
Neuropathogenesis of Encephalitic Alphaviruses in Non-Human Primate and Mouse Models of Infection



Abstract
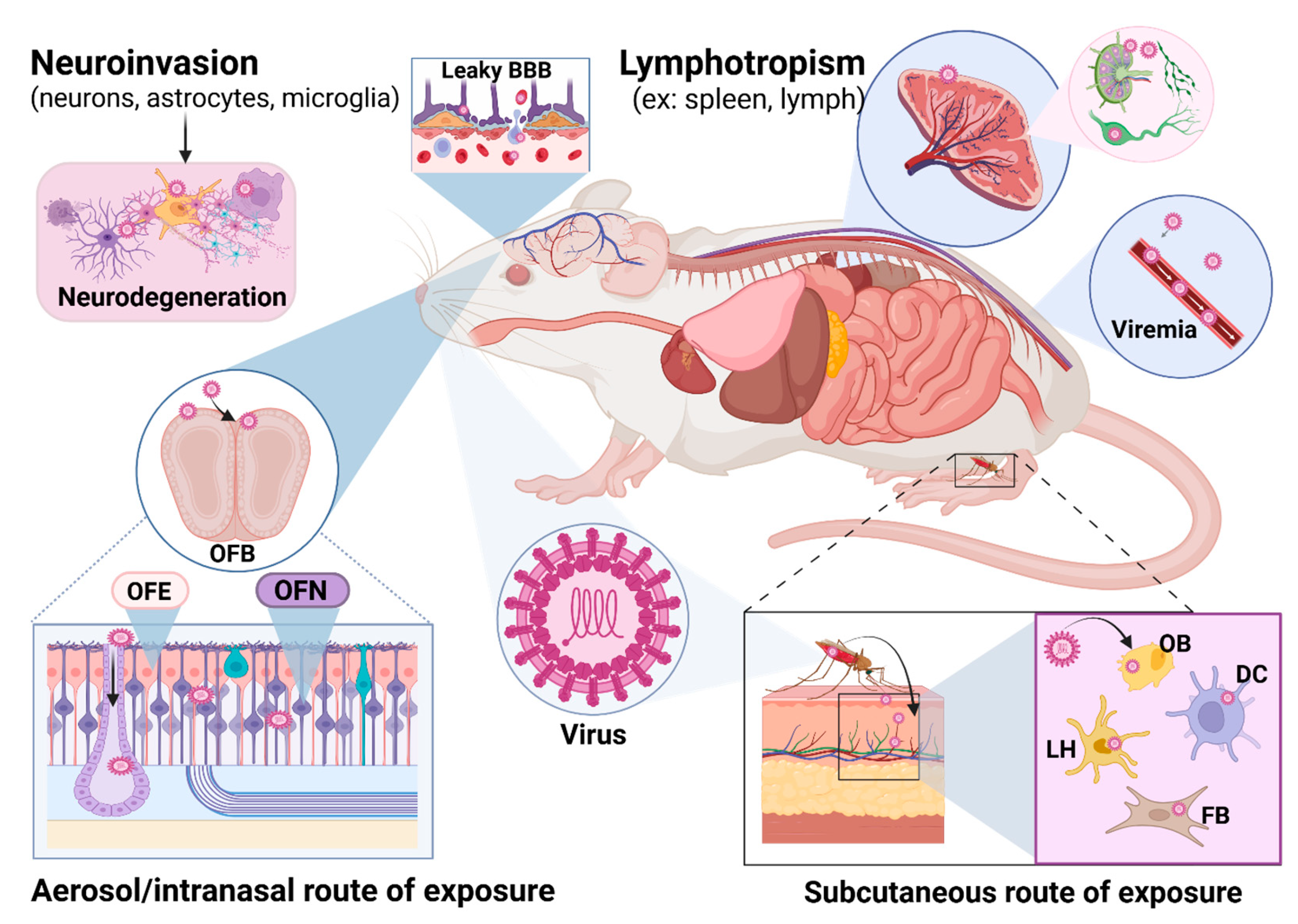
:1. Introduction

{kind=link}
{kind=link}
{kind=link}
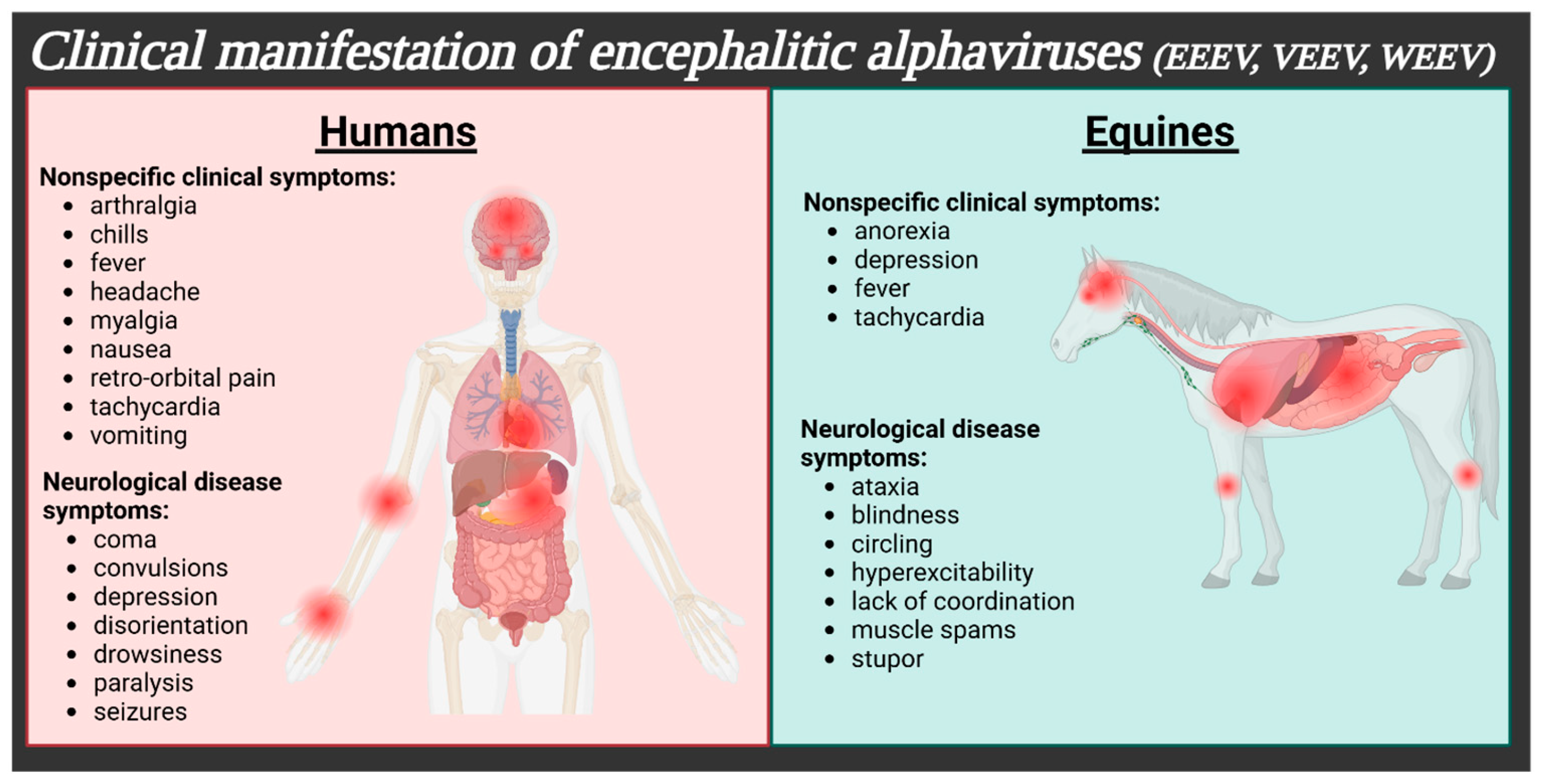
| Virus | Clinical Symptoms | Neurological Symptoms | % Mortality | % Survivors w/Sequelae | Neurological Sequelae | Refs. |
|---|---|---|---|---|---|---|
| EEEV | fever, headache, malaise, myalgia, nausea, and/or vomiting | behavioral changes, coma, and/or drowsiness | 30–75% | 50–90% | behavioral changes, convulsions, intellectual disability, paralysis, and/or seizures | [1,4,7,8] |
| VEEV | arthralgia, chills, fever, headache, malaise, myalgia, photophobia, retro-orbital pain, vomiting, and/or diarrhea | ataxia, depression, disorientation, drowsiness, coma, confusion, convulsions, paralysis, and/or seizures | <1% (up to 10% of adult neurological cases, 35% children) | 4–14% | altered personality, coma, confusion, convulsions, depression, emotional instability, photophobia, intellectual disability, paralysis, epilepsy, recurrent headaches, seizures, and/or somnolence | [1,4,7] |
| WEEV | fever, headache, malaise, myalgia, nausea, and/or vomiting | agitation, coma, confusion, drowsiness, light sensitivity, and/or seizures | 3–15% | 15–30% | coma, confusion, visual disturbances, emotional instability/behavioral changes, intellectual disability, photophobia, seizures, somnolence, taste distortion, spastic paresis, and/or respiratory distress | [1,4,7] |
2. Overview of Neuroinvasion and Neurovirulence
3. Eastern Equine Encephalitis Virus (EEEV)
3.1. Geography, Epidemiology, Clinical Disease
3.2. Animal Models, CNS Disease, and Neuropathology
4. Venezuelan Equine Encephalitis Virus (VEEV)
4.1. Geography, Epidemiology, Clinical Disease
4.2. Animal Models, CNS Disease, and Neuropathology
| Animal Model | Viral Strain | Infection Route | % Mortality | Clinical Observations | Neuropathological Features | Refs. | |
|---|---|---|---|---|---|---|---|
| Species | Strain | ||||||
| NHP | Cynomolgus macaques | TrD, INH-9813 | AE * | 0% | Viremia, fever dehydration, lymphopenia, lethargy, dehydration, Ataxia, photophobia, nystagmus, tremors, hyperactivity, twitching | Vasculitis, leukocyte infiltration, lyphoplasmatic meningoencephalitis, gliosis | [72,74,80] |
| NHP | Cynomolgus macaques | INH-9813 | SC ** | 0% | Fever, viremia | None | [4] |
| Mice | C3H/HeN, | TC83 | IN *** | 80% [76], 60% [81] | Weight loss, ruffled fur, viremia, hunched, lethargic | Perivascular cuffing, meningitis, encephalitis, gliosis | [76,81] |
| Mice | C57BL/6 | TC83 | IN | 10–20% | Weight loss, ruffled fur, viremia | Glia cell activation, prepulse inhibition | [20,77,78,79] |
| Mice | C57BL/6, BALB/c, NIH Swiss | TrD, ZPC-738 | IN | 100% | Weight loss, viremia, ruffled fur, hunched posture, ataxia, altered gate, paralysis | Mononuclear cell infilaration, meningitis, encephalitis, microglia activation, neuron cell death | [82,83,84] |
| Mice | CD-1 | V3000, INH9813 | AE | 100% with ≥10–100 PFU | Weight loss, ruffled fur, hunched, ataxia, serizures, | Not described | [52,85] |
| Mice | C57BL/6, CD-1, BALB/c | TrD, V3000, ZPC-738, INH9813 | SC | 100% | Weight loss, viremia, ruffled fur, hunched, lethargic, paralysis | Neuronal death, spongiosis, and gliosis | [52,54,86,87] |
| Host Protein | Function | Impact on VEEV Pathogenesis | Ref. |
|---|---|---|---|
| iNOS | Enzyme that produces nitric oxide (NO), which can be a neurotoxin | Increased survival time in iNOS KO mice | [103] |
| TNFα | Proinflammatory cytokine | Increased survival time in TNFα KO mice | [103] |
| Cav-1 | Main component of caveolae, which are specialized structures in the plasma membrane that facilitate transport | Decreased neuroinvasion in Cav-1 KO mice | [88] |
| TLR4 | Pattern recognition receptor | Decreased BBB permeability in TLR4 KO mice | [92] |
