The Role of Caspase-1 and Caspase-4 in Modulating Gingival Epithelial Cell Responses to Aggregatibacter actinomycetemcomitans Infection


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Material and Methods
2.1. Cell Culture and Bacteria
2.2. Targeted Olink Protein Analysis
2.3. Measurement of LDH Release
2.4. Proliferation Assay
2.5. Colonization Assay
2.6. Invasion Assay
2.7. Statistical Analysis
3. Results
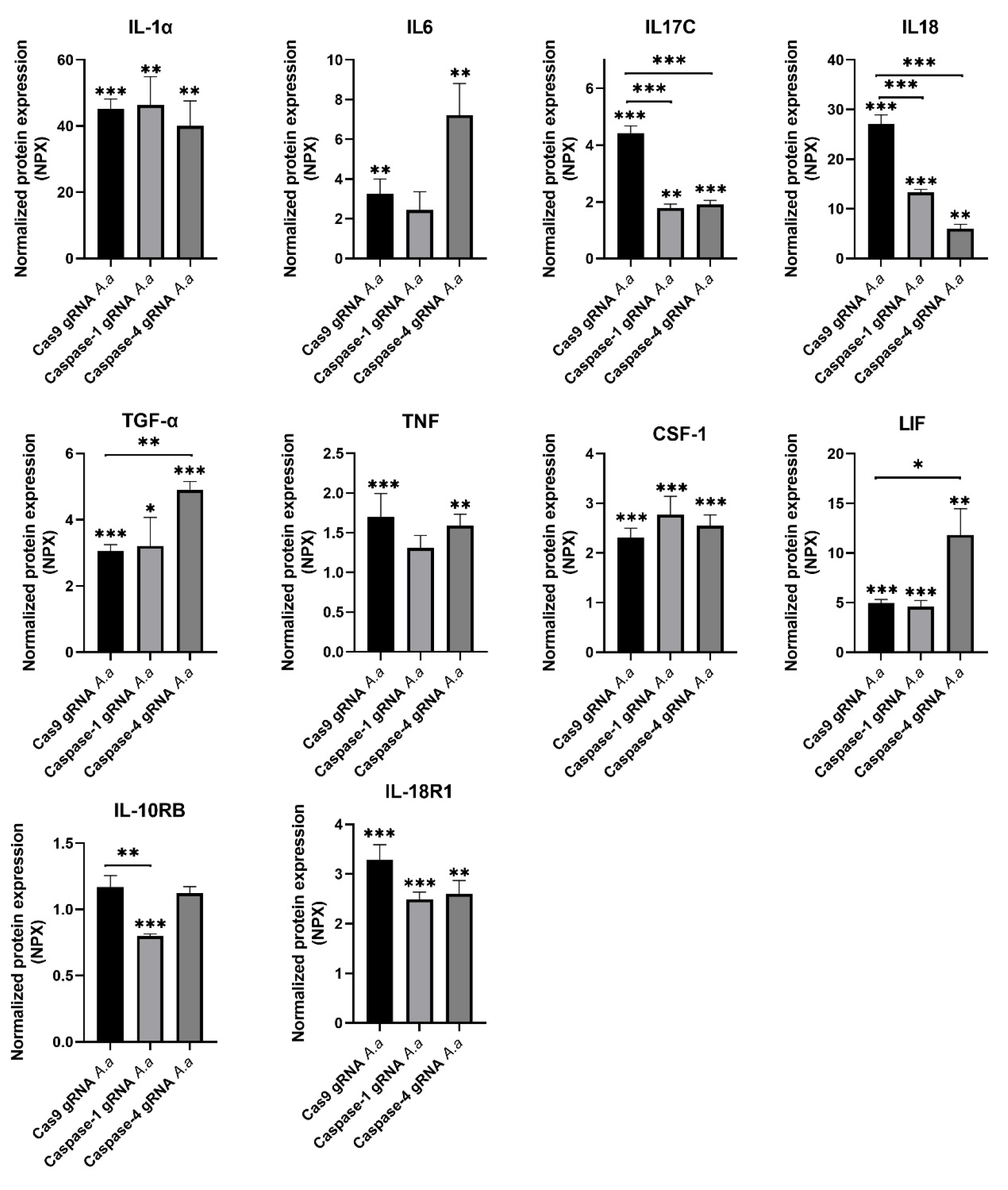
3.1. Differentially Regulated Cytokines and Cytokine Receptors in Caspase-1-and Caspase-4-Deficient Cells
3.2. Chemokine Regulation in Caspase-1-and Caspase-4-Deficient Cells
3.3. Differentially Regulated Inflammatory Mediators in Caspase-1-and Caspase-4-Deficient Cells
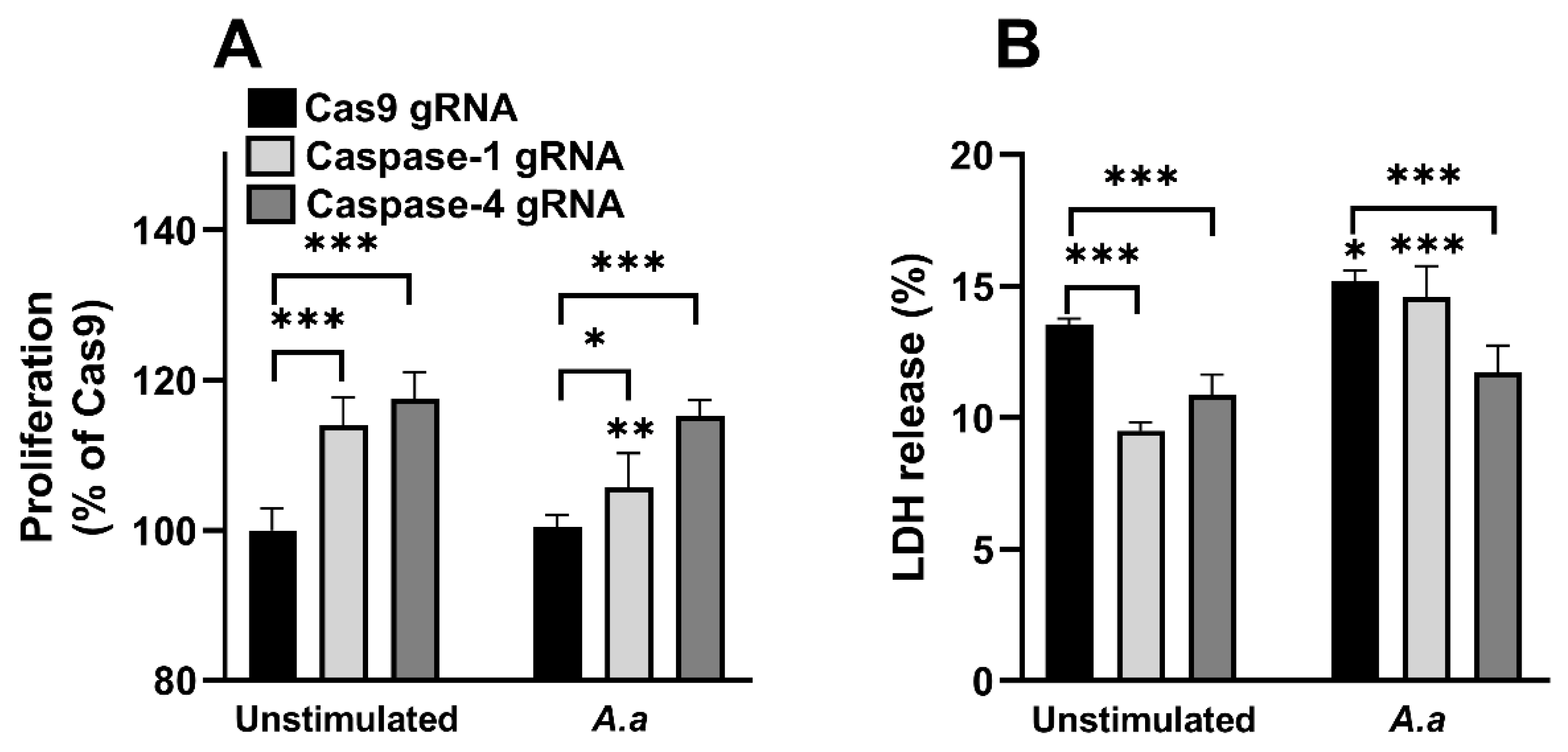
3.4. Altered Proliferation and Cell Viability in Caspase-1-and Caspase-4-Deficient Cells
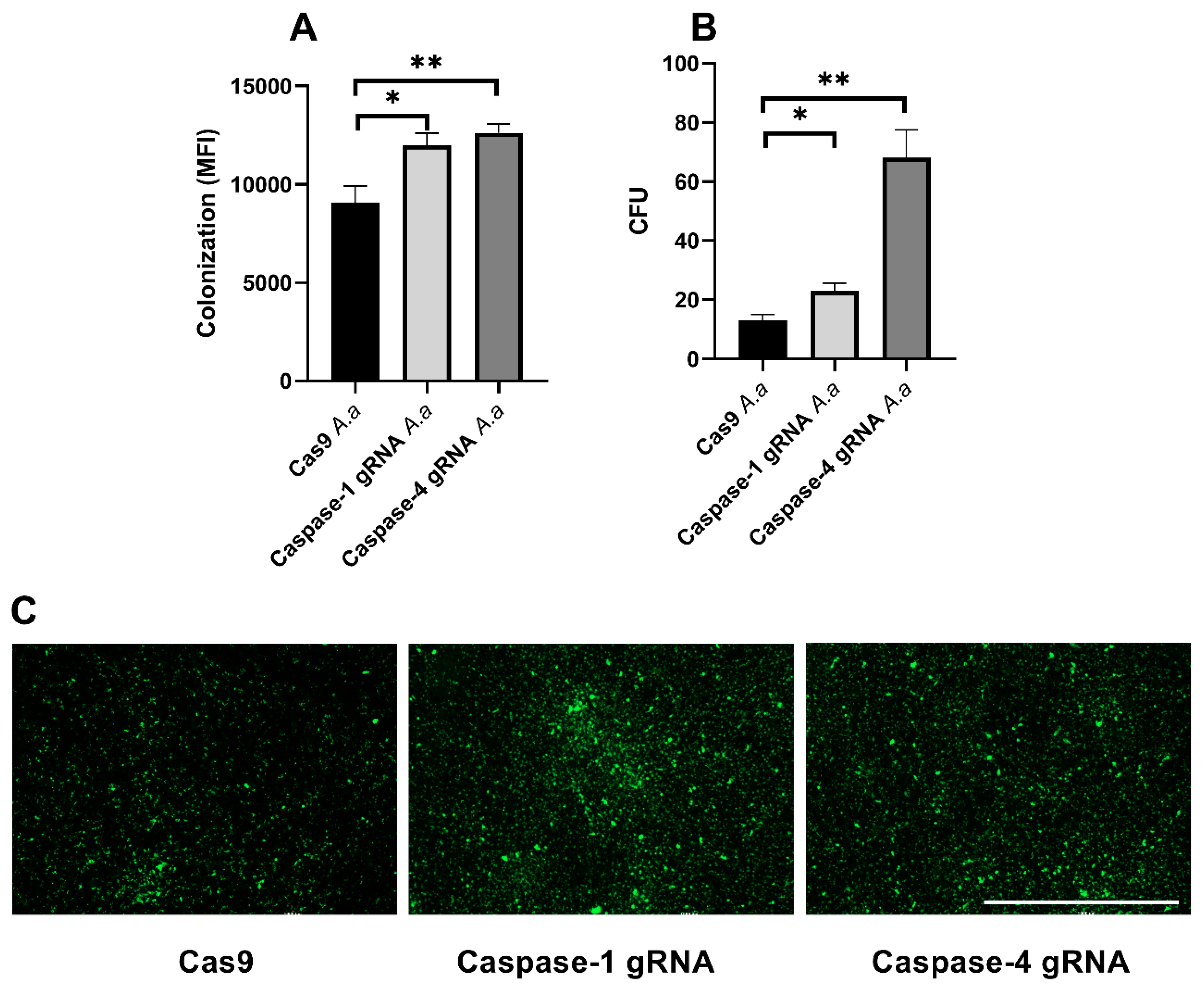
3.5. Caspase-1 and Caspase-4 Protects Gingival Epithelial Cells Against Colonization
4. Discussion
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Könönen, E.; Gursoy, M.; Gursoy, U. Periodontitis: A Multifaceted Disease of Tooth-Supporting Tissues. J. Clin. Med. 2019, 8, 1135. [Google Scholar] [CrossRef] [PubMed]
- Darveau, R.P. Periodontitis: A polymicrobial disruption of host homeostasis. Nat. Rev. Microbiol. 2010, 8, 481–490. [Google Scholar] [CrossRef] [PubMed]
- Eskan, M.A.; Hajishengallis, G.; Kinane, D.F. Differential activation of human gingival epithelial cells and monocytes by Porphyromonas gingivalis fimbriae. Infect. Immun. 2007, 75, 892–898. [Google Scholar] [CrossRef]
