
Genomic Characterization of Potential Opportunistic Zoonotic Streptococcus parasuis Isolated in China
Abstract
1. Introduction
2. Materials and Methods
2.1. Bacterial Isolation and Identification
2.2. Phylogenetic Analysis
2.3. DNA Extraction
2.4. Genome Sequencing, Assembly, and Annotation
2.5. Comparative Genomic Analysis
2.6. Mice Survival Test
3. Results
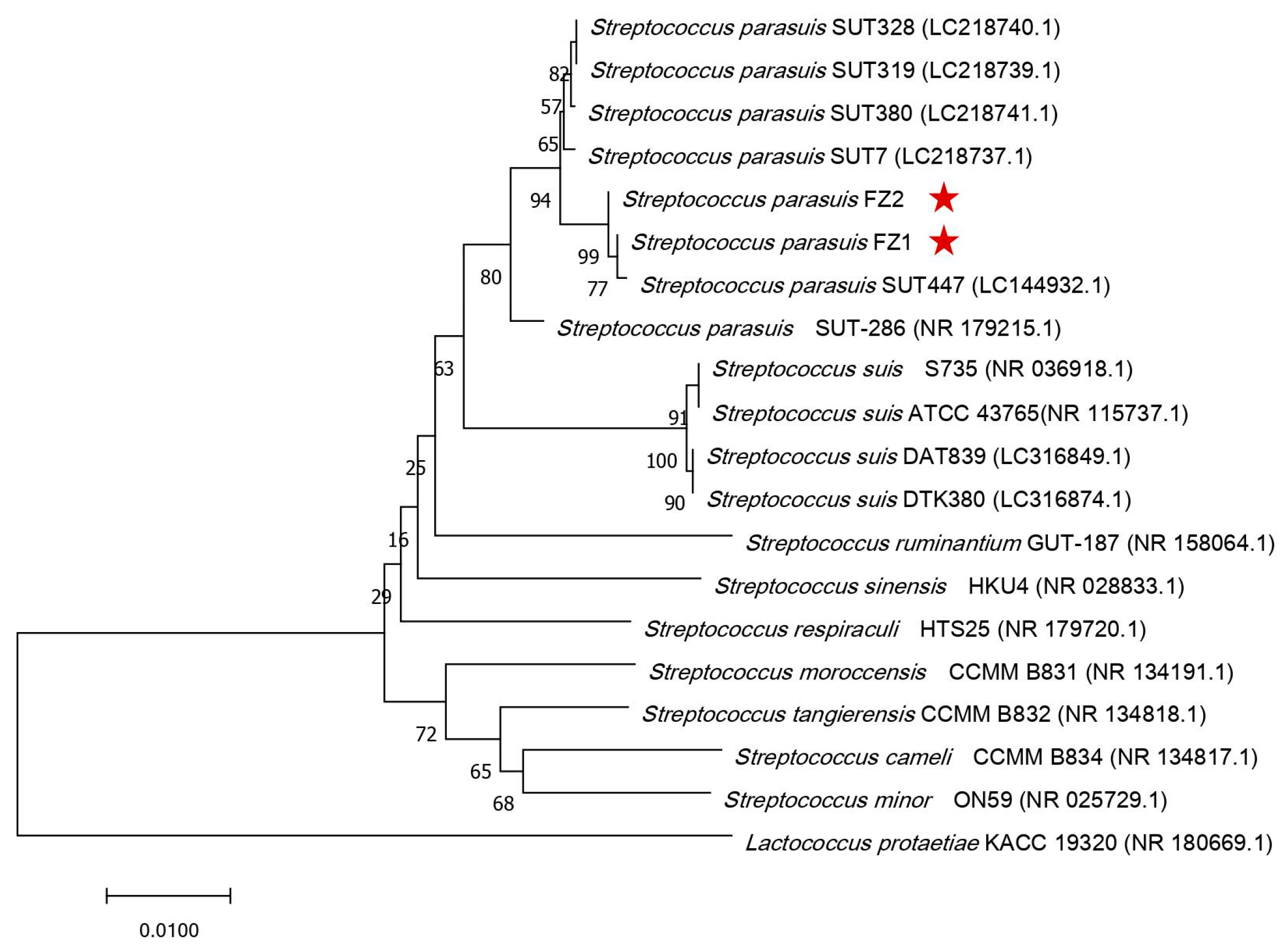
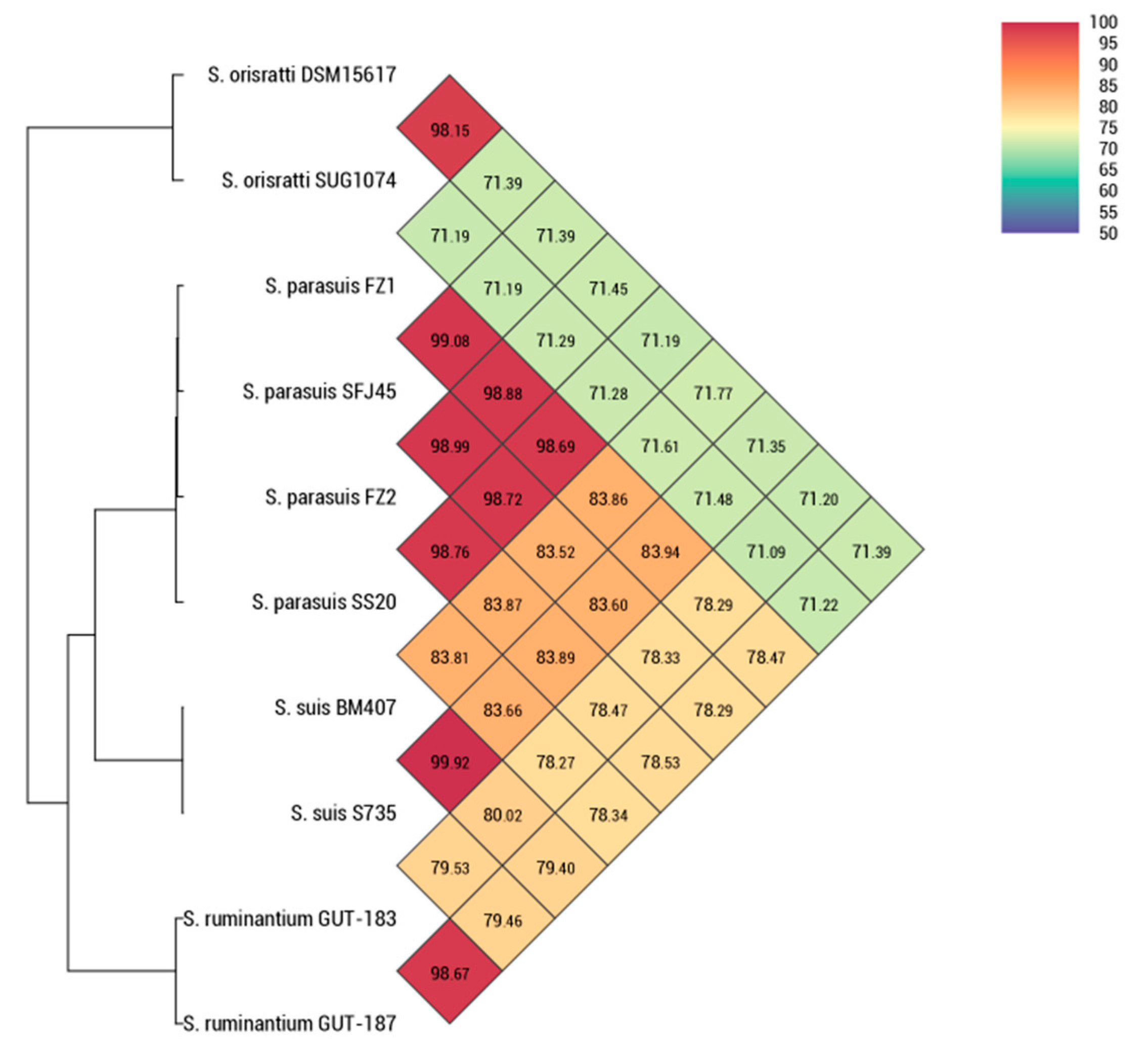
3.1. Bacterial Characteristics and Phylogenetic Analysis of S. parasuis FZ1 and FZ2
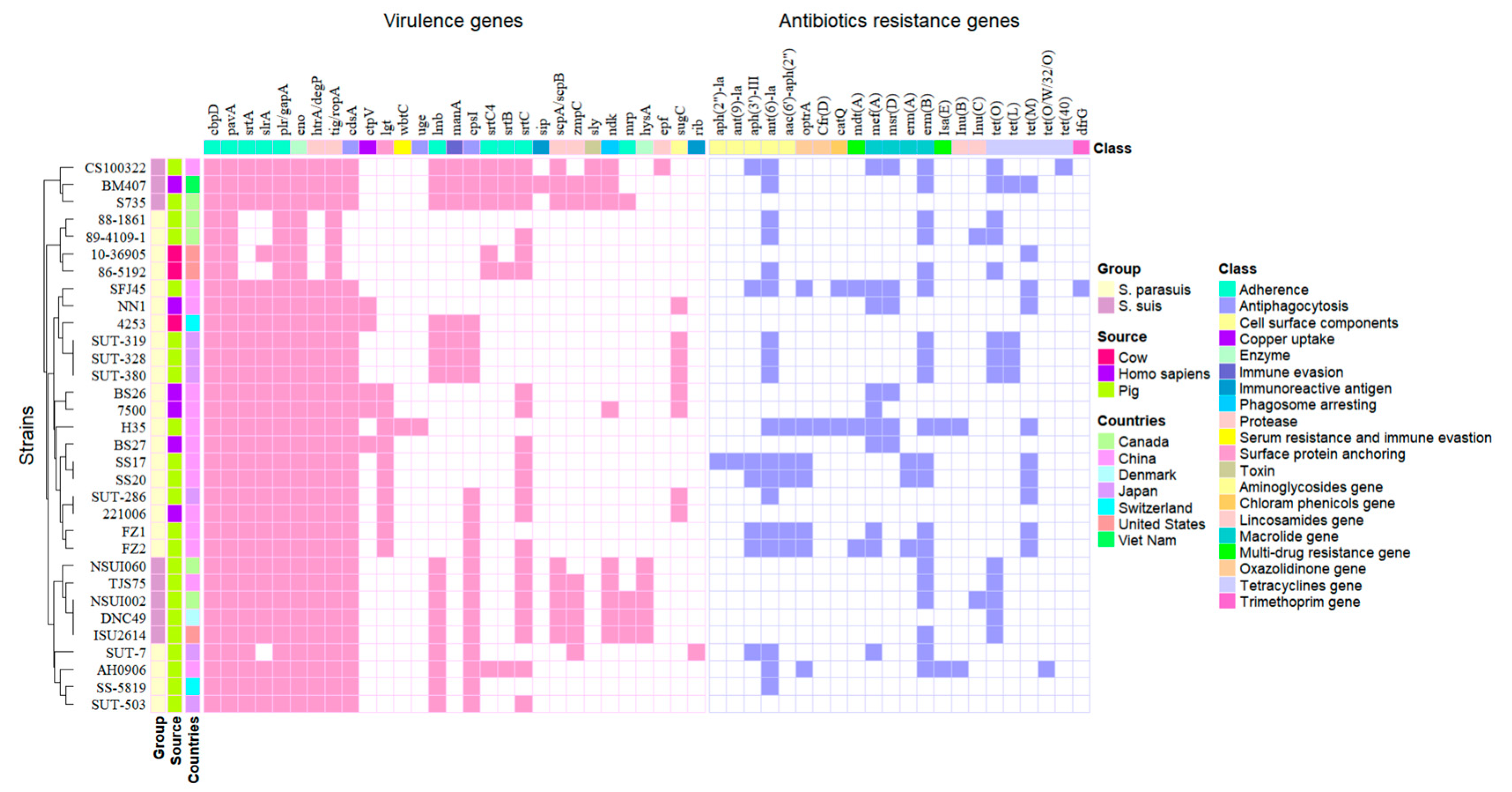
3.2. Genomic Features and Gene Functional Analysis of S. parasuis Isolates
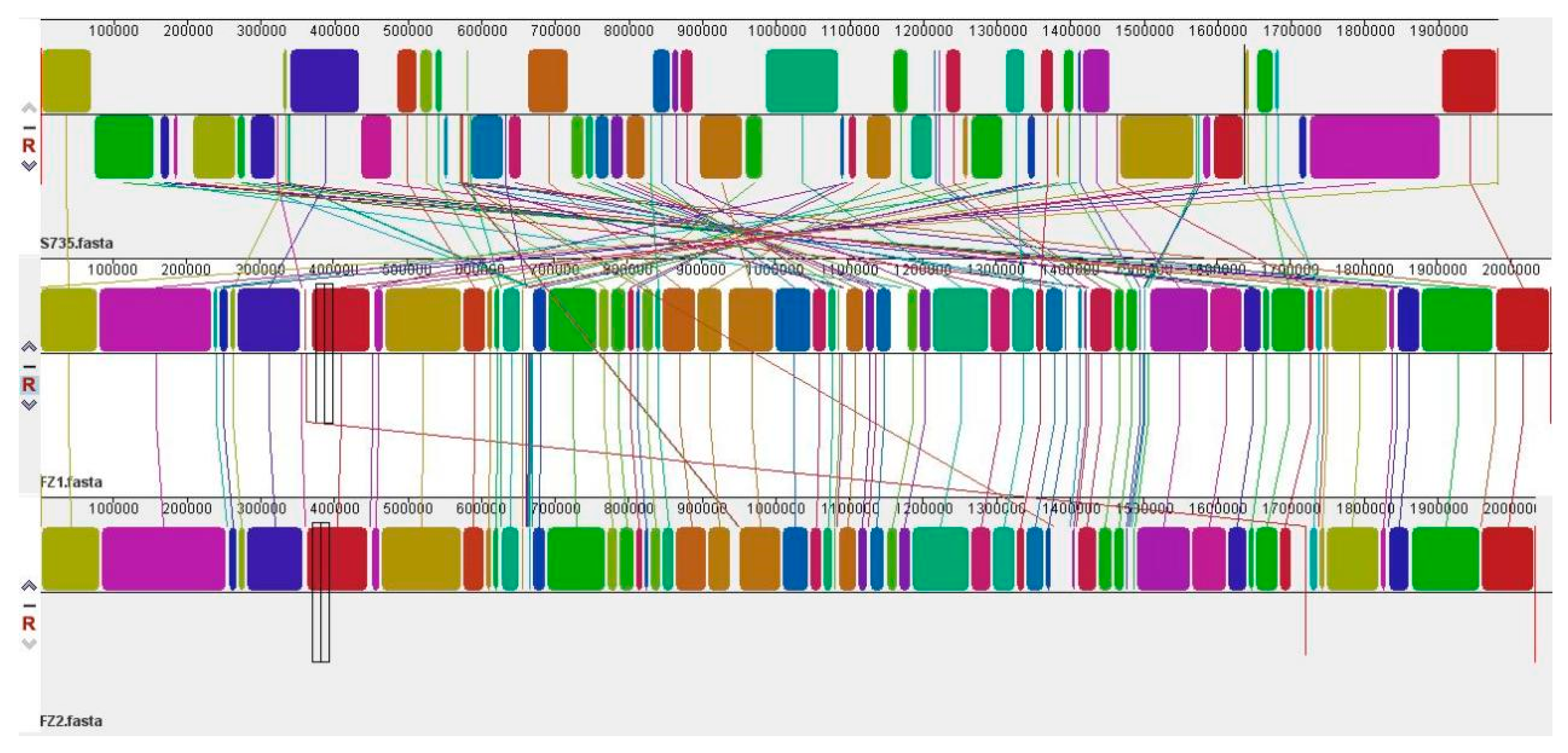
3.3. Comparative Genomic Analysis of S. parasuis FZ1 and S. parasuis FZ2
3.4. Differential Survival Rates in Mice Infected with S. parasuis FZ1 and S. parasuis FZ2
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Goyette-Desjardins, G.; Auger, J.P.; Xu, J.; Segura, M.; Gottschalk, M. Streptococcus suis, an important pig pathogen and emerging zoonotic agent-an update on the worldwide distribution based on serotyping and sequence typing. Emerg. Microbes Infect. 2014, 3, e45. [Google Scholar] [CrossRef] [PubMed]
- Guo, G.; Du, D.; Yu, Y.; Zhang, Y.; Qian, Y.; Zhang, W. Pan-genome analysis of Streptococcus suis serotype 2 revealed genomic diversity among strains of different virulence. Transbound. Emerg. Dis. 2021, 68, 637–647. [Google Scholar] [CrossRef]
- Wang, J.P.; Yi, X.L.; Liang, P.J.; Tao, Y.M.H.; Wang, Y.; Jin, D.; Luo, B.; Yang, J.; Zheng, H. Investigation of the Genomic and Pathogenic Features of the Potentially Zoonotic. Pathogens 2021, 10, 834. [Google Scholar] [CrossRef] [PubMed]
- Yamada, R.; Tien, L.H.T.; Arai, S.; Tohya, M.; Ishida-Kuroki, K.; Nomoto, R.; Kim, H.; Suzuki, E.; Osawa, R.; Watanabe, T.; et al. Development of PCR for identifying Streptococcus parasuis, a close relative of Streptococcus suis. J. Vet. Med. Sci. 2018, 80, 1101–1107. [Google Scholar] [CrossRef] [PubMed]