| LDLRAD3 | Entry receptor for VEEV | Decreased viral replication and 100% survival in LDLRAD3-KO mice | [86] |
| ICAM-1 | Adhesion molecule involved in leukocyte migration at the BBB | Decreased neuroinflammation (perivascular cuffing and neuronal necrosis) in ICAM-1 KO mice | [104] |
| IFNα/β | Antiviral cytokine | Increased mortality in IFNAR KO mice | [97] |
5. Western Equine Encephalitis Virus (WEEV)
5.1. Basic Background (Geography, Epidemiology, Clinical Disease)
5.2. Animal Models, CNS Disease, and Neuropathology
| Animal Model | Viral Strain | Infection Route | % Mortality | Clinical Observations | Neuropathological Features | Refs. | |
|---|---|---|---|---|---|---|---|
| Species | Strain | ||||||
| NHP | Cynomolgus macaques | CBA-87, Fleming | AE * | 9–100% | Fever, antisocial behavior, lethargy, reduced appetite, tremors | Encephalitis, occipital meningitis, focal hemorrhage in the frontal lobe and brainstem, perivascular cuffing and infiltrating neutrophils. Antigen in neurons and microglia of cortex. | [80,114,115] |
| NHP | Cynomolgus macaques | Fleming | SC ** | 0% | Minor weight loss | No lesions or viral antigen in the brain. | [4] |
| Mice | BALB/c | Fleming, McMillian (McM), Imperial 181 (IMP) | AE | 0–100% | Weight loss, ruffled fur, hunched posture, lethargy, closed eyes, labored breathing, mobility issues. Hyperreactivity, circling seizures, tremors, spinning, fixed gaze, obsessive grooming, and/or twitching | Encephalitis, focal leptomeningitis in the cerebrum, perivascular cuffing, infiltrating neutrophils, gliosis, neuronal death. Antigen in neurons and neuronal processes of substantia nigra, CVOs, glial cells, and astrocytes. | [108,118] |
| Mice | CD-1, BALB/c, C57BL/6 and astrocyte specific NFκB KO mice | McM, McM-fLuc, CBA-87, Mn548, B11, Mn520 | IN *** | 100% | Ruffled fur, hunched posture, lethargy, labored breathing. Depression, motor deficits, ataxia, unresponsive to visual stimuli during late stage of infection, seizures | Necrosis and secondary demyelination in olfactory bulb, hypothalamus, midbrain, hippocampus; gliosis, infiltrating neutrophils, perivascular cuffing. Antigen in neurons of hippocampus and nigrostriatal pathway. Loss of neurons in hippocampus. | [119,120,121] |
| Mice | CD-1, C57BL/6 BALB/c, A/J, DBA/2, BALB/cBy | McM, IMP, CBA-87 | SC | 20–100% (McM, CBA-87), 0% (IMP) | Seizures, viremia | Meningoencephalitis in the hypothalamus, pineal gland, and the area postrema; perivascular cuffing and infiltrating neutrophils; necrosis in hippocampus, gliosis in frontal and cerebral cortex. | [21,116,117,122] |
| Host Protein | Function | Impact on VEEV Pathogenesis | Ref. |
|---|---|---|---|
| Cav-1 | Main component of caveolae, which are specialized structures in the plasma membrane that facilitate transport | Decreased neuroinvasion in Cav-1 KO mice | [88] |
| PCDH10 | Cell adhesion molecule and entry receptor for WEEV | Treatment with PCDH10 decoy (PCDH10EC1–Fc) protects mice from WEEV lethal challenge | [17] |
| VLDLR | Entry receptor for WEEV | Increased survival in VLDLR KO mice | [58] |
| NFκB | Transcription factor that regulates inflammation and apoptotic signaling | Decreased α-synuclein aggregation, neuronal loss, and gliosis in astrocyte-specific NFκB KO mice | [119] |
6. Discussion and Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ronca, S.E.; Dineley, K.T.; Paessler, S. Neurological Sequelae Resulting from Encephalitic Alphavirus Infection. Front. Microbiol. 2016, 7, 959. [Google Scholar] [CrossRef]
- Powers, A.M. Resurgence of Interest in Eastern Equine Encephalitis Virus Vaccine Development. J. Med. Entomol. 2022, 59, 20–26. [Google Scholar] [CrossRef]
- Kim, A.S.; Diamond, M.S. A molecular understanding of alphavirus entry and antibody protection. Nat. Rev. Microbiol. 2023, 21, 396–407. [Google Scholar] [CrossRef] [PubMed]
