
Blood Transcriptome Profiling Highlights the Role of Intestinal Bacterial Translocation in Severe COVID-19

 , , ,
, , ,  ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. Study Design
2.2. RNA Extraction Library Preparation
2.3. Bioinformatics
2.4. Estimation of Cell Populations
2.5. Quantitative PCR-Data Analysis
3. Results
3.1. GO Term Enrichment Analysis
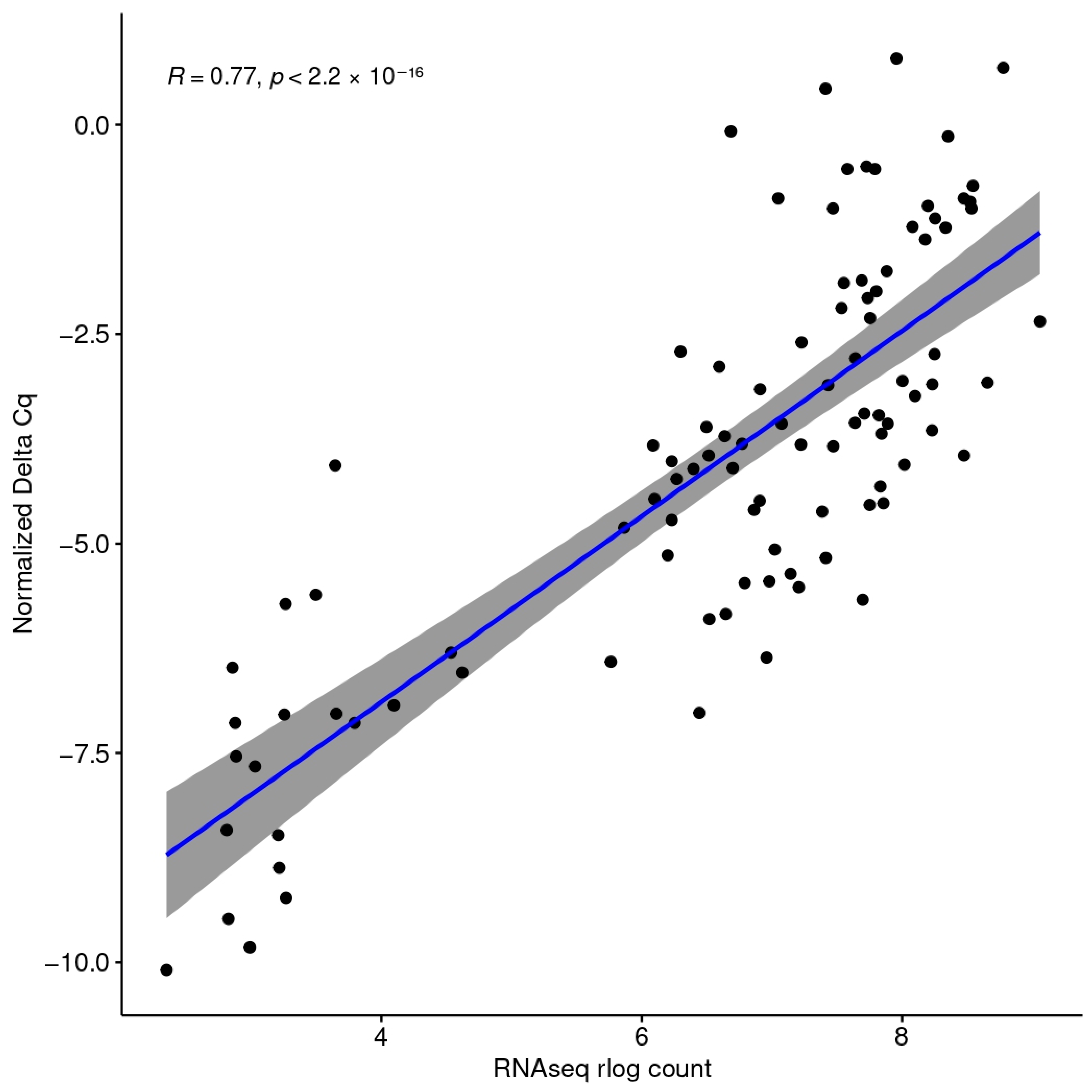
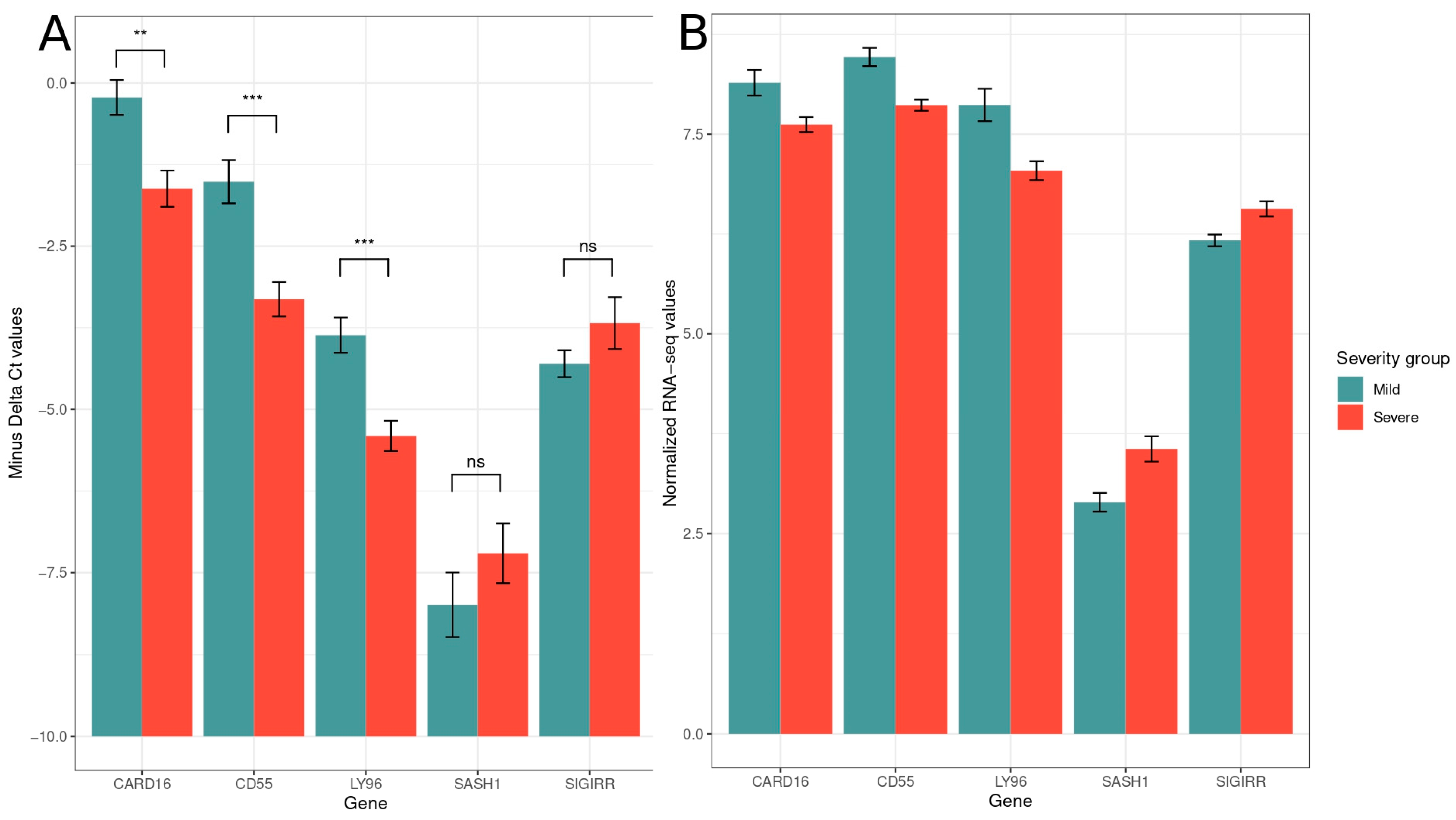

3.2. Digital Cytometry
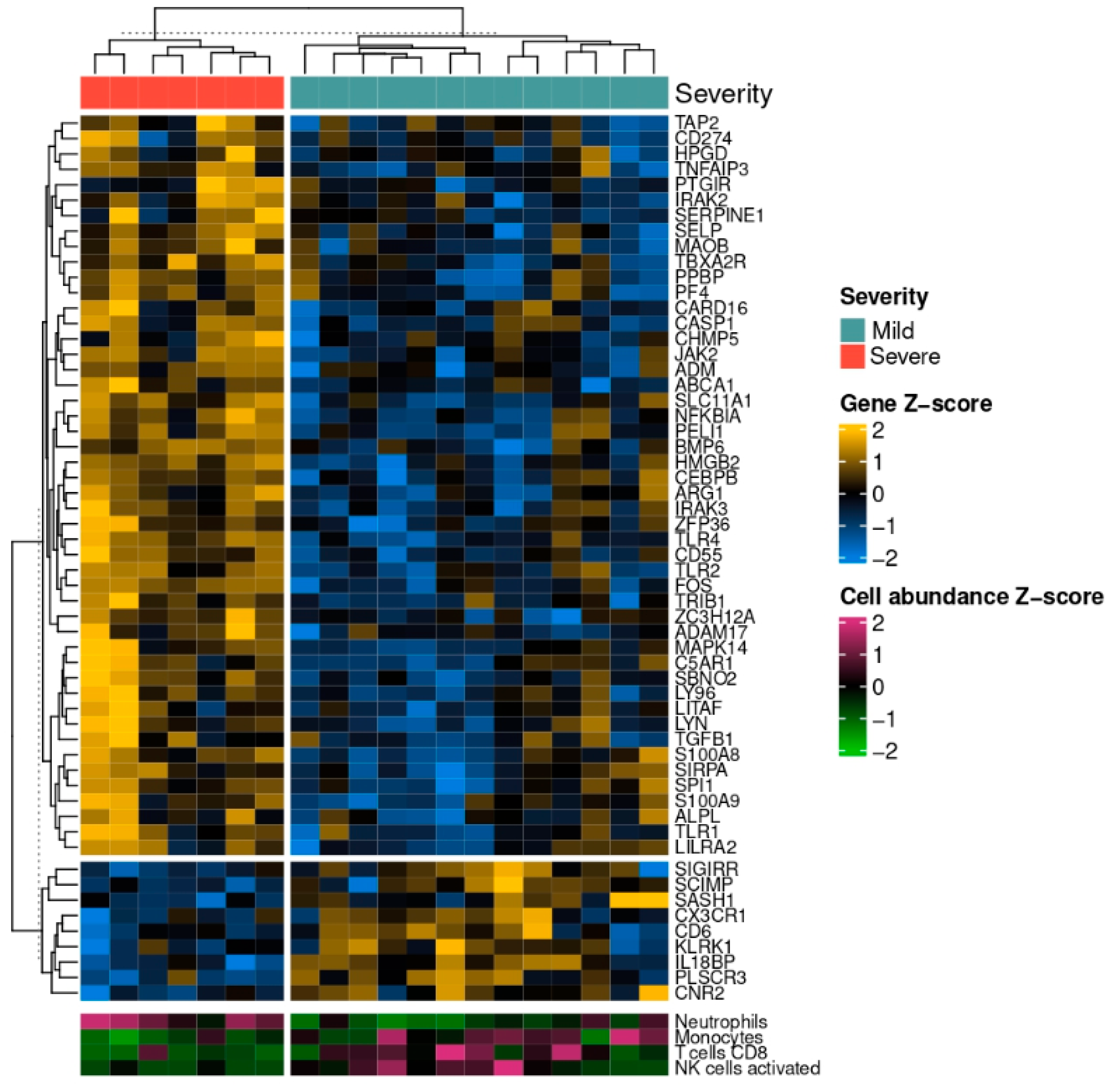
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhou, P.; Yang, X.L.; Wang, X.G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.R.; Zhu, Y.; Li, B.; Huang, C.L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef]
- WHO Director-General’s Remarks at the Media Briefing on 2019-nCoV on 11 February 2020. Available online: https://www.who.int/director-general/speeches/detail/who-director-general-s-remarks-at-the-media-briefing-on-2019-ncov-on-11-february-2020 (accessed on 18 December 2023).
- Lauer, S.A.; Grantz, K.H.; Bi, Q.; Jones, F.K.; Zheng, Q.; Meredith, H.R.; Azman, A.S.; Reich, N.G.; Lessler, J. The Incubation Period of Coronavirus Disease 2019 (COVID-19) From Publicly Reported Confirmed Cases: Estimation and Application. Ann. Intern. Med. 2020, 172, 577–582. [Google Scholar] [CrossRef] [PubMed]
- Israfil, S.M.H.; Sarker, M.d.M.R.; Rashid, P.T.; Talukder, A.A.; Kawsar, K.A.; Khan, F.; Akhter, S.; Poh, C.L.; Mohamed, I.N.; Ming, L.C.; et al. Clinical Characteristics and Diagnostic Challenges of COVID−19: An Update From the Global Perspective. Front. Public Health 2021, 8, 567395. Available online: https://www.frontiersin.org/articles/10.3389/fpubh.2020.567395 (accessed on 21 December 2023). [CrossRef] [PubMed]