- Nomoto, R.; Maruyama, F.; Ishida, S.; Tohya, M.; Sekizaki, T.; Osawa, R. Reappraisal of the taxonomy of Streptococcus suis serotypes 20, 22 and 26: Streptococcus parasuis sp. nov. Int. J. Syst. Evol. Microbiol. 2015, 65 Pt 2, 438–443. [Google Scholar] [CrossRef]
- Wei, Z.; Li, R.; Zhang, A.; He, H.; Hua, Y.; Xia, J.; Cai, X.; Chen, H.; Jin, M. Characterization of Streptococcus suis isolates from the diseased pigs in China between 2003 and 2007. Vet. Microbiol. 2009, 137, 196–201. [Google Scholar] [CrossRef]
- Gottschalk, M.; Lacouture, S.; Bonifait, L.; Roy, D.; Fittipaldi, N.; Grenier, D. Characterization of Streptococcus suis isolates recovered between 2008 and 2011 from diseased pigs in Québec, Canada. Vet. Microbiol. 2013, 162, 819–825. [Google Scholar] [CrossRef]
- Gottschalk, M.; Higgins, R.; Jacques, M.; Mittal, K.R.; Henrichsen, J. Description of 14 new capsular types of Streptococcus suis. J. Clin. Microbiol. 1989, 27, 2633–2636. [Google Scholar] [CrossRef]
- Qi, K.; Yi, X.; Wang, M.; Wang, J.; Sun, H.; Liang, P.; Xu, J.; Zheng, H. Streptococcus parasuis, an Emerging Zoonotic Pathogen, Possesses the Capacity to Induce Cerebral Inflammatory Responses. Pathogens 2023, 12, 600. [Google Scholar] [CrossRef]
- Han, N.; Li, J.; Wan, P.; Pan, Y.; Xu, T.; Xiong, W.; Zeng, Z. Co-Existence of Oxazolidinone Resistance Genes cfr(D) and optrA on Two Streptococcus parasuis Isolates from Swine. Antibiotics 2023, 12, 825. [Google Scholar] [CrossRef]
- Heuer, H.; Krsek, M.; Baker, P.; Smalla, K.; Wellington, E.M. Analysis of actinomycete communities by specific amplification of genes encoding 16S rRNA and gel-electrophoretic separation in denaturing gradients. Appl. Environ. Microbiol. 1997, 63, 3233–3241. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [PubMed]