- Smith, D.R.; Schmaljohn, C.S.; Badger, C.; Ostrowski, K.; Zeng, X.; Grimes, S.D.; Rayner, J.O. Comparative pathology study of Venezuelan, eastern, and western equine encephalitis viruses in non-human primates. Antivir. Res. 2020, 182, 104875. [Google Scholar] [CrossRef]
- Steele, K.E.; Twenhafel, N.A. REVIEW PAPER: Pathology of animal models of alphavirus encephalitis. Vet. Pathol. 2010, 47, 790–805. [Google Scholar] [CrossRef] [PubMed]
- Select Agents and Toxins List. CDC. Available online: https://www.selectagents.gov/sat/list.htm (accessed on 28 December 2024).
- Reyna, R.A.; Weaver, S.C. Sequelae and Animal Modeling of Encephalitic Alphavirus Infections. Viruses 2023, 15, 382. [Google Scholar] [CrossRef] [PubMed]
- Ladzinski, A.T.; Tai, A.; Rumschlag, M.T.; Smith, C.S.; Mehta, A.; Boapimp, P.; Edewaard, E.J.; Douce, R.W.; Morgan, L.F.; Wang, M.S.; et al. Clinical Characteristics of the 2019 Eastern Equine Encephalitis Outbreak in Michigan. Open Forum Infect. Dis. 2023, 10, ofad206. [Google Scholar] [CrossRef] [PubMed]
- U.S. Food and Drug Administration. Animal Rule Information. 2024. Available online: https://www.fda.gov/emergency-preparedness-and-response/preparedness-research/animal-rule-information (accessed on 28 December 2024).
- Rusnak, J.M.; Glass, P.J.; Weaver, S.C.; Sabourin, C.L.; Glenn, A.M.; Klimstra, W.; Badorrek, C.S.; Nasar, F.; Ward, L.A. Approach to Strain Selection and the Propagation of Viral Stocks for Venezuelan Equine Encephalitis Virus Vaccine Efficacy Testing under the Animal Rule. Viruses 2019, 11, 807. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R. Understanding and managing acute encephalitis. F1000Res 2020, 9, 60. [Google Scholar] [CrossRef]
- Kafai, N.M.; Diamond, M.S.; Fox, J.M. Distinct Cellular Tropism and Immune Responses to Alphavirus Infection. Annu. Rev. Immunol. 2022, 40, 615–649. [Google Scholar] [CrossRef] [PubMed]
- Cao, D.; Ma, B.; Cao, Z.; Xu, X.; Zhang, X.; Xiang, Y. The receptor VLDLR binds Eastern Equine Encephalitis virus through multiple distinct modes. Nat. Commun. 2024, 15, 6866. [Google Scholar] [CrossRef] [PubMed]
- Yang, P.; Li, W.; Fan, X.; Pan, J.; Mann, C.J.; Varnum, H.; Clark, L.E.; Clark, S.A.; Coscia, A.; Basu, H.; et al. Structural basis for VLDLR recognition by eastern equine encephalitis virus. Nat. Commun. 2024, 15, 6548. [Google Scholar] [CrossRef] [PubMed]
- Adams, L.J.; Raju, S.; Ma, H.; Gilliland, T., Jr.; Reed, D.S.; Klimstra, W.B.; Fremont, D.H.; Diamond, M.S. Structural and functional basis of VLDLR usage by Eastern equine encephalitis virus. Cell 2024, 187, 360–374.e19. [Google Scholar] [CrossRef]
- Ma, H.; Kim, A.S.; Kafai, N.M.; Earnest, J.T.; Shah, A.P.; Case, J.B.; Basore, K.; Gilliland, T.C.; Sun, C.; Nelson, C.A.; et al. LDLRAD3 is a receptor for Venezuelan equine encephalitis virus. Nature 2020, 588, 308–314. [Google Scholar] [CrossRef]
- Li, W.; Plante, J.A.; Lin, C.; Basu, H.; Plung, J.S.; Fan, X.; Boeckers, J.M.; Oros, J.; Buck, T.K.; Anekal, P.V.; et al. Shifts in receptors during submergence of an encephalitic arbovirus. Nature 2024, 632, 614–621. [Google Scholar] [CrossRef]
- Ma, H.; Adams, L.J.; Raju, S.; Sariol, A.; Kafai, N.M.; Janova, H.; Klimstra, W.B.; Fremont, D.H.; Diamond, M.S. The low-density lipoprotein receptor promotes infection of multiple encephalitic alphaviruses. Nat. Commun. 2024, 15, 246. [Google Scholar] [CrossRef]
- Klein, R.S.; Garber, C.; Funk, K.E.; Salimi, H.; Soung, A.; Kanmogne, M.; Manivasagam, S.; Agner, S.; Cain, M. Neuroinflammation During RNA Viral Infections. Annu. Rev. Immunol. 2019, 37, 73–95. [Google Scholar] [CrossRef]
- Cain, M.D.; Salimi, H.; Gong, Y.; Yang, L.; Hamilton, S.L.; Heffernan, J.R.; Hou, J.; Miller, M.J.; Klein, R.S. Virus entry and replication in the brain precedes blood-brain barrier disruption during intranasal alphavirus infection. J. Neuroimmunol. 2017, 308, 118–130. [Google Scholar] [CrossRef]