- McClure, R.; Massari, P. TLR-Dependent Human Mucosal Epithelial Cell Responses to Microbial Pathogens. Front. Immunol. 2014, 5, 386. [Google Scholar] [CrossRef]
- Asikainen, S.; Chen, C. Oral ecology and person-to-person transmission of Actinobacillus actinomycetemcomitans and Porphyromonas gingivalis. Periodontol. 2000 1999, 20, 65–81. [Google Scholar] [CrossRef]
- Nørskov-Lauritsen, N.; Claesson, R.; Jensen, A.B.; Åberg, C.H.; Haubek, D. Aggregatibacter actinomycetemcomitans: Clinical Significance of a Pathobiont Subjected to Ample Changes in Classification and Nomenclature. Pathogens 2019, 8, 243. [Google Scholar] [CrossRef]
- Kachlany, S.C. Aggregatibacter actinomycetemcomitans leukotoxin: From threat to therapy. J. Dent. Res. 2010, 89, 561–570. [Google Scholar] [CrossRef]
- Haubek, D. The highly leukotoxic JP2 clone of Aggregatibacter actinomycetemcomitans: Evolutionary aspects, epidemiology and etiological role in aggressive periodontitis. APMIS Suppl. 2010, 130, 1–53. [Google Scholar] [CrossRef]
- Åberg, C.H.; Kwamin, F.; Claesson, R.; Johansson, A.; Haubek, D. Presence of JP2 and Non-JP2 Genotypes of Aggregatibacter actinomycetemcomitans and attachment loss in adolescents in Ghana. J. Periodontol. 2012, 83, 1520–1528. [Google Scholar] [CrossRef]
- Demirel, K.J.; Wu, R.; Guimaraes, A.N.; Demirel, I. The role of NLRP3 in regulating gingival epithelial cell responses evoked by Aggregatibacter actinomycetemcomitans. Cytokine 2023, 169, 156316. [Google Scholar] [CrossRef]