- Koren, S.; Walenz, B.P.; Berlin, K.; Miller, J.R.; Bergman, N.H.; Phillippy, A.M. Canu: Scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 2017, 27, 722–736. [Google Scholar] [CrossRef] [PubMed]
- Walker, B.J.; Abeel, T.; Shea, T.; Priest, M.; Abouelliel, A.; Sakthikumar, S.; Cuomo, C.A.; Zeng, Q.; Wortman, J.; Young, S.K.; et al. Pilon: An integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS ONE 2014, 9, e112963. [Google Scholar] [CrossRef]
- Luo, R.; Liu, B.; Xie, Y.; Li, Z.; Huang, W.; Yuan, J.; He, G.; Chen, Y.; Pan, Q.; Liu, Y.; et al. Erratum: SOAPdenovo2: An empirically improved memory-efficient short-read de novo assembler. GigaScience 2015, 4, 30. [Google Scholar] [CrossRef] [PubMed]
- Krzywinski, M.; Schein, J.; Birol, I.; Connors, J.; Gascoyne, R.; Horsman, D.; Jones, S.J.; Marra, M.A. Circos: An information aesthetic for comparative genomics. Genome Res. 2009, 19, 1639–1645. [Google Scholar] [CrossRef]
- Lagesen, K.; Hallin, P.; Rødland, E.A.; Staerfeldt, H.H.; Rognes, T.; Ussery, D.W. RNAmmer: Consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 2007, 35, 3100–3108. [Google Scholar] [CrossRef]
- Chan, P.P.; Lin, B.Y.; Mak, A.J.; Lowe, T.M. tRNAscan-SE 2.0: Improved detection and functional classification of transfer RNA genes. Nucleic Acids Res. 2021, 49, 9077–9096. [Google Scholar] [CrossRef]
- Lee, I.; Kim, Y.O.; Park, S.C.; Chun, J. OrthoANI: An improved algorithm and software for calculating average nucleotide identity. Int. J. Syst. Evol. Microbiol. 2016, 66, 1100–1103. [Google Scholar] [CrossRef]
- Jolley, K.A.; Bray, J.E.; Maiden, M.C.J. Open-access bacterial population genomics: BIGSdb software, the PubMLST.org website and their applications. Wellcome Open Res. 2018, 3, 124. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Zheng, D.; Jin, Q.; Chen, L.; Yang, J. VFDB 2019: A comparative pathogenomic platform with an interactive web interface. Nucleic Acids Res. 2019, 47, D687–D692. [Google Scholar] [CrossRef]
