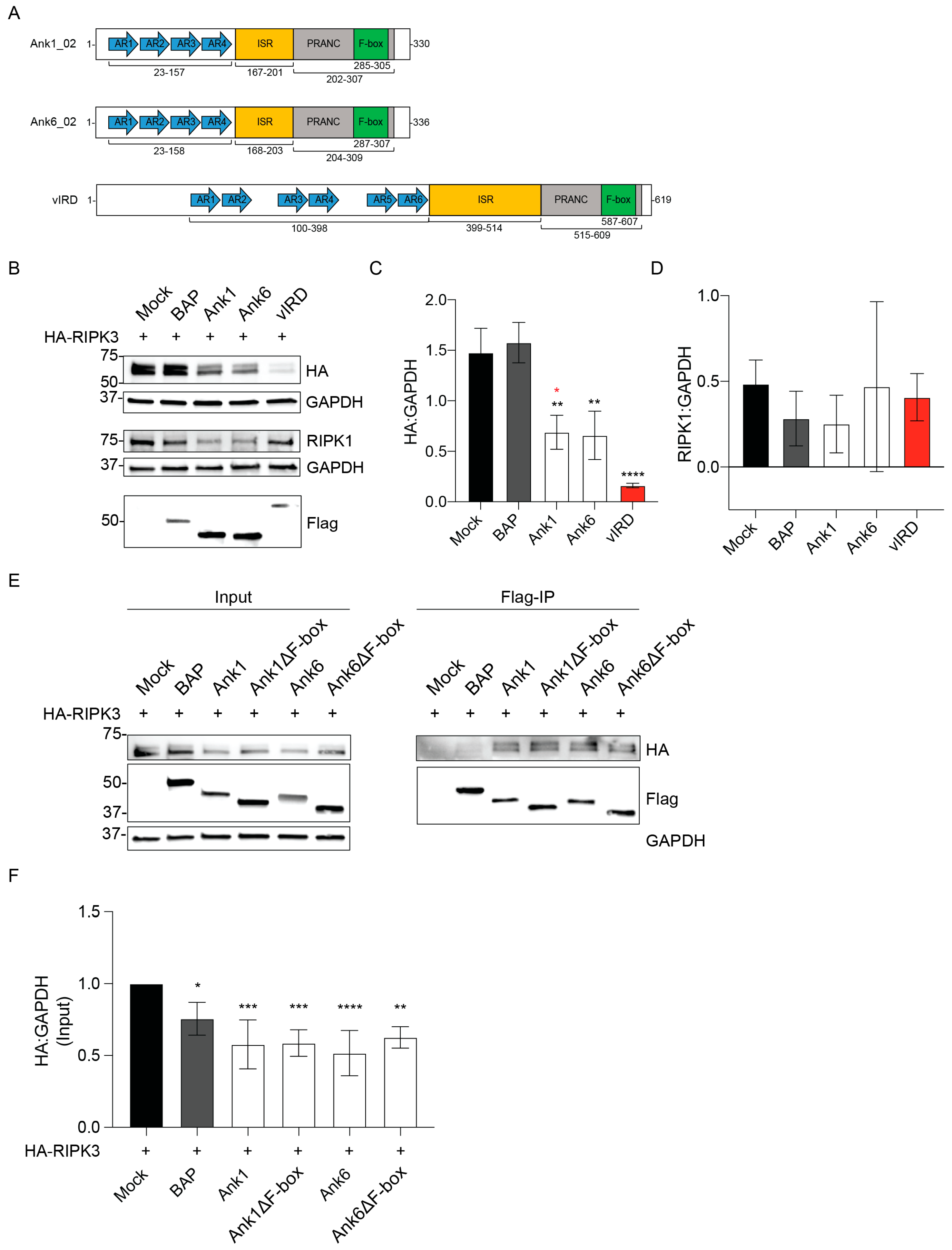
3.1. Ectopically Expressed O. tsutsugamushi Ank1 and Ank6 Reduce RIPK3 Cellular Levels
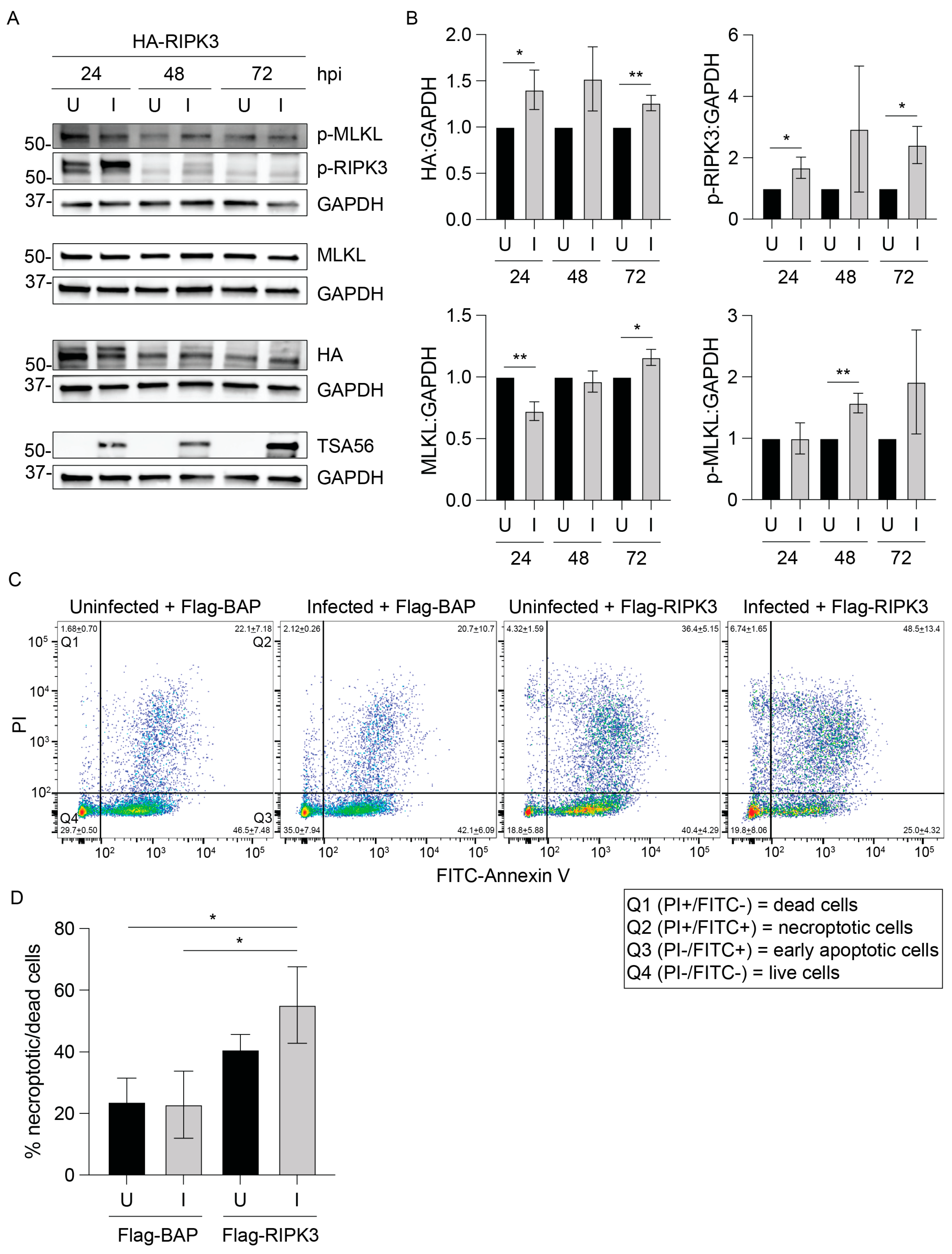
3.2. RIPK3 Levels Are Lower in O. tsutsugamushi-Infected Cells
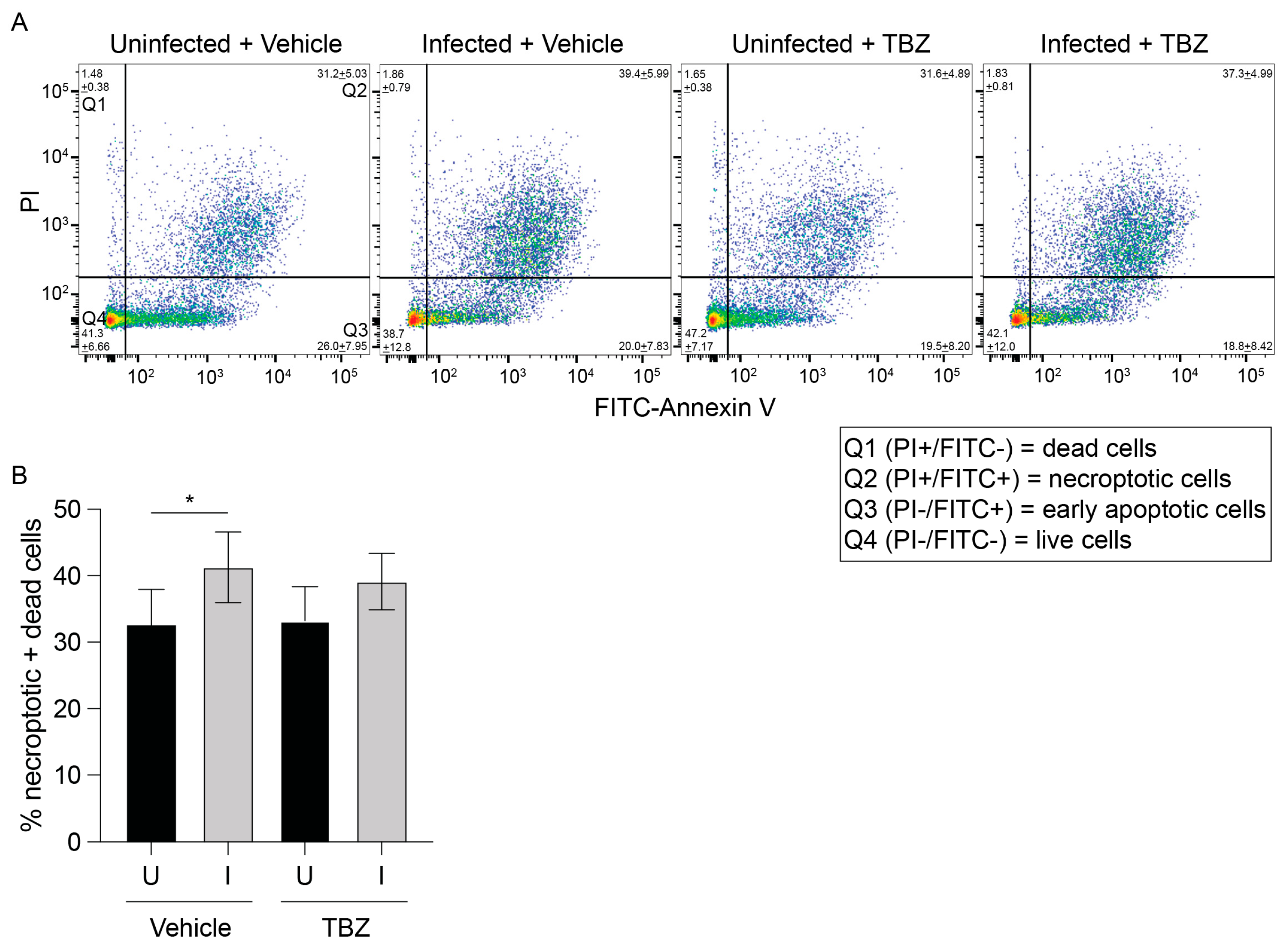
3.3. O. tsutsugamushi Fails to Inhibit Necroptosis Induced by RIPK3 Overexpression
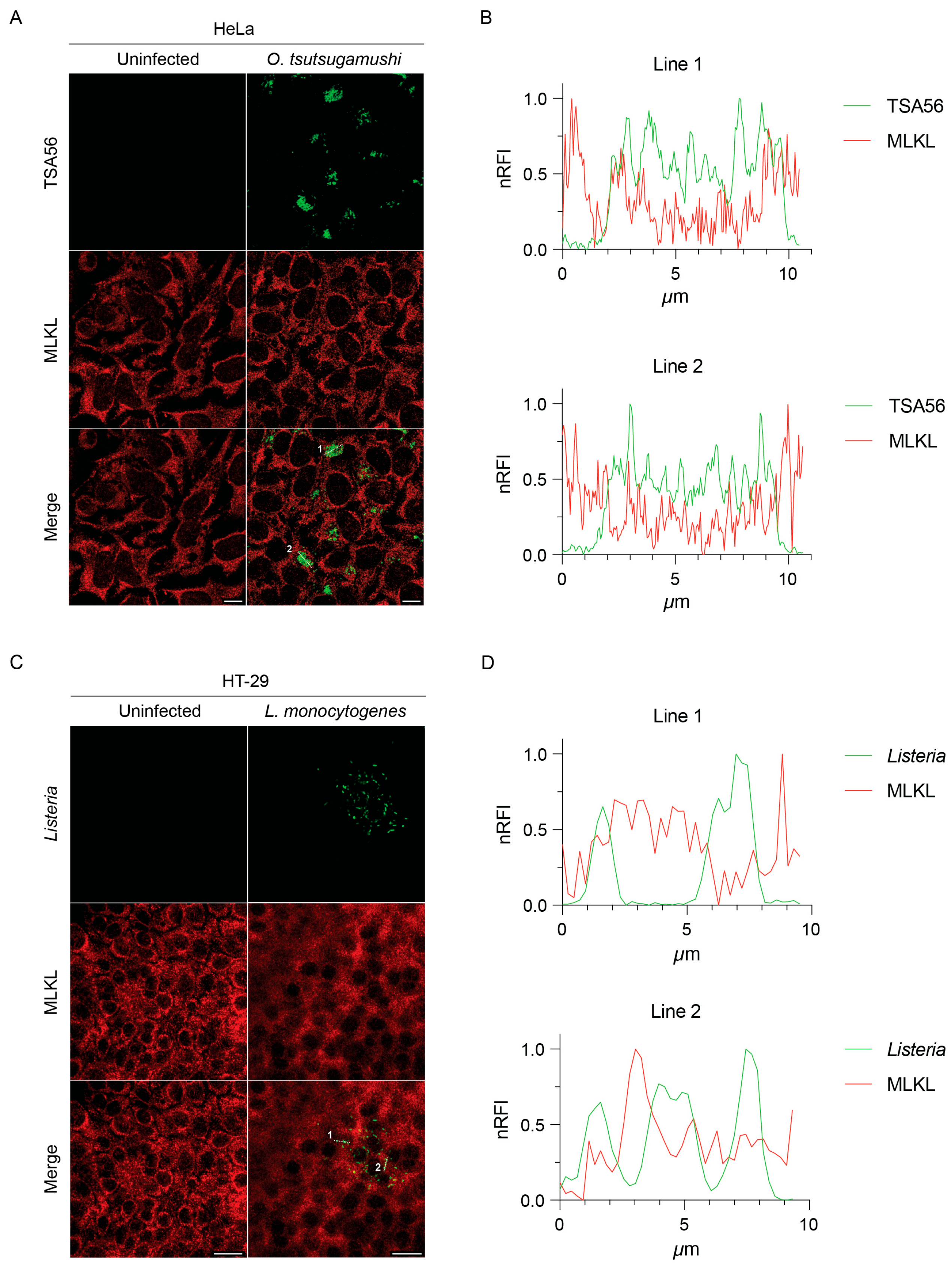
3.4. MLKL Does Not Associate with Intracytosolic O. tsutsugamushi
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| PCD | Programmed cell death |
| RIPK1 | Serine/threonine kinase receptor interacting protein 1 |
| RIPK3 | Serine/threonine kinase receptor interacting protein 3 |
| MLKL | Mixed-lineage kinase domain-like |
| AR | Ankyrin repeat |
| vIRD | Viral inducer of RIPK3 degradation |
| SCF | Skp1/cullin-1/F-box |
| NF-κB | Nuclear factor kappa-B |
| hpi | Hours post infection |
| MOI | Multiplicity of infection |
| TSA56 | 56 kDa type-specific antigen |
| HA | Hemagglutinin |
| BAP | Bacterial alkaline phosphatase |
| DPBS | Dulbecco’s phosphate-buffered saline |
| EDTA | Ethylenediaminetetraacetic acid |
| BHI | Brain heart infusion |
| OD | Optical density |
| PCR | Polymerase chain reaction |
| DNA | Deoxyribonucleic acid |
| PBS | Phosphate-buffered saline |
| TBS | Tris-buffered saline |
| RIPA | Radioimmunoprecipitation assay |
| NP-40 | Nonidet P-40 |
| SDS-PAGE | Sodium deoxycholate–polyacrylamide gel electrophoresis |
| GAPDH | Glyceraldehyde-3-phosphate dehydrogenase |
| HRP | Horseradish peroxidase |
| TBZ | TNF + BV6 + zVAD-fmk |
| TNF | Tumor necrosis factor |
| BSA | Bovine serum albumin |
| PI | Propidium iodide |
| ANOVA | Analysis of variance |
| DAMP | Damage-associated molecular pattern |
| IL-1 | Interleukin-1 |
| IL-6 | Interleukin-6 |
| IL-33 | Interleukin-33 |
| CCL2 | C-C motif chemokine ligand 2 |
| NLRP3 | NOD-, LRR- and pyrin-domain-containing protein 3 |
| Ulk1 | Unc-51-like autophagy activating kinase 1 |
| Atg5 | Autophagy-related 5 |
References
- Nailwal, H.; Chan, F.K. Necroptosis in anti-viral inflammation. Cell Death Differ. 2019, 26, 4–13. [Google Scholar] [CrossRef]
- He, S.; Han, J. Manipulation of Host Cell Death Pathways by Herpes Simplex Virus. Curr. Top. Microbiol. Immunol. 2023, 442, 85–103. [Google Scholar] [CrossRef] [PubMed]
- Koehler, H.S.; Jacobs, B.L. Subversion of Programed Cell Death by Poxviruses. Curr. Top. Microbiol. Immunol. 2023, 442, 105–131. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Nailwal, H.; Rector, J.; Rahman, M.M.; Sam, R.; McFadden, G.; Chan, F.K. A class of viral inducer of degradation of the necroptosis adaptor RIPK3 regulates virus-induced inflammation. Immunity 2021, 54, 247–258.e7. [Google Scholar] [CrossRef]
- Jorgensen, I.; Rayamajhi, M.; Miao, E.A. Programmed cell death as a defence against infection. Nat. Rev. Immunol. 2017, 17, 151–164. [Google Scholar] [CrossRef] [PubMed]
- Shlomovitz, I.; Zargarian, S.; Erlich, Z.; Edry-Botzer, L.; Gerlic, M. Distinguishing Necroptosis from Apoptosis. Methods Mol. Biol. 2018, 1857, 35–51. [Google Scholar] [CrossRef]