- Kelk, P.; Claesson, R.; Chen, C.; Sjöstedt, A.; Johansson, A. IL-1beta secretion induced by Aggregatibacter (Actinobacillus) actinomycetemcomitans is mainly caused by the leukotoxin. Int. J. Med. Microbiol. 2008, 298, 529–541. [Google Scholar] [CrossRef] [PubMed]
- Belibasakis, G.N.; Johansson, A. Aggregatibacter actinomycetemcomitans targets NLRP3 and NLRP6 inflammasome expression in human mononuclear leukocytes. Cytokine 2012, 59, 124–130. [Google Scholar] [CrossRef] [PubMed]
- Sollberger, G.; Strittmatter, G.E.; Kistowska, M.; French, L.E.; Beer, H.-D. Caspase-4 Is Required for Activation of Inflammasomes. J. Immunol. 2012, 188, 1992–2000. [Google Scholar] [CrossRef] [PubMed]
- Zheng, D.; Liwinski, T.; Elinav, E. Inflammasome activation and regulation: Toward a better understanding of complex mechanisms. Cell Discov. 2020, 6, 36. [Google Scholar] [CrossRef]
- Lindblad, A.; Persson, K.; Demirel, I. IL-1RA is part of the inflammasome-regulated immune response in bladder epithelial cells and influences colonization of uropathogenic. Cytokine 2019, 123, 154772. [Google Scholar] [CrossRef]
- Swedik, S.; Madola, A.; Levine, A. IL-17C in human mucosal immunity: More than just a middle child. Cytokine 2021, 146, 155641. [Google Scholar] [CrossRef]
- Ramirez-Carrozzi, V.; Sambandam, A.; Luis, E.; Lin, Z.; Jeet, S.; Lesch, J.; Hackney, J.; Kim, J.; Zhou, M.; Lai, J.; et al. IL-17C regulates the innate immune function of epithelial cells in an autocrine manner. Nat. Immunol. 2011, 12, 1159–1166. [Google Scholar] [CrossRef]