- Bortolaia, V.; Kaas, R.S.; Ruppe, E.; Roberts, M.C.; Schwarz, S.; Cattoir, V.; Philippon, A.; Allesoe, R.L.; Rebelo, A.R.; Florensa, A.F.; et al. ResFinder 4.0 for predictions of phenotypes from genotypes. J. Antimicrob. Chemother. 2020, 75, 3491–3500. [Google Scholar] [CrossRef] [PubMed]
- Couvin, D.; Bernheim, A.; Toffano-Nioche, C.; Touchon, M.; Michalik, J.; Néron, B.; Rocha, E.P.C.; Vergnaud, G.; Gautheret, D.; Pourcel, C. CRISPRCasFinder, an update of CRISRFinder, includes a portable version, enhanced performance and integrates search for Cas proteins. Nucleic Acids Res. 2018, 46, W246–W251. [Google Scholar] [CrossRef]
- Wishart, D.S.; Han, S.; Saha, S.; Oler, E.; Peters, H.; Grant, J.R.; Stothard, P.; Gautam, V. PHASTEST: Faster than PHASTER, better than PHAST. Nucleic Acids Res. 2023, 51, W443–W450. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Liu, G.; Liu, M.; Tai, C.; Deng, Z.; Song, J.; Ou, H.Y. ICEberg 3.0: Functional categorization and analysis of the integrative and conjugative elements in bacteria. Nucleic Acids Res. 2024, 52, D732–D737. [Google Scholar] [CrossRef]
- Bertelli, C.; Laird, M.R.; Williams, K.P.; Lau, B.Y.; Hoad, G.; Winsor, G.L.; Brinkman, F.S.L. IslandViewer 4: Expanded prediction of genomic islands for larger-scale datasets. Nucleic Acids Res. 2017, 45, W30–W35. [Google Scholar] [CrossRef]
- Chaudhari, N.M.; Gupta, V.K.; Dutta, C. BPGA—An ultra-fast pan-genome analysis pipeline. Sci. Rep. 2016, 6, 24373. [Google Scholar] [CrossRef]
- Darling, A.E.; Mau, B.; Perna, N.T. progressiveMauve: Multiple genome alignment with gene gain, loss and rearrangement. PLoS ONE 2010, 5, e11147. [Google Scholar] [CrossRef]
- Guo, G.; Wang, Z.; Li, Q.; Yu, Y.; Li, Y.; Tan, Z.; Zhang, W. Genomic characterization of Streptococcus parasuis, a close relative of Streptococcus suis and also a potential opportunistic zoonotic pathogen. BMC Genom. 2022, 23, 469. [Google Scholar] [CrossRef]