- Yang, J.; Zheng, Y.; Gou, X.; Pu, K.; Chen, Z.; Guo, Q.; Ji, R.; Wang, H.; Wang, Y.; Zhou, Y.; et al. Prevalence of comorbidities and its effects in patients infected with SARS-CoV-2: A systematic review and meta-analysis. Int. J. Infect. Dis. Off. Publ. Int. Soc. Infect. Dis. 2020, 94, 91–95. [Google Scholar] [CrossRef]
- Diamond, M.S.; Kanneganti, T.D. Innate immunity: The first line of defense against SARS-CoV-2. Nat. Immunol. 2022, 23, 165–176. [Google Scholar] [CrossRef]
- Ragab, D.; Salah Eldin, H.; Taeimah, M.; Khattab, R.; Salem, R. The COVID-19 Cytokine Storm; What We Know So Far. Front. Immunol. 2020, 11, 1446. [Google Scholar] [CrossRef] [PubMed]
- Giron, L.B.; Dweep, H.; Yin, X.; Wang, H.; Damra, M.; Goldman, A.R.; Gorman, N.; Palmer, C.S.; Tang, H.Y.; Shaikh, M.W.; et al. Plasma Markers of Disrupted Gut Permeability in Severe COVID-19 Patients. Front. Immunol. 2021, 12, 1996. [Google Scholar]
- Nagpal, R.; Yadav, H. Bacterial Translocation from the Gut to the Distant Organs: An Overview. Ann. Nutr. Metab. 2017, 71 (Suppl. S1), 11–16. [Google Scholar] [CrossRef]
- Fasano, A.; Not, T.; Wang, W.; Uzzau, S.; Berti, I.; Tommasini, A.; Goldblum, S.E. Zonulin, a newly discovered modulator of intestinal permeability, and its expression in coeliac disease. Lancet 2000, 355, 1518–1519. [Google Scholar] [CrossRef]
- Brogna, C.; Brogna, B.; Bisaccia, D.R.; Lauritano, F.; Marino, G.; Montano, L.; Cristoni, S.; Prisco, M.; Piscopo, M. Could SARS-CoV-2 Have Bacteriophage Behavior or Induce the Activity of Other Bacteriophages? Vaccines 2022, 10, 708. [Google Scholar] [CrossRef]