- Phillips, A.T.; Rico, A.B.; Stauft, C.B.; Hammond, S.L.; Aboellail, T.A.; Tjalkens, R.B.; Olson, K.E. Entry Sites of Venezuelan and Western Equine Encephalitis Viruses in the Mouse Central Nervous System following Peripheral Infection. J. Virol. 2016, 90, 5785–5796. [Google Scholar] [CrossRef]
- Sharma, A.; Knollmann-Ritschel, B. Current Understanding of the Molecular Basis of Venezuelan Equine Encephalitis Virus Pathogenesis and Vaccine Development. Viruses 2019, 11, 164. [Google Scholar] [CrossRef]
- Gardner, C.L.; Burke, C.W.; Tesfay, M.Z.; Glass, P.J.; Klimstra, W.B.; Ryman, K.D. Eastern and Venezuelan equine encephalitis viruses differ in their ability to infect dendritic cells and macrophages: Impact of altered cell tropism on pathogenesis. J. Virol. 2008, 82, 10634–10646. [Google Scholar] [CrossRef]
- Griffin, D.E.; Levine, B.; Tyor, W.R.; Tucker, P.C.; Hardwick, J.M. Age-dependent susceptibility to fatal encephalitis: Alphavirus infection of neurons. Arch. Virol. Suppl. 1994, 9, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Kraft, A.D.; Harry, G.J. Features of microglia and neuroinflammation relevant to environmental exposure and neurotoxicity. Int. J. Environ. Res. Public Health 2011, 8, 2980–3018. [Google Scholar] [CrossRef] [PubMed]
- Gao, C.; Jiang, J.; Tan, Y.; Chen, S. Microglia in neurodegenerative diseases: Mechanism and potential therapeutic targets. Signal Transduct. Target. Ther. 2023, 8, 359. [Google Scholar] [CrossRef] [PubMed]
- Hatton, C.F.; Duncan, C.J.A. Microglia Are Essential to Protective Antiviral Immunity: Lessons From Mouse Models of Viral Encephalitis. Front. Immunol. 2019, 10, 2656. [Google Scholar] [CrossRef] [PubMed]
- Puentes-Orozco, M.; Albarracin, S.L.; Velasquez, M.M. Neuroinflammation and major depressive disorder: Astrocytes at the crossroads. Front. Cell Neurosci. 2024, 18, 1504555. [Google Scholar] [CrossRef]
- Grist, J.J.; Marro, B.; Lane, T.E. Neutrophils and viral-induced neurologic disease. Clin. Immunol. 2018, 189, 52–56. [Google Scholar] [CrossRef]
- Ludlow, M.; Kortekaas, J.; Herden, C.; Hoffmann, B.; Tappe, D.; Trebst, C.; Griffin, D.E.; Brindle, H.E.; Solomon, T.; Brown, A.S.; et al. Neurotropic virus infections as the cause of immediate and delayed neuropathology. Acta Neuropathol. 2016, 131, 159–184. [Google Scholar] [CrossRef]
- Stobierski, M.G.; Signs, K.; Dinh, E.; Cooley, T.M.; Melotti, J.; Schalow, M.; Patterson, J.S.; Bolin, S.R.; Walker, E.D. Eastern Equine Encephalomyelitis in Michigan: Historical Review of Equine, Human, and Wildlife Involvement, Epidemiology, Vector Associations, and Factors Contributing to Endemicity. J. Med. Entomol. 2022, 59, 27–40. [Google Scholar] [CrossRef]
- Arechiga-Ceballos, N.; Aguilar-Setien, A. Alphaviral equine encephalomyelitis (Eastern, Western and Venezuelan). Rev. Sci. Tech. 2015, 34, 491–501. [Google Scholar] [CrossRef]
- Case Definition for Eastern Equine Encephalitis; United States Department of Agriculture: Washington, DC, USA, 2011.
- Lindsey, N.P.; Staples, J.E.; Fischer, M. Eastern Equine Encephalitis Virus in the United States, 2003–2016. Am. J. Trop. Med. Hyg. 2018, 98, 1472–1477. [Google Scholar] [CrossRef] [PubMed]
- Brown, S.C.; Cormier, J.; Tuan, J.; Lier, A.J.; McGuone, D.; Armstrong, P.M.; Kaddouh, F.; Parikh, S.; Landry, M.L.; Gobeske, K.T. Four Human Cases of Eastern Equine Encephalitis in Connecticut, USA, during a Larger Regional Outbreak, 2019. Emerg. Infect. Dis. 2021, 27, 2042–2051. [Google Scholar] [CrossRef] [PubMed]
- Schooley, M.; Sullivan, M.; Moller, L. Mosquito-Borne Virus EEE Kills New Hampshire Man. 2024. Available online: https://www.cbsnews.com/boston/news/hampstead-new-hampshire-eee-death/ (accessed on 28 December 2024).
- Binnicker, M. New Hampshire Man Dies from EEE. What You Need to Know About This Mosquito-Borne Infection. 2024. Available online: https://www.forbes.com/sites/matthewbinnicker/2024/08/27/what-is-eee-the-potentially-deadly-virus-in-the-northeastern-us/ (accessed on 28 December 2024).