- Fälker, S.; Nelson, A.L.; Morfeldt, E.; Jonas, K.; Hultenby, K.; Ries, J.; Melefors, O.; Normark, S.; Henriques-Normark, B. Sortase-mediated assembly and surface topology of adhesive pneumococcal pili. Mol. Microbiol. 2008, 70, 595–607. [Google Scholar] [CrossRef]
- Ward, S.K.; Abomoelak, B.; Hoye, E.A.; Steinberg, H.; Talaat, A.M. CtpV: A putative copper exporter required for full virulence of Mycobacterium tuberculosis. Mol. Microbiol. 2010, 77, 1096–1110. [Google Scholar] [CrossRef]
- Kalscheuer, R.; Weinrick, B.; Veeraraghavan, U.; Besra, G.S.; Jacobs, W.R., Jr. Trehalose-recycling ABC transporter LpqY-SugA-SugB-SugC is essential for virulence of Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. USA 2010, 107, 21761–21766. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.; Liu, D.P.; Li, K.X.; Hu, S.N.; He, Z.L. Pan-Genome Analysis of Reveals Key Factors Influencing Genomic Plasticity. Microbiol. Spectr. 2022, 10, e0311722. [Google Scholar] [CrossRef] [PubMed]
- West, P.T.; Chanin, R.B.; Bhatt, A.S. From genome structure to function: Insights into structural variation in microbiology. Curr. Opin. Microbiol. 2022, 69, 102192. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Yang, Q.; Schwarz, S.; Yang, W.; Xu, Q.; Wang, L.; Liu, S.; Zhang, W. Identification of a Streptococcus parasuis isolate co-harbouring the oxazolidinone resistance genes cfr(D) and optrA. J. Antimicrob. Chemother. 2021, 76, 3059–3061. [Google Scholar] [CrossRef]
- Gu, Y.; Shen, S.; Han, B.; Tian, X.; Yang, F.; Zhang, K. Family livestock waste: An ignored pollutant resource of antibiotic resistance genes. Ecotoxicol. Environ. Saf. 2020, 197, 110567. [Google Scholar] [CrossRef]
- He, Y.; Yuan, Q.; Mathieu, J.; Stadler, L.; Senehi, N.; Sun, R.; Alvarez, P.J.J. Antibiotic resistance genes from livestock waste: Occurrence, dissemination, and treatment. Npj Clean. Water 2020, 3, 4. [Google Scholar] [CrossRef]