- Brogna, C.; Viduto, V.; Fabrowski, M.; Cristoni, S.; Marino, G.; Montano, L.; Piscopo, M. The importance of the gut microbiome in the pathogenesis and transmission of SARS-CoV-2: Someone on Earth: “we moved at the speed of Science!”—Science from the center of the Universe: “Hey man, I’m still waiting for you in the 50s!”. Gut Microbes 2023, 15, 2244718. [Google Scholar] [CrossRef] [PubMed]
- Petrillo, M.; Querci, M.; Brogna, C.; Ponti, J.; Cristoni, S.; Markov, P.V.; Valsesia, A.; Leoni, G.; Benedetti, A.; Wiss, T.; et al. Evidence of SARS-CoV-2 Bacteriophage Potential in Human Gut Microbiota. F1000Research 2022, 11, 292. Available online: https://f1000research.com/articles/11-292/v1 (accessed on 8 April 2025). [CrossRef]
- Brogna, C.; Costanzo, V.; Brogna, B.; Bisaccia, D.R.; Brogna, G.; Giuliano, M.; Montano, L.; Viduto, V.; Cristoni, S.; Fabrowski, M.; et al. Analysis of Bacteriophage Behavior of a Human RNA Virus, SARS-CoV-2, through the Integrated Approach of Immunofluorescence Microscopy, Proteomics and D-Amino Acid Quantification. Int. J. Mol. Sci. 2023, 24, 3929. [Google Scholar] [CrossRef]
- Brogna, C.; Cristoni, S.; Petrillo, M.; Bisaccia, D.R.; Lauritano, F.; Montano, L.; Prisco, M.; Piscopo, M. The first report on detecting SARS-CoV-2 inside bacteria of the human gut microbiome: A case series on asymptomatic family members and a child with COVID-19. F1000Research 2024, 11, 135. [Google Scholar] [CrossRef]
- Zari, A.; Redwan, E.M.; Raszek, M.; Cowley, D.; Hromić-Jahjefendić, A.; Uversky, V.N.; Fabrowski, M.; Brogna, C.; Piscopo, M.; Rubio-Casillas, A. Interplay between Multisystem Inflammatory Syndrome in Children, Interleukin 6, Microbiome, and Gut Barrier Integrity. Immuno 2024, 4, 226–246. [Google Scholar] [CrossRef]