- Vogel, P.; Kell, W.M.; Fritz, D.L.; Parker, M.D.; Schoepp, R.J. Early events in the pathogenesis of eastern equine encephalitis virus in mice. Am. J. Pathol. 2005, 166, 159–171. [Google Scholar] [CrossRef]
- Morens, D.M.; Folkers, G.K.; Fauci, A.S. Eastern Equine Encephalitis Virus—Another Emergent Arbovirus in the United States. N. Engl. J. Med. 2019, 381, 1989–1992. [Google Scholar] [CrossRef] [PubMed]
- Zubair, A.S.; McAlpine, L.S.; Gobeske, K.T. Virology, ecology, epidemiology, pathology, and treatment of eastern equine encephalitis. J. Neurol. Sci. 2024, 457, 122886. [Google Scholar] [CrossRef] [PubMed]
- Wilcox, D.R.; Collens, S.I.; Solomon, I.H.; Mateen, F.J.; Mukerji, S.S. Eastern equine encephalitis and use of IV immunoglobulin therapy and high-dose steroids. Neurol. Neuroimmunol. Neuroinflamm 2021, 8, e917. [Google Scholar] [CrossRef]
- Adams, A.P.; Aronson, J.F.; Tardif, S.D.; Patterson, J.L.; Brasky, K.M.; Geiger, R.; de la Garza, M.; Carrion, R., Jr.; Weaver, S.C. Common marmosets (Callithrix jacchus) as a nonhuman primate model to assess the virulence of eastern equine encephalitis virus strains. J. Virol. 2008, 82, 9035–9042. [Google Scholar] [CrossRef]
- Dexter, E.P.; Dexter, D.D.; Lindsay, C.W.; Ross, R.R.; Lutwick, L. Case of fatal eastern equine encephalitis. IDCases 2021, 26, e01288. [Google Scholar] [CrossRef]
- Deresiewicz, R.L.; Thaler, S.J.; Hsu, L.; Zamani, A.A. Clinical and neuroradiographic manifestations of eastern equine encephalitis. N. Engl. J. Med. 1997, 336, 1867–1874. [Google Scholar] [CrossRef]
- Feemster, R.F. Outbreak of Encephalitis in Man Due to the Eastern Virus of Equine Encephalomyelitis. Am. J. Public Health Nations Health 1938, 28, 1403–1410. [Google Scholar] [CrossRef]
- Del Piero, F.; Wilkins, P.A.; Dubovi, E.J.; Biolatti, B.; Cantile, C. Clinical, pathologic, immunohistochemical, and virologic findings of eastern equine encephalomyelitis in two horses. Vet. Pathol. 2001, 38, 451–456. [Google Scholar] [CrossRef]
- Williams, J.A.; Long, S.Y.; Zeng, X.; Kuehl, K.; Babka, A.M.; Davis, N.M.; Liu, J.; Trefry, J.C.; Daye, S.; Facemire, P.R.; et al. Eastern equine encephalitis virus rapidly infects and disseminates in the brain and spinal cord of cynomolgus macaques following aerosol challenge. PLoS Negl. Trop. Dis. 2022, 16, e0010081. [Google Scholar] [CrossRef] [PubMed]
- Porter, A.I.; Erwin-Cohen, R.A.; Twenhafel, N.; Chance, T.; Yee, S.B.; Kern, S.J.; Norwood, D.; Hartman, L.J.; Parker, M.D.; Glass, P.J.; et al. Characterization and pathogenesis of aerosolized eastern equine encephalitis in the common marmoset (Callithrix jacchus). Virol. J. 2017, 14, 25. [Google Scholar] [CrossRef] [PubMed]
- Albe, J.R.; Ma, H.; Gilliland, T.H.; McMillen, C.M.; Gardner, C.L.; Boyles, D.A.; Cottle, E.L.; Dunn, M.D.; Lundy, J.D.; O’Malley, K.J.; et al. Physiological and immunological changes in the brain associated with lethal eastern equine encephalitis virus in macaques. PLoS Pathog. 2021, 17, e1009308. [Google Scholar] [CrossRef] [PubMed]
- Honnold, S.P.; Mossel, E.C.; Bakken, R.R.; Fisher, D.; Lind, C.M.; Cohen, J.W.; Eccleston, L.T.; Spurgers, K.B.; Erwin-Cohen, R.; Bradfute, S.B.; et al. Eastern equine encephalitis virus in mice I: Clinical course and outcome are dependent on route of exposure. Virol. J. 2015, 12, 152. [Google Scholar] [CrossRef]
- Honnold, S.P.; Mossel, E.C.; Bakken, R.R.; Lind, C.M.; Cohen, J.W.; Eccleston, L.T.; Spurgers, K.B.; Erwin-Cohen, R.; Glass, P.J.; Maheshwari, R.K. Eastern equine encephalitis virus in mice II: Pathogenesis is dependent on route of exposure. Virol. J. 2015, 12, 154. [Google Scholar] [CrossRef]
- Gardner, C.L.; Sun, C.; Dunn, M.D.; Gilliland, T.C., Jr.; Trobaugh, D.W.; Terada, Y.; Reed, D.S.; Hartman, A.L.; Klimstra, W.B. In Vitro and In Vivo Phenotypes of Venezuelan, Eastern and Western Equine Encephalitis Viruses Derived from cDNA Clones of Human Isolates. Viruses 2022, 15, 5. [Google Scholar] [CrossRef]
- Phelps, A.L.; O’Brien, L.M.; Eastaugh, L.S.; Davies, C.; Lever, M.S.; Ennis, J.; Zeitlin, L.; Nunez, A.; Ulaeto, D.O. Aerosol infection of Balb/c mice with eastern equine encephalitis virus; susceptibility and lethality. Virol. J. 2019, 16, 2. [Google Scholar] [CrossRef]
- Phelps, A.L.; Salguero, F.J.; Hunter, L.; Stoll, A.L.; Jenner, D.C.; O’Brien, L.M.; Williamson, E.D.; Lever, M.S.; Laws, T.R. Tumour Necrosis Factor-alpha, Chemokines, and Leukocyte Infiltrate Are Biomarkers for Pathology in the Brains of Venezuelan Equine Encephalitis (VEEV)-Infected Mice. Viruses 2023, 15, 1307. [Google Scholar] [CrossRef]
- Elmore, S. Apoptosis: A review of programmed cell death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef]
- Chen, T.; Dai, Y.; Hu, C.; Lin, Z.; Wang, S.; Yang, J.; Zeng, L.; Li, S.; Li, W. Cellular and molecular mechanisms of the blood-brain barrier dysfunction in neurodegenerative diseases. Fluids Barriers CNS 2024, 21, 60. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Li, M.; Yang, L.; Qin, W.; Yang, S.; Yuan, J.; Jiang, T.; Hu, W. The relationship between blood-brain barrier permeability and enlarged perivascular spaces: A cross-sectional study. Clin. Interv. Aging 2019, 14, 871–878. [Google Scholar] [CrossRef] [PubMed]
- Palakurty, S.; Raju, S.; Sariol, A.; Chong, Z.; Wagoner, N.; Ma, H.; Zimmerman, O.; Adams, L.J.; Carmona, C.; Liu, Z.; et al. The VLDLR entry receptor is required for the pathogenesis of multiple encephalitic alphaviruses. Cell Rep. 2024, 43, 114809. [Google Scholar] [CrossRef] [PubMed]
- Deuker, L.; Doeller, C.F.; Fell, J.; Axmacher, N. Human neuroimaging studies on the hippocampal CA3 region—Integrating evidence for pattern separation and completion. Front. Cell Neurosci. 2014, 8, 64. [Google Scholar] [CrossRef]