- Xia, X.; Lei, L.; Wang, S.; Hu, J.; Zhang, G. Necroptosis and its role in infectious diseases. Apoptosis 2020, 25, 169–178. [Google Scholar] [CrossRef]
- Abarca, K.; Martínez-Valdebenito, C.; Angulo, J.; Jiang, J.; Farris, C.M.; Richards, A.L.; Acosta-Jamett, G.; Weitzel, T. Molecular Description of a Novel Orientia Species Causing Scrub Typhus in Chile. Emerg. Infect. Dis. 2020, 26, 2148–2156. [Google Scholar] [CrossRef]
- Izzard, L.; Fuller, A.; Blacksell, S.D.; Paris, D.H.; Richards, A.L.; Aukkanit, N.; Nguyen, C.; Jiang, J.; Fenwick, S.; Day, N.P.; et al. Isolation of a novel Orientia species (O. chuto sp. nov.) from a patient infected in Dubai. J. Clin. Microbiol. 2010, 48, 4404–4409. [Google Scholar] [CrossRef]
- Kocher, C.; Jiang, J.; Morrison, A.C.; Castillo, R.; Leguia, M.; Loyola, S.; Ampuero, J.S.; Cespedes, M.; Halsey, E.S.; Bausch, D.G.; et al. Serologic Evidence of Scrub Typhus in the Peruvian Amazon. Emerg. Infect. Dis. 2017, 23, 1389–1391. [Google Scholar] [CrossRef]
- Luce-Fedrow, A.; Lehman, M.L.; Kelly, D.J.; Mullins, K.; Maina, A.N.; Stewart, R.L.; Ge, H.; John, H.S.; Jiang, J.; Richards, A.L. A Review of Scrub Typhus (Orientia tsutsugamushi and Related Organisms): Then, Now, and Tomorrow. Trop. Med. Infect. Dis. 2018, 3, 8. [Google Scholar] [CrossRef] [PubMed]
- Paris, D.H.; Phetsouvanh, R.; Tanganuchitcharnchai, A.; Jones, M.; Jenjaroen, K.; Vongsouvath, M.; Ferguson, D.P.; Blacksell, S.D.; Newton, P.N.; Day, N.P.; et al. Orientia tsutsugamushi in human scrub typhus eschars shows tropism for dendritic cells and monocytes rather than endothelium. PLoS Negl. Trop. Dis. 2012, 6, e1466. [Google Scholar] [CrossRef] [PubMed]
- Salje, J. Orientia tsutsugamushi: A neglected but fascinating obligate intracellular bacterial pathogen. PLoS Pathog. 2017, 13, e1006657. [Google Scholar] [CrossRef]
- Silva de la Fuente, M.C.; Perez, C.; Martinez-Valdebenito, C.; Perez, R.; Vial, C.; Stekolnikov, A.; Abarca, K.; Weitzel, T.; Acosta-Jamett, G. Eco-epidemiology of rodent-associated trombiculid mites and infection with Orientia spp. in Southern Chile. PLoS Negl. Trop. Dis. 2023, 17, e0011051. [Google Scholar] [CrossRef]
- Weitzel, T.; Dittrich, S.; López, J.; Phuklia, W.; Martinez-Valdebenito, C.; Velásquez, K.; Blacksell, S.D.; Paris, D.H.; Abarca, K. Endemic Scrub Typhus in South America. N. Engl. J. Med. 2016, 375, 954–961. [Google Scholar] [CrossRef] [PubMed]
- Weitzel, T.; Martínez-Valdebenito, C.; Acosta-Jamett, G.; Jiang, J.; Richards, A.L.; Abarca, K. Scrub Typhus in Continental Chile, 2016–2018(1). Emerg. Infect. Dis. 2019, 25, 1214–1217. [Google Scholar] [CrossRef]
- Weitzel, T.; Silva-de la Fuente, M.C.; Martínez-Valdebenito, C.; Stekolnikov, A.A.; Pérez, C.; Pérez, R.; Vial, C.; Abarca, K.; Acosta-Jamett, G. Novel vector of scrub typhus in subantarctic Chile—Evidence from human exposure. Clin. Infect. Dis. An. Off. Publ. Infect. Dis. Soc. Am. 2021, 74, 1862–1865. [Google Scholar] [CrossRef]