- DePamphilis, M.L.; Adler, J. Attachment of Flagellar Basal Bodies to the Cell Envelope: Specific Attachment to the Outer, Lipopolysaccharide Membrane and the Cytoplasmic Membrane. J. Bacteriol. 1971, 105, 396–407. [Google Scholar] [CrossRef]
- Poltorak, A.; He, X.; Smirnova, I.; Liu, M.Y.; Van Huffel, C.; Du, X.; Birdwell, D.; Alejos, E.; Silva, M.; Galanos, C.; et al. Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: Mutations in Tlr4 gene. Science 1998, 282, 2085–2088. [Google Scholar] [CrossRef]
- Lu, Y.C.; Yeh, W.C.; Ohashi, P.S. LPS/TLR4 signal transduction pathway. Cytokine 2008, 42, 145–151. [Google Scholar] [CrossRef] [PubMed]
- Kermali, M.; Khalsa, R.K.; Pillai, K.; Ismail, Z.; Harky, A. The role of biomarkers in diagnosis of COVID-19—A systematic review. Life Sci. 2020, 254, 117788. [Google Scholar] [CrossRef]
- Wargodsky, R.; Cruz, P.D.; LaFleur, J.; Yamane, D.; Kim, J.S.; Benjenk, I.; Heinz, E.; Irondi, O.O.; Farrar, K.; Toma, I.; et al. RNA Sequencing in COVID-19 patients identifies neutrophil activation biomarkers as a promising diagnostic platform for infections. PLoS ONE 2022, 17, e0261679. [Google Scholar] [CrossRef]
- Papadopoulou, G.; Manoloudi, E.; Repousi, N.; Skoura, L.; Hurst, T.; Karamitros, T. Molecular and Clinical Prognostic Biomarkers of COVID-19 Severity and Persistence. Pathogens 2022, 11, 311. Available online: https://pubmed.ncbi.nlm.nih.gov/35335635/ (accessed on 26 April 2022). [CrossRef]
- COVID-19-Therapeutikos-Algorithmos-20210222. Available online: https://eody.gov.gr/wp-content/uploads/2021/03/covid-19-therapeutikos-algorithmos-20210222.pdf (accessed on 15 February 2024).