- Cebesoy, E.I.; Altaca, M.; Kocak-Oztug, N.A.; Bingül, I.; Cifcibasi, E. Associations between interleukin-10,-12, and-18 and periodontal health and disease: A cross-sectional study. Clin. Oral Investig. 2024, 28, 458. [Google Scholar] [CrossRef]
- Raja, S.; Byakod, G.; Pudakalkatti, P. Growth factors in periodontal regeneration. Int. J. Dent. Hyg. 2009, 7, 82–89. [Google Scholar] [CrossRef]
- Mogi, M.; Otogoto, J.; Ota, N.; Inagaki, H.; Minami, M.; Kojima, K. Interleukin 1 beta, interleukin 6, beta 2-microglobulin, and transforming growth factor-alpha in gingival crevicular fluid from human periodontal disease. Arch. Oral Biol. 1999, 44, 535–539. [Google Scholar] [CrossRef]
- Liang, Y.; Zhou, Y.; Jiang, T.; Zhang, Z.; Wang, S.; Wang, Y. Expression of LIF and LIFR in periodontal tissue during orthodontic tooth movement. Angle Orthod. 2011, 81, 600–608. [Google Scholar] [CrossRef]
- Nicola, N.A.; Babon, J.J. Leukemia inhibitory factor (LIF). Cytokine Growth Factor Rev. 2015, 26, 533–544. [Google Scholar] [CrossRef] [PubMed]
- Hughes, C.E.; Nibbs, R.J.B. A guide to chemokines and their receptors. FEBS J. 2018, 285, 2944–2971. [Google Scholar] [CrossRef]
- Xuan, W.; Qu, Q.; Zheng, B.; Xiong, S.; Fan, G.-H. The chemotaxis of M1 and M2 macrophages is regulated by different chemokines. J. Leukoc. Biol. 2015, 97, 61–69. [Google Scholar] [CrossRef]
- Sokol, C.L.; Luster, A.D. The chemokine system in innate immunity. Cold Spring Harb. Perspect. Biol. 2015, 7, a016303. [Google Scholar] [CrossRef]