- Richardson, E.A.; Garshong, R.; Chen, K.; Crossley, D.; McLean, B.S.; Wasserberg, G.; Apperson, C.S.; Roe, R.M.; Ponnusamy, L. Orientia, Rickettsia, and the microbiome in rodent attached chiggers in North Carolina, USA. PLoS ONE 2024, 19, e0311698. [Google Scholar] [CrossRef]
- Chen, K.; Travanty, N.V.; Garshong, R.; Crossley, D.; Wasserberg, G.; Apperson, C.S.; Roe, R.M.; Ponnusamy, L. Detection of Orientia spp. Bacteria in Field-Collected Free-Living Eutrombicula Chigger Mites, United States. Emerg. Infect. Dis. 2023, 29, 1676–1679. [Google Scholar] [CrossRef]
- Kim, M.K.; Kee, S.H.; Cho, K.A.; Chung, M.H.; Lim, B.U.; Chang, W.H.; Kang, J.S. Apoptosis of endothelial cell line ECV304 persistently infected with Orientia tsutsugamushi. Microbiol. Immunol. 1999, 43, 751–757. [Google Scholar] [CrossRef]
- Kim, M.K.; Seong, S.Y.; Seoh, J.Y.; Han, T.H.; Song, H.J.; Lee, J.E.; Shin, J.H.; Lim, B.U.; Kang, J.S. Orientia tsutsugamushi inhibits apoptosis of macrophages by retarding intracellular calcium release. Infect. Immun. 2002, 70, 4692–4696. [Google Scholar] [CrossRef]
- Tay, S.T.; Rohani, M.Y.; Ho, T.M.; Devi, S. In vitro demonstration of the hemolytic, cytotoxic activities and induction of apoptosis in Orientia tsutsugamushi infected L929 mouse fibroblast cells. Southeast. Asian J. Trop. Med. Public Health 2003, 34, 352–356. [Google Scholar]
- Allen, P.E.; Adcox, H.E.; Siff, T.E.; Gupta, S.; Hunt, J.R.; Carlyon, J.A. Orientia tsutsugamushi alters the intranuclear balance of cullin-1 and c-MYC to inhibit apoptosis. Infect. Immun. 2025, 93, e0055924. [Google Scholar] [CrossRef]
- Kee, S.H.; Cho, K.A.; Kim, M.K.; Lim, B.U.; Chang, W.H.; Kang, J.S. Disassembly of focal adhesions during apoptosis of endothelial cell line ECV304 infected with Orientia tsutsugamushi. Microb. Pathog. 1999, 27, 265–271. [Google Scholar] [CrossRef]
- Rahman, M.M.; McFadden, G. Modulation of NF-kappaB signalling by microbial pathogens. Nat. Rev. Microbiol. 2011, 9, 291–306. [Google Scholar] [CrossRef] [PubMed]
- Adcox, H.E.; Hunt, J.R.; Allen, P.E.; Siff, T.E.; Rodino, K.G.; Ottens, A.K.; Carlyon, J.A. Orientia tsutsugamushi Ank5 promotes NLRC5 cytoplasmic retention and degradation to inhibit MHC class I expression. Nat. Commun. 2024, 15, 8069. [Google Scholar] [CrossRef] [PubMed]
- Evans, S.M.; Rodino, K.G.; Adcox, H.E.; Carlyon, J.A. Orientia tsutsugamushi uses two Ank effectors to modulate NF-κB p65 nuclear transport and inhibit NF-κB transcriptional activation. PLoS Pathog. 2018, 14, e1007023. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Hunt, J.R.; Adcox, H.E.; Andersen, S.E.; Gumpf, J.J.; Green, R.S.; Beyer, A.R.; Evans, S.M.; VieBrock, L.; Read, C.B.; et al. Functional Characterization of Non-Ankyrin Repeat Domains of Orientia tsutsugamushi Ank Effectors Reveals Their Importance for Molecular Pathogenesis. Infect. Immun. 2022, 90, e0062821. [Google Scholar] [CrossRef] [PubMed]
- Brady, G.; Bowie, A.G. Innate immune activation of NFκB and its antagonism by poxviruses. Cytokine Growth Factor Rev. 2014, 25, 611–620. [Google Scholar] [CrossRef]
- Gupta, T.; Chahota, R. Unique ankyrin repeat proteins in the genome of poxviruses-Boon or Wane, a critical review. Gene 2024, 927, 148759. [Google Scholar] [CrossRef]