- Malli, F.; Lampropoulos, I.C.; Perlepe, G.; Papagiannis, D.; Gourgoulianis, K.I. Analysis of SARS-CoV-2 Cases, COVID-19 Outcomes and Vaccinations, during the Different SARS-CoV-2 Variants in Greece. Vaccines 2023, 11, 126. [Google Scholar] [CrossRef] [PubMed]
- SourceForge. BBMap. 2023. Available online: https://sourceforge.net/projects/bbmap/ (accessed on 15 February 2024).
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef] [PubMed]
- QuantSeq 3‘ mRNA-Seq Integrated Data Analysis. Available online: https://www.lexogen.com/wp-content/uploads/2021/05/015UG108V0311_QuantSeq-Data-Analysis-Pipeline_2021-05-04.pdf (accessed on 15 February 2024).
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq—A Python framework to work with high-throughput sequencing data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.; Hu, E.; Xu, S.; Chen, M.; Guo, P.; Dai, Z.; Feng, T.; Zhou, L.; Tang, W.; Zhan, L.; et al. clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. Innovation 2021, 2, 100141. [Google Scholar] [CrossRef]
- Newman, A.M.; Steen, C.B.; Liu, C.L.; Gentles, A.J.; Chaudhuri, A.A.; Scherer, F.; Khodadoust, M.S.; Esfahani, M.S.; Luca, B.A.; Steiner, D.; et al. Determining cell type abundance and expression from bulk tissues with digital cytometry. Nat. Biotechnol. 2019, 37, 773–782. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2−ΔΔCT Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Jeanty, C.; Longrois, D.; Mertes, P.-M.; Wagner, D.R.; Devaux, Y. An Optimized Protocol for Microarray Validation by Quantitative PCR Using Amplified Amino Allyl Labeled RNA. BMC Genom. 2010, 11, 542. [Google Scholar] [CrossRef]
- Kwon, N.; Lee, K.E.; Singh, M.; Kang, S.G. Suitable Primers for GAPDH Reference Gene Amplification in Quantitative RT-PCR Analysis of Human Gene Expression. Gene Rep. 2021, 24, 101272. [Google Scholar] [CrossRef]
- Helms, J.; Tacquard, C.; Severac, F.; Leonard-Lorant, I.; Ohana, M.; Delabranche, X.; Merdji, H.; Clere-Jehl, R.; Schenck, M.; Fagot Gandet, F.; et al. High risk of thrombosis in patients with severe SARS-CoV-2 infection: A multicenter prospective cohort study. Intensive Care Med. 2020, 46, 1089–1098. [Google Scholar] [CrossRef] [PubMed]
- Nicolai, L.; Leunig, A.; Brambs, S.; Kaiser, R.; Weinberger, T.; Weigand, M.; Muenchhoff, M.; Hellmuth, J.C.; Ledderose, S.; Schulz, H.; et al. Immunothrombotic Dysregulation in COVID-19 Pneumonia Is Associated with Respiratory Failure and Coagulopathy. Circulation 2020, 142, 1176–1189. [Google Scholar] [CrossRef]
- Al-Samkari, H.; Karp Leaf, R.S.; Dzik, W.H.; Carlson, J.C.T.; Fogerty, A.E.; Waheed, A.; Goodarzi, K.; Bendapudi, P.K.; Bornikova, L.; Gupta, S.; et al. COVID-19 and coagulation: Bleeding and thrombotic manifestations of SARS-CoV-2 infection. Blood 2020, 136, 489–500. [Google Scholar] [CrossRef] [PubMed]
- Shimazu, R.; Akashi, S.; Ogata, H.; Nagai, Y.; Fukudome, K.; Miyake, K.; Kimoto, M. MD-2, a Molecule that Confers Lipopolysaccharide Responsiveness on Toll-like Receptor 4. J. Exp. Med. 1999, 189, 1777–1782. [Google Scholar] [CrossRef]
- Chang, M.; Jin, W.; Sun, S.C. Peli1 facilitates TRIF-dependent Toll-like receptor signaling and proinflammatory cytokine production. Nat. Immunol. 2009, 10, 1089–1095. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.; Woodward, T.; Amar, S. A PTP4A3 peptide PIMAP39 modulates TNF-alpha levels and endotoxic shock. J. Innate Immun. 2010, 2, 43–55. [Google Scholar] [CrossRef]