- Jian, Z.H.; Zeng, L.; Xu, T.J.; Sun, S.; Yan, S.X.; Yang, L.; Huang, Y.; Jia, J.J.; Dou, T.F. Antibiotic resistance genes in bacteria: Occurrence, spread, and control. J. Basic. Microb. 2021, 61, 1049–1070. [Google Scholar] [CrossRef]
- Huang, J.H.; Ma, J.L.; Shang, K.X.; Hu, X.; Liang, Y.; Li, D.W.; Wu, Z.W.; Dai, L.; Chen, L.; Wang, L.P. Evolution and Diversity of the Antimicrobial Resistance Associated Mobilome in: A Probable Mobile Genetic Elements Reservoir for Other Streptococci. Front. Cell. Infect. Microbiol. 2016, 6, 118. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | Strains | Source | Location | Year | Accession No. | Complete? | Length |
|---|---|---|---|---|---|---|---|
| Streptococcus parasuis | SUT-380 | Pig | Japan | 2013 | AP024277.1 | Y | 2,109,881 |
| Streptococcus parasuis | SUT-503 | Pig | Japan | 2014 | AP024280.1 | Y | 2,065,066 |
| Streptococcus parasuis | SUT-286 | Pig | Japan | 2013 | AP024276.1 | Y | 2,197,342 |
| Streptococcus parasuis | SUT-7 | Pig | Japan | 2012 | AP024275.1 | Y | 2,202,836 |
| Streptococcus parasuis | BS27 | Homo sapiens | China | 2018 | JAETXU000000000.1 | N | 1,909,795 |
| Streptococcus parasuis | BS26 | Homo sapiens | China | 2018 | CP069079.1 | Y | 1,932,292 |
| Streptococcus parasuis | H35 | Pig | China | 2018 | CP076721.1 | Y | 2,186,318 |
| Streptococcus parasuis | 4253 | Cow | Switzerland | 2018 | SHGT00000000.1 | N | 1,881,656 |
| Streptococcus parasuis | 86–5192 | Cow | United States | 1980 | ALLG00000000.1 | N | 2,110,166 |
| Streptococcus parasuis | 88–1861 | Pig | Canada | 1980 | ALLW00000000.1 | N | 2,272,254 |
| Streptococcus parasuis | 89–4109-1 | Pig | Canada | 1980 | ALLL00000000.1 | N | 2,176,728 |
| Streptococcus parasuis | SUT-319 | Pig | Japan | / | DRX016753 | N | 2,129,893 |
| Streptococcus parasuis | SUT-328 | Pig | Japan | / | BOJH00000000.1 | N | 2,126,590 |
| Streptococcus parasuis | 10–36,905 | Cow | United States | 2010 | WNXH00000000.1 | N | 2,148,541 |