- Reus, J.B.; Rex, E.A.; Gammon, D.B. How to Inhibit Nuclear Factor-Kappa B Signaling: Lessons from Poxviruses. Pathogens 2022, 11, 1061. [Google Scholar] [CrossRef] [PubMed]
- Herbert, M.H.; Squire, C.J.; Mercer, A.A. Poxviral ankyrin proteins. Viruses 2015, 7, 709–738. [Google Scholar] [CrossRef]
- Beyer, A.R.; VieBrock, L.; Rodino, K.G.; Miller, D.P.; Tegels, B.K.; Marconi, R.T.; Carlyon, J.A. Orientia tsutsugamushi Strain Ikeda Ankyrin Repeat-Containing Proteins Recruit SCF1 Ubiquitin Ligase Machinery via Poxvirus-Like F-Box Motifs. J. Bacteriol. 2015, 197, 3097–3109. [Google Scholar] [CrossRef]
- Wangsanut, T.; Brann, K.R.; Adcox, H.E.; Carlyon, J.A. Orientia tsutsugamushi modulates cellular levels of NF-kappaB inhibitor p105. PLoS Negl. Trop. Dis. 2021, 15, e0009339. [Google Scholar] [CrossRef]
- Ohashi, N.; Koyama, Y.; Urakami, H.; Fukuhara, M.; Tamura, A.; Kawamori, F.; Yamamoto, S.; Kasuya, S.; Yoshimura, K. Demonstration of antigenic and genotypic variation in Orientia tsutsugamushi which were isolated in Japan, and their classification into type and subtype. Microbiol. Immunol. 1996, 40, 627–638. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, S.E.; Chiarelli, T.J.; Park, M.A.; Carlyon, J.A. Orientia tsutsugamushi infection reduces host gluconeogenic but not glycolytic substrates. Infect. Immun. 2024, 92, e0028424. [Google Scholar] [CrossRef]
- Bishop, D.K.; Hinrichs, D.J. Adoptive transfer of immunity to Listeria monocytogenes. The influence of in vitro stimulation on lymphocyte subset requirements. J. Immunol. 1987, 139, 2005–2009. [Google Scholar] [CrossRef] [PubMed]
- VieBrock, L.; Evans, S.M.; Beyer, A.R.; Larson, C.L.; Beare, P.A.; Ge, H.; Singh, S.; Rodino, K.G.; Heinzen, R.A.; Richards, A.L.; et al. Orientia tsutsugamushi ankyrin repeat-containing protein family members are Type 1 secretion system substrates that traffic to the host cell endoplasmic reticulum. Front. Cell. Infect. Microbiol. 2014, 4, 186. [Google Scholar] [CrossRef]
- Seo, J.; Lee, E.W.; Sung, H.; Seong, D.; Dondelinger, Y.; Shin, J.; Jeong, M.; Lee, H.K.; Kim, J.H.; Han, S.Y.; et al. CHIP controls necroptosis through ubiquitylation- and lysosome-dependent degradation of RIPK3. Nat. Cell Biol. 2016, 18, 291–302. [Google Scholar] [CrossRef]
- Beyer, A.R.; Rodino, K.G.; VieBrock, L.; Green, R.S.; Tegels, B.K.; Oliver, L.D., Jr.; Marconi, R.T.; Carlyon, J.A. Orientia tsutsugamushi Ank9 is a multifunctional effector that utilizes a novel GRIP-like Golgi localization domain for Golgi-to-endoplasmic reticulum trafficking and interacts with host COPB2. Cell. Microbiol. 2017, 19. [Google Scholar] [CrossRef]
- Costigan, A.; Hollville, E.; Martin, S.J. Discriminating Between Apoptosis, Necrosis, Necroptosis, and Ferroptosis by Microscopy and Flow Cytometry. Curr. Protoc. 2023, 3, e951. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Joshi, K.; Denning, M.F.; Zhang, J. RIPK3 signaling and its role in the pathogenesis of cancers. Cell. Mol. Life Sci. 2021, 78, 7199–7217. [Google Scholar] [CrossRef]