- Oliva, A.; Cammisotto, V.; Cangemi, R.; Ferro, D.; Miele, M.C.; De Angelis, M.; Cancelli, F.; Pignatelli, P.; Venditti, M.; Pugliese, F.; et al. Low-Grade Endotoxemia and Thrombosis in COVID-19. Clin. Transl. Gastroenterol. 2021, 12, e00348. [Google Scholar] [CrossRef]
- Oliva, A.; Miele, M.C.; Di Timoteo, F.; De Angelis, M.; Mauro, V.; Aronica, R.; Al Ismail, D.; Ceccarelli, G.; Pinacchio, C.; D’Ettorre, G.; et al. Persistent Systemic Microbial Translocation and Intestinal Damage During Coronavirus Disease-19. Front. Immunol. 2021, 12, 708149. Available online: https://www.frontiersin.org/articles/10.3389/fimmu.2021.708149 (accessed on 21 February 2024). [CrossRef]
- Basting, C.M.; Langat, R.; Broedlow, C.A.; Guerrero, C.R.; Bold, T.D.; Bailey, M.; Velez, A.; Schroeder, T.; Short-Miller, J.; Cromarty, R.; et al. SARS-CoV-2 infection is associated with intestinal permeability, systemic inflammation, and microbial dysbiosis in hospitalized patients. Microbiol. Spectr. 2024, 12, e00680-24. [Google Scholar] [CrossRef]
- Sørensen, O.E.; Follin, P.; Johnsen, A.H.; Calafat, J.; Tjabringa, G.S.; Hiemstra, P.S.; Borregaard, N. Human cathelicidin, hCAP-18, is processed to the antimicrobial peptide LL-37 by extracellular cleavage with proteinase 3. Blood 2001, 97, 3951–3959. [Google Scholar] [CrossRef]
- Ericksen, B.; Wu, Z.; Lu, W.; Lehrer, R.I. Antibacterial Activity and Specificity of the Six Human α-Defensins. Antimicrob. Agents Chemother. 2005, 49, 269–275. [Google Scholar] [CrossRef]
- Theprungsirikul, J.; Skopelja-Gardner, S.; Rigby, W.F.C. Killing three birds with one BPI: Bactericidal, opsonic, and anti-inflammatory functions. J. Transl. Autoimmun. 2021, 4, 100105. [Google Scholar] [CrossRef] [PubMed]
- Gazzano-Santoro, H.; Parent, J.B.; Grinna, L.; Horwitz, A.; Parsons, T.; Theofan, G.; Elsbach, P.; Weiss, J.; Conlon, P.J. High-affinity binding of the bactericidal/permeability-increasing protein and a recombinant amino-terminal fragment to the lipid A region of lipopolysaccharide. Infect. Immun. 1992, 60, 4754–4761. [Google Scholar] [CrossRef] [PubMed]
- Brenchley, J.M.; Price, D.A.; Schacker, T.W.; Asher, T.E.; Silvestri, G.; Rao, S.; Kazzaz, Z.; Bornstein, E.; Lambotte, O.; Altmann, D.; et al. Microbial translocation is a cause of systemic immune activation in chronic HIV infection. Nat. Med. 2006, 12, 1365–1371. [Google Scholar] [CrossRef] [PubMed]
- Chancharoenthana, W.; Kamolratanakul, S.; Ariyanon, W.; Thanachartwet, V.; Phumratanaprapin, W.; Wilairatana, P.; Leelahavanichkul, A. Abnormal Blood Bacteriome, Gut Dysbiosis, and Progression to Severe Dengue Disease. Front. Cell Infect. Microbiol. 2022, 12, 890817. [Google Scholar] [CrossRef]
- Zang, R.; Gomez Castro, M.F.; McCune, B.T.; Zeng, Q.; Rothlauf, P.W.; Sonnek, N.M.; Liu, Z.; Brulois, K.F.; Wang, X.; Greenberg, H.B.; et al. TMPRSS2 and TMPRSS4 promote SARS-CoV-2 infection of human small intestinal enterocytes. Sci. Immunol. 2020, 5, eabc3582. [Google Scholar] [CrossRef]
- Minami, S.; Matsumoto, N.; Omori, H.; Nakamura, Y.; Tamiya, S.; Nouda, R.; Nurdin, J.A.; Yamasaki, M.; Kotaki, T.; Kanai, Y.; et al. Effective SARS-CoV-2 replication of monolayers of intestinal epithelial cells differentiated from human induced pluripotent stem cells. Sci. Rep. 2023, 13, 11610. [Google Scholar] [CrossRef]