- Angenstein, F. A Combined fMRI and Electrophysiological Approach to Study Signal Processing and Signal Propagation in the Rodent Hippocampus. In Handbook of Behavioral Neuroscience; Manahan-Vaughan, D., Ed.; Elsevier: Amsterdam, The Netherlands, 2018; Volume 28, pp. 425–439. [Google Scholar]
- Trobaugh, D.W.; Sun, C.; Bhalla, N.; Gardner, C.L.; Dunn, M.D.; Klimstra, W.B. Cooperativity between the 3′ untranslated region microRNA binding sites is critical for the virulence of eastern equine encephalitis virus. PLoS Pathog. 2019, 15, e1007867. [Google Scholar] [CrossRef]
- Roy, C.J.; Reed, D.S.; Wilhelmsen, C.L.; Hartings, J.; Norris, S.; Steele, K.E. Pathogenesis of aerosolized Eastern Equine Encephalitis virus infection in guinea pigs. Virol. J. 2009, 6, 170. [Google Scholar] [CrossRef]
- Banner, L.R.; Moayeri, N.N.; Patterson, P.H. Leukemia inhibitory factor is expressed in astrocytes following cortical brain injury. Exp. Neurol. 1997, 147, 1–9. [Google Scholar] [CrossRef]
- Grossetete, M.; Phelps, J.; Arko, L.; Yonas, H.; Rosenberg, G.A. Elevation of matrix metalloproteinases 3 and 9 in cerebrospinal fluid and blood in patients with severe traumatic brain injury. Neurosurgery 2009, 65, 702–708. [Google Scholar] [CrossRef]
- Pan, W.; Yu, C.; Hsuchou, H.; Zhang, Y.; Kastin, A.J. Neuroinflammation facilitates LIF entry into brain: Role of TNF. Am. J. Physiol. Cell Physiol. 2008, 294, C1436–C1442. [Google Scholar] [CrossRef]
- Zheng, K.; Li, C.; Shan, X.; Liu, H.; Fan, W.; Wang, Z.; Zheng, P. Matrix metalloproteinases and their tissue inhibitors in serum and cerebrospinal fluid of patients with moderate and severe traumatic brain injury. Neurol. India 2013, 61, 606–609. [Google Scholar] [CrossRef]
- Zacks, M.A.; Paessler, S. Encephalitic alphaviruses. Vet. Microbiol. 2010, 140, 281–286. [Google Scholar] [CrossRef] [PubMed]
- Azar, S.R.; Campos, R.K.; Bergren, N.A.; Camargos, V.N.; Rossi, S.L. Epidemic Alphaviruses: Ecology, Emergence and Outbreaks. Microorganisms 2020, 8, 1167. [Google Scholar] [CrossRef] [PubMed]
- Guzman-Teran, C.; Calderon-Rangel, A.; Rodriguez-Morales, A.; Mattar, S. Venezuelan equine encephalitis virus: The problem is not over for tropical America. Ann. Clin. Microbiol. Antimicrob. 2020, 19, 19. [Google Scholar] [CrossRef] [PubMed]
- Aguilar, P.V.; Estrada-Franco, J.G.; Navarro-Lopez, R.; Ferro, C.; Haddow, A.D.; Weaver, S.C. Endemic Venezuelan equine encephalitis in the Americas: Hidden under the dengue umbrella. Future Virol. 2011, 6, 721–740. [Google Scholar] [CrossRef]
- Carrera, J.P.; Pitti, Y.; Molares-Martinez, J.C.; Casal, E.; Pereyra-Elias, R.; Saenz, L.; Guerrero, I.; Galue, J.; Rodriguez-Alvarez, F.; Jackman, C.; et al. Clinical and Serological Findings of Madariaga and Venezuelan Equine Encephalitis Viral Infections: A Follow-up Study 5 Years After an Outbreak in Panama. Open Forum Infect. Dis. 2020, 7, ofaa359. [Google Scholar] [CrossRef]
- Burke, C.W.; Froude, J.W.; Rossi, F.; White, C.E.; Moyer, C.L.; Ennis, J.; Pitt, M.L.; Streatfield, S.; Jones, R.M.; Musiychuk, K.; et al. Therapeutic monoclonal antibody treatment protects nonhuman primates from severe Venezuelan equine encephalitis virus disease after aerosol exposure. PLoS Pathog. 2019, 15, e1008157. [Google Scholar] [CrossRef]
- Rusnak, J.M.; Dupuy, L.C.; Niemuth, N.A.; Glenn, A.M.; Ward, L.A. Comparison of Aerosol- and Percutaneous-acquired Venezuelan Equine Encephalitis in Humans and Nonhuman Primates for Suitability in Predicting Clinical Efficacy under the Animal Rule. Comp. Med. 2018, 68, 380–395. [Google Scholar] [CrossRef]
- Ma, H.; Albe, J.R.; Gilliland, T.; McMillen, C.M.; Gardner, C.L.; Boyles, D.A.; Cottle, E.L.; Dunn, M.D.; Lundy, J.D.; Salama, N.; et al. Long-term persistence of viral RNA and inflammation in the CNS of macaques exposed to aerosolized Venezuelan equine encephalitis virus. PLoS Pathog. 2022, 18, e1009946. [Google Scholar] [CrossRef]
- Grieder, F.B.; Davis, B.K.; Zhou, X.D.; Chen, S.J.; Finkelman, F.D.; Gause, W.C. Kinetics of cytokine expression and regulation of host protection following infection with molecularly cloned Venezuelan equine encephalitis virus. Virology 1997, 233, 302–312. [Google Scholar] [CrossRef]
- Julander, J.G.; Skirpstunas, R.; Siddharthan, V.; Shafer, K.; Hoopes, J.D.; Smee, D.F.; Morrey, J.D. C3H/HeN mouse model for the evaluation of antiviral agents for the treatment of Venezuelan equine encephalitis virus infection. Antivir. Res. 2008, 78, 230–241. [Google Scholar] [CrossRef]
- Fongsaran, C.; Dineley, K.T.; Paessler, S.; Cisneros, I.E. VEEV TC-83 Triggers Dysregulation of the Tryptophan-Kynurenine Pathway in the Central Nervous System That Correlates with Cognitive Impairment in Tg2576 Mice. Pathogens 2024, 13, 397. [Google Scholar] [CrossRef] [PubMed]
- Fongsaran, C.; Jirakanwisal, K.; Peng, B.H.; Fracassi, A.; Taglialatela, G.; Dineley, K.T.; Paessler, S.; Cisneros, I.E. Arbovirus infection increases the risk for the development of neurodegenerative disease pathology in the murine model. Brain Behav. Immun. Health 2024, 38, 100780. [Google Scholar] [CrossRef] [PubMed]
- Ronca, S.E.; Smith, J.; Koma, T.; Miller, M.M.; Yun, N.; Dineley, K.T.; Paessler, S. Mouse Model of Neurological Complications Resulting from Encephalitic Alphavirus Infection. Front. Microbiol. 2017, 8, 188. [Google Scholar] [CrossRef] [PubMed]
- Ko, S.Y.; Akahata, W.; Yang, E.S.; Kong, W.P.; Burke, C.W.; Honnold, S.P.; Nichols, D.K.; Huang, Y.S.; Schieber, G.L.; Carlton, K.; et al. A virus-like particle vaccine prevents equine encephalitis virus infection in nonhuman primates. Sci. Transl. Med. 2019, 11, eaav3113. [Google Scholar] [CrossRef]