- Yuan, J.; Ofengeim, D. A guide to cell death pathways. Nat. Rev. Mol. Cell Biol. 2024, 25, 379–395. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Wang, H.; Wang, Z.; He, S.; Chen, S.; Liao, D.; Wang, L.; Yan, J.; Liu, W.; Lei, X.; et al. Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell 2012, 148, 213–227. [Google Scholar] [CrossRef]
- Su, Z.; Yang, Z.; Xie, L.; DeWitt, J.P.; Chen, Y. Cancer therapy in the necroptosis era. Cell Death Differ. 2016, 23, 748–756. [Google Scholar] [CrossRef]
- Onizawa, M.; Oshima, S.; Schulze-Topphoff, U.; Oses-Prieto, J.A.; Lu, T.; Tavares, R.; Prodhomme, T.; Duong, B.; Whang, M.I.; Advincula, R.; et al. The ubiquitin-modifying enzyme A20 restricts ubiquitination of the kinase RIPK3 and protects cells from necroptosis. Nat. Immunol. 2015, 16, 618–627. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Xiang, Y.; Liu, S.; Li, C.; Dong, J.; Kong, X.; Ji, X.; Cheng, X.; Zhang, L. RIPK3 signaling and its role in regulated cell death and diseases. Cell Death Discov. 2024, 10, 200. [Google Scholar] [CrossRef] [PubMed]
- Moriwaki, K.; Chan, F.K. The Inflammatory Signal Adaptor RIPK3: Functions Beyond Necroptosis. Int. Rev. Cell Mol. Biol. 2017, 328, 253–275. [Google Scholar] [CrossRef]
- Sai, K.; Parsons, C.; House, J.S.; Kathariou, S.; Ninomiya-Tsuji, J. Necroptosis mediators RIPK3 and MLKL suppress intracellular Listeria replication independently of host cell killing. J. Cell Biol. 2019, 218, 1994–2005. [Google Scholar] [CrossRef]
- Yu, X.; Yuan, J.; Shi, L.; Dai, S.; Yue, L.; Yan, M. Necroptosis in bacterial infections. Front. Immunol. 2024, 15, 1394857. [Google Scholar] [CrossRef]
- Wu, X.; Zhao, X.; Li, F.; Wang, Y.; Ou, Y.; Zhang, H.; Li, X.; Wu, X.; Wang, L.; Li, M.; et al. MLKL-mediated endothelial necroptosis drives vascular damage and mortality in systemic inflammatory response syndrome. Cell. Mol. Immunol. 2024, 21, 1309–1321. [Google Scholar] [CrossRef] [PubMed]
- Shelite, T.R.; Liang, Y.; Wang, H.; Mendell, N.L.; Trent, B.J.; Sun, J.; Gong, B.; Xu, G.; Hu, H.; Bouyer, D.H.; et al. IL-33-Dependent Endothelial Activation Contributes to Apoptosis and Renal Injury in Orientia tsutsugamushi-Infected Mice. PLoS Negl. Trop. Dis. 2016, 10, e0004467. [Google Scholar] [CrossRef]
- Soong, L.; Mendell, N.L.; Olano, J.P.; Rockx-Brouwer, D.; Xu, G.; Goez-Rivillas, Y.; Drom, C.; Shelite, T.R.; Valbuena, G.; Walker, D.H.; et al. An Intradermal Inoculation Mouse Model for Immunological Investigations of Acute Scrub Typhus and Persistent Infection. PLoS Negl. Trop. Dis. 2016, 10, e0004884. [Google Scholar] [CrossRef] [PubMed]
- Astrup, E.; Janardhanan, J.; Otterdal, K.; Ueland, T.; Prakash, J.A.; Lekva, T.; Strand, O.A.; Abraham, O.C.; Thomas, K.; Damas, J.K.; et al. Cytokine network in scrub typhus: High levels of interleukin-8 are associated with disease severity and mortality. PLoS Negl. Trop. Dis. 2014, 8, e2648. [Google Scholar] [CrossRef]