- Molnarfi, N.; Benkhoucha, M.; Funakoshi, H.; Nakamura, T.; Lalive, P.H. Hepatocyte growth factor: A regulator of inflammation and autoimmunity. Autoimmun. Rev. 2015, 14, 293–303. [Google Scholar] [CrossRef]
- Wu, J.; Wang, Y. Role of TNFSF9 bidirectional signal transduction in antitumor immunotherapy. Eur. J. Pharmacol. 2022, 928, 175097. [Google Scholar] [CrossRef]
- Kesavardhana, S.; Malireddi, R.S.; Kanneganti, T.D. Caspases in Cell Death, Inflammation, and Pyroptosis. Annu. Rev. Immunol. 2020, 38, 567–595. [Google Scholar] [CrossRef]
- Chen, X.; Lu, W.; Wu, D. Sirtuin 2 (SIRT2): Confusing Roles in the Pathophysiology of Neurological Disorders. Front. Neurosci. 2021, 15, 614107. [Google Scholar] [CrossRef]
- Bhagavatham, S.K.S.; Khanchandani, P.; Kannan, V.; Potikuri, D.; Sridharan, D.; Pulukool, S.K.; Naik, A.A.; Dandamudi, R.B.; Divi, S.M.; Pargaonkar, A.; et al. Adenosine deaminase modulates metabolic remodeling and orchestrates joint destruction in rheumatoid arthritis. Sci. Rep. 2021, 11, 15129. [Google Scholar] [CrossRef]
- Bednash, J.S.; Johns, F.; Patel, N.; Smail, T.R.; Londino, J.D.; Mallampalli, R.K. The deubiquitinase STAMBP modulates cytokine secretion through the NLRP3 inflammasome. Cell. Signal. 2021, 79, 109859. [Google Scholar] [CrossRef] [PubMed]
- Heymann, M.C.; Winkler, S.; Luksch, H.; Flecks, S.; Franke, M.; Ruß, S.; Özen, S.; Yilmaz, E.; Klein, C.; Kallinich, T.; et al. Human Procaspase-1 Variants with Decreased Enzymatic Activity Are Associated with Febrile Episodes and May Contribute to Inflammation via RIP2 and NF-kB Signaling. J. Immunol. 2014, 192, 4379–4385. [Google Scholar] [CrossRef] [PubMed]