- Stanifer, M.L.; Kee, C.; Cortese, M.; Zumaran, C.M.; Triana, S.; Mukenhirn, M.; Kraeusslich, H.-G.; Alexandrov, T.; Bartenschlager, R.; Boulant, S. Critical Role of Type III Interferon in Controlling SARS-CoV-2 Infection in Human Intestinal Epithelial Cells. Cell Rep. 2020, 32, 107863. [Google Scholar] [CrossRef]
- Sato, J.; Kanazawa, A.; Ikeda, F.; Yoshihara, T.; Goto, H.; Abe, H.; Komiya, K.; Kawaguchi, M.; Shimizu, T.; Ogihara, T.; et al. Gut Dysbiosis and Detection of “Live Gut Bacteria” in Blood of Japanese Patients with Type 2 Diabetes. Diabetes Care 2014, 37, 2343–2350. [Google Scholar] [CrossRef]
- Thaiss, C.A.; Levy, M.; Grosheva, I.; Zheng, D.; Soffer, E.; Blacher, E.; Braverman, S.; Tengeler, A.C.; Barak, O.; Elazar, M.; et al. Hyperglycemia drives intestinal barrier dysfunction and risk for enteric infection. Science 2018, 359, 1376–1383. [Google Scholar] [CrossRef]
- Vemuri, R.; Ruggiero, A.; Whitfield, J.M.; Dugan, G.O.; Cline, J.M.; Block, M.R.; Guo, H.; Kavanagh, K. Hypertension promotes microbial translocation and dysbiotic shifts in the fecal microbiome of nonhuman primates. Am. J. Physiol-Heart Circ. Physiol. 2022, 322, H474–H485. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Goel, R.; Kumar, A.; Qi, Y.; Lobaton, G.; Hosaka, K.; Mohammed, M.; Handberg, E.M.; Richards, E.M.; Pepine, C.J.; et al. Imbalance of gut microbiome and intestinal epithelial barrier dysfunction in patients with high blood pressure. Clin. Sci. 2018, 132, 701–718. [Google Scholar] [CrossRef]
- Assimakopoulos, S.F.; Eleftheriotis, G.; Lagadinou, M.; Karamouzos, V.; Dousdampanis, P.; Siakallis, G.; Marangos, M. SARS-CoV-2-Induced Viral Sepsis: The Role of Gut Barrier Dysfunction. Microorganisms 2022, 10, 1050. Available online: https://www.mdpi.com/2076-2607/10/5/1050 (accessed on 21 February 2024). [CrossRef] [PubMed]
- Li, J.; Zhang, K.; Zhang, Y.; Gu, Z.; Huang, C. Neutrophils in COVID-19: Recent insights and advances. Virol. J. 2023, 20, 169. [Google Scholar] [CrossRef] [PubMed]
- Shiomi, H.; Shimizu, T.; Endo, Y.; Murata, S.; Kurumi, Y.; Uji, Y.; Tani, T. Relations Among Circulating Monocytes, Dendritic Cells, and Bacterial Translocation in Patients with Intestinal Obstruction. World J. Surg. 2007, 31, 1212. [Google Scholar] [CrossRef]
- Knoll, R.; Schultze, J.L.; Schulte-Schrepping, J. Monocytes and Macrophages in COVID-19. Front. Immunol. 2021, 12, 720109. [Google Scholar] [CrossRef]
- Osman, M.; Faridi, R.M.; Sligl, W.; Shabani-Rad, M.T.; Dharmani-Khan, P.; Parker, A.; Kalra, A.; Tripathi, M.B.; Storek, J.; Cohen Tervaert, J.W.; et al. Impaired natural killer cell counts and cytolytic activity in patients with severe COVID-19. Blood Adv. 2020, 4, 5035–5039. [Google Scholar] [CrossRef]
- Le, T.; Aronow, R.A.; Kirshtein, A.; Shahriyari, L. A review of digital cytometry methods: Estimating the relative abundance of cell types in a bulk of cells. Briefings Bioinform. 2021, 22, bbaa219. [Google Scholar] [CrossRef]
- Li, Z.; Jin, P.; Xiang, R.; Li, X.; Shen, J.; He, M.; Liu, X.; Zhu, H.; Wu, S.; Dong, F.; et al. A CD8+ T cell related immune score predicts survival and refines the risk assessment in acute myeloid leukemia. Front. Immunol. 2024, 15, 1408109. Available online: https://www.frontiersin.org/journals/immunology/articles/10.3389/fimmu.2024.1408109/full (accessed on 9 April 2025). [CrossRef]