- Kramme, S.; An le, V.; Khoa, N.D.; Trin le, V.; Tannich, E.; Rybniker, J.; Fleischer, B.; Drosten, C.; Panning, M. Orientia tsutsugamushi bacteremia and cytokine levels in Vietnamese scrub typhus patients. J. Clin. Microbiol. 2009, 47, 586–589. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Koehler, H.S.; Mocarski, E.S.; Dix, R.D. RIPK3 and caspase 8 collaborate to limit herpes simplex encephalitis. PLoS Pathog. 2022, 18, e1010857. [Google Scholar] [CrossRef]
- Downey, J.; Pernet, E.; Coulombe, F.; Allard, B.; Meunier, I.; Jaworska, J.; Qureshi, S.; Vinh, D.C.; Martin, J.G.; Joubert, P.; et al. RIPK3 interacts with MAVS to regulate type I IFN-mediated immunity to Influenza A virus infection. PLoS Pathog. 2017, 13, e1006326. [Google Scholar] [CrossRef]
- Daniels, B.P.; Snyder, A.G.; Olsen, T.M.; Orozco, S.; Oguin, T.H., 3rd; Tait, S.W.G.; Martinez, J.; Gale, M., Jr.; Loo, Y.M.; Oberst, A. RIPK3 Restricts Viral Pathogenesis via Cell Death-Independent Neuroinflammation. Cell 2017, 169, 301–313.e311. [Google Scholar] [CrossRef]
- Cervantes, P.W.; Martorelli Di Genova, B.; Erazo Flores, B.J.; Knoll, L.J. RIPK3 Facilitates Host Resistance to Oral Toxoplasma gondii Infection. Infect. Immun. 2021, 89, e00021-21. [Google Scholar] [CrossRef]
- Bian, P.; Ye, C.; Zheng, X.; Luo, C.; Yang, J.; Li, M.; Wang, Y.; Yang, J.; Zhou, Y.; Zhang, F.; et al. RIPK3 Promotes JEV Replication in Neurons via Downregulation of IFI44L. Front. Microbiol. 2020, 11, 368. [Google Scholar] [CrossRef]
- Nogusa, S.; Thapa, R.J.; Dillon, C.P.; Liedmann, S.; Oguin, T.H., 3rd; Ingram, J.P.; Rodriguez, D.A.; Kosoff, R.; Sharma, S.; Sturm, O.; et al. RIPK3 Activates Parallel Pathways of MLKL-Driven Necroptosis and FADD-Mediated Apoptosis to Protect against Influenza A Virus. Cell Host Microbe 2016, 20, 13–24. [Google Scholar] [CrossRef]
- Mohamed, M.R.; Rahman, M.M.; Lanchbury, J.S.; Shattuck, D.; Neff, C.; Dufford, M.; van Buuren, N.; Fagan, K.; Barry, M.; Smith, S.; et al. Proteomic screening of variola virus reveals a unique NF-kappaB inhibitor that is highly conserved among pathogenic orthopoxviruses. Proc. Natl. Acad. Sci. USA 2009, 106, 9045–9050. [Google Scholar] [CrossRef]
- Lawlor, K.E.; Khan, N.; Mildenhall, A.; Gerlic, M.; Croker, B.A.; D’Cruz, A.A.; Hall, C.; Kaur Spall, S.; Anderton, H.; Masters, S.L.; et al. RIPK3 promotes cell death and NLRP3 inflammasome activation in the absence of MLKL. Nat. Commun. 2015, 6, 6282. [Google Scholar] [CrossRef]
- Najjar, M.; Saleh, D.; Zelic, M.; Nogusa, S.; Shah, S.; Tai, A.; Finger, J.N.; Polykratis, A.; Gough, P.J.; Bertin, J.; et al. RIPK1 and RIPK3 Kinases Promote Cell-Death-Independent Inflammation by Toll-like Receptor 4. Immunity 2016, 45, 46–59. [Google Scholar] [CrossRef]
- Al-Moujahed, A.; Tian, B.; Efstathiou, N.E.; Konstantinou, E.K.; Hoang, M.; Lin, H.; Miller, J.W.; Vavvas, D.G. Receptor interacting protein kinase 3 (RIP3) regulates iPSCs generation through modulating cell cycle progression genes. Stem Cell Res. 2019, 35, 101387. [Google Scholar] [CrossRef]
- Wu, X.; Tian, L.; Li, J.; Zhang, Y.; Han, V.; Li, Y.; Xu, X.; Li, H.; Chen, X.; Chen, J.; et al. Investigation of receptor interacting protein (RIP3)-dependent protein phosphorylation by quantitative phosphoproteomics. Mol. Cell. Proteom. 2012, 11, 1640–1651. [Google Scholar] [CrossRef]