- Lakshmanan, U.; Porter, A.G. Caspase-4 interacts with TNF receptor-associated factor 6 and mediates lipopolysaccharide-induced NF-κB-dependent production of IL-8 and CC chemokine ligand 4 (macrophage-inflammatory protein-1β). J. Immunol. 2007, 179, 8480–8490. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Demirel, K.J.; Neves Guimaraes, A.; Demirel, I. The Role of Caspase-1 and Caspase-4 in Modulating Gingival Epithelial Cell Responses to Aggregatibacter actinomycetemcomitans Infection. Pathogens 2025, 14, 295. https://doi.org/10.3390/pathogens14030295
Demirel KJ, Neves Guimaraes A, Demirel I. The Role of Caspase-1 and Caspase-4 in Modulating Gingival Epithelial Cell Responses to Aggregatibacter actinomycetemcomitans Infection. Pathogens. 2025; 14(3):295. https://doi.org/10.3390/pathogens14030295
Chicago/Turabian StyleDemirel, Kartheyaene Jayaprakash, Alessandra Neves Guimaraes, and Isak Demirel. 2025. "The Role of Caspase-1 and Caspase-4 in Modulating Gingival Epithelial Cell Responses to Aggregatibacter actinomycetemcomitans Infection" Pathogens 14, no. 3: 295. https://doi.org/10.3390/pathogens14030295
APA StyleDemirel, K. J., Neves Guimaraes, A., & Demirel, I. (2025). The Role of Caspase-1 and Caspase-4 in Modulating Gingival Epithelial Cell Responses to Aggregatibacter actinomycetemcomitans Infection. Pathogens, 14(3), 295. https://doi.org/10.3390/pathogens14030295