- Carey, B.D.; Akhrymuk, I.; Dahal, B.; Pinkham, C.L.; Bracci, N.; Finstuen-Magro, S.; Lin, S.C.; Lehman, C.W.; Sokoloski, K.J.; Kehn-Hall, K. Protein Kinase C subtype delta interacts with Venezuelan equine encephalitis virus capsid protein and regulates viral RNA binding through modulation of capsid phosphorylation. PLoS Pathog. 2020, 16, e1008282. [Google Scholar] [CrossRef]
- Bennett, A.M.; Elvin, S.J.; Wright, A.J.; Jones, S.M.; Phillpotts, R.J. An immunological profile of Balb/c mice protected from airborne challenge following vaccination with a live attenuated Venezuelan equine encephalitis virus vaccine. Vaccine 2000, 19, 337–347. [Google Scholar] [CrossRef]
- Cain, M.D.; Klein, N.R.; Jiang, X.; Salimi, H.; Wu, Q.; Miller, M.J.; Klimstra, W.B.; Klein, R.S. Post-exposure intranasal IFNalpha suppresses replication and neuroinvasion of Venezuelan Equine Encephalitis virus within olfactory sensory neurons. J. Neuroinflamm. 2024, 21, 24. [Google Scholar] [CrossRef]
- Paessler, S.; Ni, H.; Petrakova, O.; Fayzulin, R.Z.; Yun, N.; Anishchenko, M.; Weaver, S.C.; Frolov, I. Replication and clearance of Venezuelan equine encephalitis virus from the brains of animals vaccinated with chimeric SIN/VEE viruses. J. Virol. 2006, 80, 2784–2796. [Google Scholar] [CrossRef]
- Kafai, N.M.; Williamson, L.E.; Binshtein, E.; Sukupolvi-Petty, S.; Gardner, C.L.; Liu, J.; Mackin, S.; Kim, A.S.; Kose, N.; Carnahan, R.H.; et al. Neutralizing antibodies protect mice against Venezuelan equine encephalitis virus aerosol challenge. J. Exp. Med. 2022, 219, e20212532. [Google Scholar] [CrossRef]
- Kafai, N.M.; Janova, H.; Cain, M.D.; Alippe, Y.; Muraro, S.; Sariol, A.; Elam-Noll, M.; Klein, R.S.; Diamond, M.S. Entry receptor LDLRAD3 is required for Venezuelan equine encephalitis virus peripheral infection and neurotropism leading to pathogenesis in mice. Cell Rep. 2023, 42, 112946. [Google Scholar] [CrossRef]
- Sharma, A.; Bhattacharya, B.; Puri, R.K.; Maheshwari, R.K. Venezuelan equine encephalitis virus infection causes modulation of inflammatory and immune response genes in mouse brain. BMC Genom. 2008, 9, 289. [Google Scholar] [CrossRef]
- Salimi, H.; Cain, M.D.; Jiang, X.; Roth, R.A.; Beatty, W.L.; Sun, C.; Klimstra, W.B.; Hou, J.; Klein, R.S. Encephalitic Alphaviruses Exploit Caveola-Mediated Transcytosis at the Blood-Brain Barrier for Central Nervous System Entry. mBio 2020, 11, e02731-19. [Google Scholar] [CrossRef] [PubMed]
- Vogel, P.; Abplanalp, D.; Kell, W.; Ibrahim, M.S.; Downs, M.B.; Pratt, W.D.; Davis, K.J. Venezuelan equine encephalitis in BALB/c mice: Kinetic analysis of central nervous system infection following aerosol or subcutaneous inoculation. Arch. Pathol. Lab. Med. 1996, 120, 164–172. [Google Scholar] [PubMed]
- Ryzhikov, A.B.; Ryabchikova, E.I.; Sergeev, A.N.; Tkacheva, N.V. Spread of Venezuelan equine encephalitis virus in mice olfactory tract. Arch. Virol. 1995, 140, 2243–2254. [Google Scholar] [CrossRef] [PubMed]
- Schafer, A.; Brooke, C.B.; Whitmore, A.C.; Johnston, R.E. The role of the blood-brain barrier during Venezuelan equine encephalitis virus infection. J. Virol. 2011, 85, 10682–10690. [Google Scholar] [CrossRef]
- Hollidge, B.S.; Cohen, C.A.; Akuoku Frimpong, J.; Badger, C.V.; Dye, J.M.; Schmaljohn, C.S. Toll-like receptor 4 mediates blood-brain barrier permeability and disease in C3H mice during Venezuelan equine encephalitis virus infection. Virulence 2021, 12, 430–443. [Google Scholar] [CrossRef]
- Lehman, C.W.; Smith, A.; Kelly, J.; Jacobs, J.L.; Dinman, J.D.; Kehn-Hall, K. EGR1 Upregulation during Encephalitic Viral Infections Contributes to Inflammation and Cell Death. Viruses 2022, 14, 1210. [Google Scholar] [CrossRef]
- Williams, E.P.; Xue, Y.; Lee, J.; Fitzpatrick, E.A.; Kong, Y.; Reichard, W.; Writt, H.; Jonsson, C.B. Deep spatial profiling of Venezuelan equine encephalitis virus reveals increased genetic diversity amidst neuroinflammation and cell death during brain infection. J. Virol. 2023, 97, e0082723. [Google Scholar] [CrossRef]
- Gorelkin, L. Venezuelan equine encephalomyelitis in an adult animal host. An electron microscopic study. Am. J. Pathol. 1973, 73, 425–442. [Google Scholar]
- Lundberg, L.; Carey, B.; Kehn-Hall, K. Venezuelan Equine Encephalitis Virus Capsid-The Clever Caper. Viruses 2017, 9, 279. [Google Scholar] [CrossRef]
- Grieder, F.B.; Vogel, S.N. Role of interferon and interferon regulatory factors in early protection against Venezuelan equine encephalitis virus infection. Virology 1999, 257, 106–118. [Google Scholar] [CrossRef] [PubMed]
- Lukaszewski, R.A.; Brooks, T.J. Pegylated alpha interferon is an effective treatment for virulent venezuelan equine encephalitis virus and has profound effects on the host immune response to infection. J. Virol. 2000, 74, 5006–5015. [Google Scholar] [CrossRef] [PubMed]
- Charles, P.C.; Trgovcich, J.; Davis, N.L.; Johnston, R.E. Immunopathogenesis and immune modulation of Venezuelan equine encephalitis virus-induced disease in the mouse. Virology 2001, 284, 190–202. [Google Scholar] [CrossRef] [PubMed]
- Yun, N.E.; Peng, B.H.; Bertke, A.S.; Borisevich, V.; Smith, J.K.; Smith, J.N.; Poussard, A.L.; Salazar, M.; Judy, B.M.; Zacks, M.A.; et al. CD4+ T cells provide protection against acute lethal encephalitis caused by Venezuelan equine encephalitis virus. Vaccine 2009, 27, 4064–4073. [Google Scholar] [CrossRef]
- Brooke, C.B.; Deming, D.J.; Whitmore, A.C.; White, L.J.; Johnston, R.E. T cells facilitate recovery from Venezuelan equine encephalitis virus-induced encephalomyelitis in the absence of antibody. J. Virol. 2010, 84, 4556–4568. [Google Scholar] [CrossRef]
- Taylor, K.; Kolokoltsova, O.; Ronca, S.E.; Estes, M.; Paessler, S. Live, Attenuated Venezuelan Equine Encephalitis Virus Vaccine (TC83) Causes Persistent Brain Infection in Mice with Non-functional alphabeta T-Cells. Front. Microbiol. 2017, 8, 81. [Google Scholar] [CrossRef]