- Yang, Z.; Wang, Y.; Zhang, Y.; He, X.; Zhong, C.Q.; Ni, H.; Chen, X.; Liang, Y.; Wu, J.; Zhao, S.; et al. RIP3 targets pyruvate dehydrogenase complex to increase aerobic respiration in TNF-induced necroptosis. Nat. Cell Biol. 2018, 20, 186–197. [Google Scholar] [CrossRef]
- Zhang, D.W.; Shao, J.; Lin, J.; Zhang, N.; Lu, B.J.; Lin, S.C.; Dong, M.Q.; Han, J. RIP3, an energy metabolism regulator that switches TNF-induced cell death from apoptosis to necrosis. Science 2009, 325, 332–336. [Google Scholar] [CrossRef]
- Harris, K.G.; Morosky, S.A.; Drummond, C.G.; Patel, M.; Kim, C.; Stolz, D.B.; Bergelson, J.M.; Cherry, S.; Coyne, C.B. RIP3 Regulates Autophagy and Promotes Coxsackievirus B3 Infection of Intestinal Epithelial Cells. Cell Host Microbe 2015, 18, 221–232. [Google Scholar] [CrossRef]
- Torii, S.; Yamaguchi, H.; Nakanishi, A.; Arakawa, S.; Honda, S.; Moriwaki, K.; Nakano, H.; Shimizu, S. Identification of a phosphorylation site on Ulk1 required for genotoxic stress-induced alternative autophagy. Nat. Commun. 2020, 11, 1754. [Google Scholar] [CrossRef]
- Choi, J.H.; Cheong, T.C.; Ha, N.Y.; Ko, Y.; Cho, C.H.; Jeon, J.H.; So, I.; Kim, I.K.; Choi, M.S.; Kim, I.S.; et al. Orientia tsutsugamushi subverts dendritic cell functions by escaping from autophagy and impairing their migration. PLoS Negl. Trop. Dis. 2013, 7, e1981. [Google Scholar] [CrossRef] [PubMed]
- Ko, Y.; Choi, J.H.; Ha, N.Y.; Kim, I.S.; Cho, N.H.; Choi, M.S. Active escape of Orientia tsutsugamushi from cellular autophagy. Infect. Immun. 2013, 81, 552–559. [Google Scholar] [CrossRef]
- Samson, A.L.; Fitzgibbon, C.; Patel, K.M.; Hildebrand, J.M.; Whitehead, L.W.; Rimes, J.S.; Jacobsen, A.V.; Horne, C.R.; Gavin, X.J.; Young, S.N.; et al. A toolbox for imaging RIPK1, RIPK3, and MLKL in mouse and human cells. Cell Death Differ. 2021, 28, 2126–2144. [Google Scholar] [CrossRef]
- Adcox, H.E.; Hatke, A.L.; Andersen, S.E.; Gupta, S.; Otto, N.B.; Weber, M.M.; Marconi, R.T.; Carlyon, J.A. Orientia tsutsugamushi Nucleomodulin Ank13 Exploits the RaDAR Nuclear Import Pathway to Modulate Host Cell Transcription. mBio 2021, 12, e0181621. [Google Scholar] [CrossRef]
- Rodino, K.G.; Adcox, H.E.; Martin, R.K.; Patel, V.; Conrad, D.H.; Carlyon, J.A. The Obligate Intracellular Bacterium Orientia tsutsugamushi Targets NLRC5 To Modulate the Major Histocompatibility Complex Class I Pathway. Infect. Immun. 2019, 87, e00876-18. [Google Scholar] [CrossRef] [PubMed]
- Rodino, K.G.; VieBrock, L.; Evans, S.M.; Ge, H.; Richards, A.L.; Carlyon, J.A. Orientia tsutsugamushi Modulates Endoplasmic Reticulum-Associated Degradation to Benefit Its Growth. Infect. Immun. 2018, 86, e00596-17. [Google Scholar] [CrossRef] [PubMed]
- Min, C.K.; Kwon, Y.J.; Ha, N.Y.; Cho, B.A.; Kim, J.M.; Kwon, E.K.; Kim, Y.S.; Choi, M.S.; Kim, I.S.; Cho, N.H. Multiple Orientia tsutsugamushi ankyrin repeat proteins interact with SCF1 ubiquitin ligase complex and eukaryotic elongation factor 1 alpha. PLoS ONE 2014, 9, e105652. [Google Scholar] [CrossRef]