| Streptococcus parasuis | SFJ45 | Pig | China | 2017 | CP102747.1 | Y | 2,015,398 |
| Streptococcus parasuis | 7500 | Homo sapiens | China | 2022 | CP128410.1 | Y | 2,008,266 |
| Streptococcus parasuis | 221006 | Homo sapiens | China | 2022 | CP137602.1 | Y | 1,985,497 |
| Streptococcus parasuis | SS17 | Pig | China | 2021 | CP090522.1 | Y | 1,984,594 |
| Streptococcus parasuis | NN1 | Homo sapiens | China | 2020 | CP073632.1 | Y | 1,971,006 |
| Streptococcus parasuis | SS20 | Pig | China | 2021 | CP086728.1 | Y | 1,961,908 |
| Streptococcus parasuis | AH0906 | Pig | China | 2009 | JANFLX000000000.1 | N | 2,203,067 |
| Streptococcus parasuis | SS-5819 | Pig | Switzerland | 2022 | JAVIID010000001.1 | N | 2,130,485 |
| Streptococcus parasuis | FZ1 | Pig | China | 2021 | CP170759 | Y | 2,054,729 |
| Streptococcus parasuis | FZ2 | Pig | China | 2022 | CP170760 | Y | 2,032,338 |
| Streptococcus suis | BM407 | Homo sapiens | Viet Nam | 2004 | FM252032.1 | Y | 2,146,229 |
| Streptococcus suis | S735 | Pig | Canada | / | CP003736.1 | Y | 1,980,887 |
| Streptococcus suis | TJS75 | Pig | China | 2015 | CP095162.1 | Y | 2,368,195 |
| Streptococcus suis | NSUI060 | Pig | Canada | 2008 | CP012911.1 | Y | 2,285,232 |
| Streptococcus suis | ISU2614 | Pig | United States | 2014 | CP031377.1 | Y | 2,163,384 |
| Streptococcus suis | CS100322 | Pig | China | 2010 | CP024050.1 | Y | 2,137,649 |
| Streptococcus suis | DNC49 | Pig | Denmark | / | CP102140.1 | Y | 2,137,486 |
| Streptococcus suis | NSUI002 | Pig | Canada | 2008 | CP011419.1 | Y | 2,255,345 |
| Strains Name | Multi-Locus Allelic Profile | ST | Genome Accession | ||||||
|---|---|---|---|---|---|---|---|---|---|
| aroA | cpn60 | dpr | gki | mutS | recA | thrA | |||
| FZ1 | 299 | 440 | 56 | 83 | 83 | 67 | 155 | 2909 | CP170759 |
| FZ2 | 299 | 83 | 172 | 635 | 83 | 67 | 228 | 2910 | CP170760 |