- Jin, H.; Liu, Z. A benchmark for RNA-seq deconvolution analysis under dynamic testing environments. Genome Biol. 2021, 22, 102. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Severity Class | Criteria |
|---|---|
| Mild [13] | Absence or few imaging findings of pneumonia SpO2 > 94% in FiO2 21%. |
| Severe [7] | Extensive imaging findings of pneumonia One of the following findings: SpO2 < 94% in FiO2 21% PO2/FiO2 < 300 Respiratory Rate: >30 breaths/min Opacities in >50% of the lung parenchyma |
| Mild [13] | Severe [7] | |||
|---|---|---|---|---|
| n | (%) | n | (%) | |
| Sex | ||||
| Female [8] | 5 | 38.5 | 3 | 42.9 |
| Male [12] | 8 | 61.5 | 4 | 57.1 |
| Age | ||||
| <60 [14] | 10 | 76.9 | 4 | 57.1 |
| ≥60 [6] | 3 | 23.1 | 3 | 42.9 |
| Obesity | ||||
| No [17] | 12 | 92.3 | 5 | 71.4 |
| Yes [3] | 1 | 7.7 | 2 | 28.6 |
| Diabetes | ||||
| No [13] | 11 | 84.6 | 2 | 28.6 |
| Yes [7] | 2 | 15.4 | 5 | 71.4 |
| Hypertension | ||||
| No [14] | 10 | 76.9 | 4 | 57.1 |
| Yes [6] | 3 | 23.1 | 3 | 42.9 |
| Smoking | ||||
| Non-smoker [13] | 8 | 61.5 | 5 | 71.4 |
| Current-smoker [1] | 0 | 0 | 1 | 14.3 |
| Ex-smoker [6] | 5 | 38.5 | 1 | 14.3 |
| GI symptoms on admission | ||||
| No [15] | 10 | 76.9 | 5 | 71.4 |
| Yes [5] | 3 | 23.1 | 2 | 28.6 |
| Gene | Forward | Reverse | mRNA Refseq Accession | Source |
|---|---|---|---|---|
| LY96 | TGCCGAGGATCTGATGAC | ATTAGGTTGGTGTAGGATGAC | NM_015364 | Jeanty et al. BMC Genomics 2010 [33] |
| CARD16 | CGAAAGGGGCACAGGC | ATTCTGCCTTCTGGGCTTG | NM_001017534 | Origene, Cat. No. HP202556 |
| SIGIRR | CCTCCTTCACTCTTCAGAGAGC | ACGGCACTTGACATAGAGCAGG | NM_021805 | Origene, Cat. No. HP214224o |
| SASH1 | GTAACAGCGACCAGTCAGGATC | GACTTGGCAGATAGGAGGCTAG | NM_015278 | Origene, Cat. No. HP211516 |
| CD55 | CACGGAGTACACCTGTTTCCAG | CCCAAGCAAACCTGTCAACGTG | NM_000574 | Origene, Cat. No. HP200542 |
| GAPDH | TTGGCTACAGCAACAGGGTG | GGGGAGATTCAGTGTGGTGG | NM_002046 | Kwon et al. Gene Reports 2021 [34] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tremoulis, D.C.; Papadopoulou, G.; Pogka, V.; Argyraki, A.; Lourida, G.; Mentis, A.; Karamitros, T. Blood Transcriptome Profiling Highlights the Role of Intestinal Bacterial Translocation in Severe COVID-19. Pathogens 2025, 14, 381. https://doi.org/10.3390/pathogens14040381
Tremoulis DC, Papadopoulou G, Pogka V, Argyraki A, Lourida G, Mentis A, Karamitros T. Blood Transcriptome Profiling Highlights the Role of Intestinal Bacterial Translocation in Severe COVID-19. Pathogens. 2025; 14(4):381. https://doi.org/10.3390/pathogens14040381
Chicago/Turabian StyleTremoulis, Dimitrios Christos, Gethsimani Papadopoulou, Vasiliki Pogka, Aikaterini Argyraki, Giota Lourida, Andreas Mentis, and Timokratis Karamitros. 2025. "Blood Transcriptome Profiling Highlights the Role of Intestinal Bacterial Translocation in Severe COVID-19" Pathogens 14, no. 4: 381. https://doi.org/10.3390/pathogens14040381
APA StyleTremoulis, D. C., Papadopoulou, G., Pogka, V., Argyraki, A., Lourida, G., Mentis, A., & Karamitros, T. (2025). Blood Transcriptome Profiling Highlights the Role of Intestinal Bacterial Translocation in Severe COVID-19. Pathogens, 14(4), 381. https://doi.org/10.3390/pathogens14040381