- Ko, Y.; Cho, N.H.; Cho, B.A.; Kim, I.S.; Choi, M.S. Involvement of Ca2+ signaling in intracellular invasion of non-phagocytic host cells by Orientia tsutsugamushi. Microb. Pathog. 2011, 50, 326–330. [Google Scholar] [CrossRef]
- Ha, N.Y.; Cho, N.H.; Kim, Y.S.; Choi, M.S.; Kim, I.S. An autotransporter protein from Orientia tsutsugamushi mediates adherence to nonphagocytic host cells. Infect. Immun. 2011, 79, 1718–1727. [Google Scholar] [CrossRef]
- Kim, S.W.; Ihn, K.S.; Han, S.H.; Seong, S.Y.; Kim, I.S.; Choi, M.S. Microtubule- and dynein-mediated movement of Orientia tsutsugamushi to the microtubule organizing center. Infect. Immun. 2001, 69, 494–500. [Google Scholar] [CrossRef]
- Fullsack, S.; Rosenthal, A.; Wajant, H.; Siegmund, D. Redundant and receptor-specific activities of TRADD, RIPK1 and FADD in death receptor signaling. Cell Death Dis. 2019, 10, 122. [Google Scholar] [CrossRef] [PubMed]
- Karl, I.; Jossberger-Werner, M.; Schmidt, N.; Horn, S.; Goebeler, M.; Leverkus, M.; Wajant, H.; Giner, T. TRAF2 inhibits TRAIL- and CD95L-induced apoptosis and necroptosis. Cell Death Dis. 2014, 5, e1444. [Google Scholar] [CrossRef] [PubMed]
- Wagner, J.; Vredevoogd, D.; Yu, X.; Lu, D.; Peeper, D.S.; Hermanns, H.M.; Wang, J.; Wajant, H.; Siegmund, D. TRAF2 and RIPK1 redundantly mediate classical NFkappaB signaling by TNFR1 and CD95-type death receptors. Cell Death Dis. 2025, 16, 35. [Google Scholar] [CrossRef] [PubMed]
- Bolik, J.; Krause, F.; Stevanovic, M.; Gandrass, M.; Thomsen, I.; Schacht, S.S.; Rieser, E.; Muller, M.; Schumacher, N.; Fritsch, J.; et al. Inhibition of ADAM17 impairs endothelial cell necroptosis and blocks metastasis. J. Exp. Med. 2022, 219, e20201039. [Google Scholar] [CrossRef]
- Zhang, L.; Wei, J.; Ren, L.; Zhang, J.; Yang, M.; Jing, L.; Wang, J.; Sun, Z.; Zhou, X. Endosulfan inducing apoptosis and necroptosis through activation RIPK signaling pathway in human umbilical vascular endothelial cells. Environ. Sci. Pollut. Res. 2017, 24, 215–225. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Siff, T.E.; Allen, P.E.; Armistead, D.L.; Hunt, J.R.; Rolland, S.J.; Agaisse, H.; Carlyon, J.A. Orientia tsutsugamushi Modulates RIPK3 Cellular Levels but Does Not Inhibit Necroptosis. Pathogens 2025, 14, 478. https://doi.org/10.3390/pathogens14050478
Siff TE, Allen PE, Armistead DL, Hunt JR, Rolland SJ, Agaisse H, Carlyon JA. Orientia tsutsugamushi Modulates RIPK3 Cellular Levels but Does Not Inhibit Necroptosis. Pathogens. 2025; 14(5):478. https://doi.org/10.3390/pathogens14050478
Chicago/Turabian StyleSiff, Thomas E., Paige E. Allen, David L. Armistead, Jason R. Hunt, Steven J. Rolland, Hervé Agaisse, and Jason A. Carlyon. 2025. "Orientia tsutsugamushi Modulates RIPK3 Cellular Levels but Does Not Inhibit Necroptosis" Pathogens 14, no. 5: 478. https://doi.org/10.3390/pathogens14050478
APA StyleSiff, T. E., Allen, P. E., Armistead, D. L., Hunt, J. R., Rolland, S. J., Agaisse, H., & Carlyon, J. A. (2025). Orientia tsutsugamushi Modulates RIPK3 Cellular Levels but Does Not Inhibit Necroptosis. Pathogens, 14(5), 478. https://doi.org/10.3390/pathogens14050478