| FZ1 | FZ2 | Does Prophages/ICEs/GIs Contain Resistance Genes? | ||
|---|---|---|---|---|
| FZ1 | FZ2 | |||
| CRISPR-Cas | Type IC | / | / | / |
| Prophages | SpsFZ1-P1 | SpsFZ2-P1 | N | N |
| SpsFZ2-P2 | Y (aac(6′)-aph(2″), ant(6)-Ia, mdt(A), erm(B)) | |||
| SpsFZ2-P3 | N | |||
| ICEs | SpsFZ1-ICE1 | / | Y (tet(M)) | / |
| GIs | SpsFZ1-GI1 | SpsFZ2-GI1 | N | N |
| SpsFZ1-GI2 | SpsFZ2-GI2 | Y (ermC, aacA-aphD, OptrA, ANT(9), bacA, aadK) | Y (ermC, aacA-aphD, OptrA, ANT(9), aadK) | |
| SpsFZ1-GI3 | SpsFZ2-GI3 | Y (ermC, mef, aadK, aph3-III) | N | |
| SpsFZ1-GI4 | SpsFZ2-GI4 | N | N | |
| SpsFZ1-GI5 | SpsFZ2-GI5 | N | N | |
| SpsFZ1-GI6 | SpsFZ2-GI6 | N | N | |
| SpsFZ1-GI7 | SpsFZ2-GI7 | N | N | |
| SpsFZ1-GI8 | SpsFZ2-GI8 | N | N | |
| SpsFZ1-GI9 | / | N | / | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, G.; Liu, Y.; Jiang, Z.; Liu, K.; Wang, X.; Hao, J.; Kong, H.; Yu, Y.; Ding, Z.; Li, M.; et al. Genomic Characterization of Potential Opportunistic Zoonotic Streptococcus parasuis Isolated in China. Pathogens 2025, 14, 395. https://doi.org/10.3390/pathogens14040395
Liu G, Liu Y, Jiang Z, Liu K, Wang X, Hao J, Kong H, Yu Y, Ding Z, Li M, et al. Genomic Characterization of Potential Opportunistic Zoonotic Streptococcus parasuis Isolated in China. Pathogens. 2025; 14(4):395. https://doi.org/10.3390/pathogens14040395
Chicago/Turabian StyleLiu, Gang, Yu Liu, Zhikang Jiang, Kang Liu, Xianwen Wang, Juyuan Hao, He Kong, Yajie Yu, Zicheng Ding, Min Li, and et al. 2025. "Genomic Characterization of Potential Opportunistic Zoonotic Streptococcus parasuis Isolated in China" Pathogens 14, no. 4: 395. https://doi.org/10.3390/pathogens14040395
APA StyleLiu, G., Liu, Y., Jiang, Z., Liu, K., Wang, X., Hao, J., Kong, H., Yu, Y., Ding, Z., Li, M., & Han, X. (2025). Genomic Characterization of Potential Opportunistic Zoonotic Streptococcus parasuis Isolated in China. Pathogens, 14(4), 395. https://doi.org/10.3390/pathogens14040395