- Schoneboom, B.A.; Catlin, K.M.; Marty, A.M.; Grieder, F.B. Inflammation is a component of neurodegeneration in response to Venezuelan equine encephalitis virus infection in mice. J. Neuroimmunol. 2000, 109, 132–146. [Google Scholar] [CrossRef]
- Sharma, A.; Bhomia, M.; Honnold, S.P.; Maheshwari, R.K. Role of adhesion molecules and inflammation in Venezuelan equine encephalitis virus infected mouse brain. Virol. J. 2011, 8, 197. [Google Scholar] [CrossRef]
- Rangel, M.V.; Sebastian, A.; Leon, N.F.; Phillips, A.M.; Gorman, B.M.; Hum, N.R.; Weilhammer, D.R. Single-cell and spatiotemporal transcriptomic profiling of brain immune infiltration following Venezuelan equine encephalitis virus infection. Front. Immunol. 2024, 15, 1497839. [Google Scholar] [CrossRef]
- Campos, A.S.; Franco, A.C.; Godinho, F.M.; Huff, R.; Candido, D.S.; da Cruz Cardoso, J.; Hua, X.; Claro, I.M.; Morais, P.; Franceschina, C.; et al. Molecular Epidemiology of Western Equine Encephalitis Virus, South America, 2023-2024. Emerg. Infect. Dis. 2024, 30, 1834–1840. [Google Scholar] [CrossRef]
- Meyer, K.F.; Haring, C.M.; Howitt, B. The Etiology of Epizootic Encephalomyelitis of Horses in the San Joaquin Valley, 1930. Science 1931, 74, 227–228. [Google Scholar] [CrossRef] [PubMed]
- Phelps, A.L.; O’Brien, L.M.; Eastaugh, L.S.; Davies, C.; Lever, M.S.; Ennis, J.; Zeitlin, L.; Nunez, A.; Ulaeto, D.O. Susceptibility and Lethality of Western Equine Encephalitis Virus in Balb/c Mice When Infected by the Aerosol Route. Viruses 2017, 9, 163. [Google Scholar] [CrossRef] [PubMed]
- Frabasile, S.; Morel, N.; Perez, R.; Marrero, L.M.; Burgueno, A.; Cortinas, M.N.; Bassetti, L.; Negro, R.; Rodriguez, S.; Bormida, V.; et al. Equine Encephalomyelitis Outbreak, Uruguay, 2023–2024. Emerg. Infect. Dis. 2025, 31, 180–183. [Google Scholar] [CrossRef]
- Vissani, M.A.; Alamos, F.; Tordoya, M.S.; Minatel, L.; Schammas, J.M.; Dus Santos, M.J.; Trono, K.; Barrandeguy, M.E.; Balasuriya, U.B.R.; Carossino, M. Outbreak of Western Equine Encephalitis Virus Infection Associated with Neurological Disease in Horses Following a Nearly 40-Year Intermission Period in Argentina. Viruses 2024, 16, 1594. [Google Scholar] [CrossRef]
- Mulder, D.W.; Parrott, M.; Thaler, M. Sequelae of western equine encephalitis. Neurology 1951, 1, 318–327. [Google Scholar] [CrossRef]
- Palmer, R.J.; Finley, K.H. Sequelae of encephalitis; report of a study after the California epidemic. Calif. Med. 1956, 84, 98–100. [Google Scholar]
- Reeves, W.C.; Hutson, G.A.; Bellamy, R.E.; Scrivani, R.P. Chronic latent infections of birds with Western equine encephalomyelitis virus. Proc. Soc. Exp. Biol. Med. 1958, 97, 733–736. [Google Scholar] [CrossRef]
- Reed, D.S.; Larsen, T.; Sullivan, L.J.; Lind, C.M.; Lackemeyer, M.G.; Pratt, W.D.; Parker, M.D. Aerosol exposure to western equine encephalitis virus causes fever and encephalitis in cynomolgus macaques. J. Infect. Dis. 2005, 192, 1173–1182. [Google Scholar] [CrossRef]
- Burke, C.W.; Erwin-Cohen, R.A.; Goodson, A.I.; Wilhelmsen, C.; Edmundson, J.A.; White, C.E.; Glass, P.J. Efficacy of Western, Eastern, and Venezuelan Equine Encephalitis (WEVEE) Virus-Replicon Particle (VRP) Vaccine against WEEV in a Non-Human Primate Animal Model. Viruses 2022, 14, 1502. [Google Scholar] [CrossRef]
- Blakely, P.K.; Delekta, P.C.; Miller, D.J.; Irani, D.N. Manipulation of host factors optimizes the pathogenesis of western equine encephalitis virus infections in mice for antiviral drug development. J. Neurovirol 2015, 21, 43–55. [Google Scholar] [CrossRef]
- Logue, C.H.; Bosio, C.F.; Welte, T.; Keene, K.M.; Ledermann, J.P.; Phillips, A.; Sheahan, B.J.; Pierro, D.J.; Marlenee, N.; Brault, A.C.; et al. Virulence variation among isolates of western equine encephalitis virus in an outbred mouse model. J. Gen. Virol. 2009, 90 Pt 8, 1848–1858. [Google Scholar] [CrossRef] [PubMed]
- Burke, C.W.; Froude, J.W.; Miethe, S.; Hulseweh, B.; Hust, M.; Glass, P.J. Human-Like Neutralizing Antibodies Protect Mice from Aerosol Exposure with Western Equine Encephalitis Virus. Viruses 2018, 10, 147. [Google Scholar] [CrossRef] [PubMed]
- Bantle, C.M.; Rocha, S.M.; French, C.T.; Phillips, A.T.; Tran, K.; Olson, K.E.; Bass, T.A.; Aboellail, T.; Smeyne, R.J.; Tjalkens, R.B. Astrocyte inflammatory signaling mediates alpha-synuclein aggregation and dopaminergic neuronal loss following viral encephalitis. Exp. Neurol. 2021, 346, 113845. [Google Scholar] [CrossRef] [PubMed]
- Nagata, L.P.; Hu, W.G.; Parker, M.; Chau, D.; Rayner, G.A.; Schmaltz, F.L.; Wong, J.P. Infectivity variation and genetic diversity among strains of Western equine encephalitis virus. J. Gen. Virol. 2006, 87 Pt 8, 2353–2361. [Google Scholar] [CrossRef]
- Phillips, A.T.; Stauft, C.B.; Aboellail, T.A.; Toth, A.M.; Jarvis, D.L.; Powers, A.M.; Olson, K.E. Bioluminescent imaging and histopathologic characterization of WEEV neuroinvasion in outbred CD-1 mice. PLoS ONE 2013, 8, e53462. [Google Scholar] [CrossRef]
- Mossel, E.C.; Ledermann, J.P.; Phillips, A.T.; Borland, E.M.; Powers, A.M.; Olson, K.E. Molecular determinants of mouse neurovirulence and mosquito infection for Western equine encephalitis virus. PLoS ONE 2013, 8, e60427. [Google Scholar] [CrossRef]
- Morrow, E.M.; Yoo, S.Y.; Flavell, S.W.; Kim, T.K.; Lin, Y.; Hill, R.S.; Mukaddes, N.M.; Balkhy, S.; Gascon, G.; Hashmi, A.; et al. Identifying autism loci and genes by tracing recent shared ancestry. Science 2008, 321, 218–223. [Google Scholar] [CrossRef]
- Julander, J.G.; Siddharthan, V.; Blatt, L.M.; Schafer, K.; Sidwell, R.W.; Morrey, J.D. Effect of exogenous interferon and an interferon inducer on western equine encephalitis virus disease in a hamster model. Virology 2007, 360, 454–460. [Google Scholar] [CrossRef]
- Wu, J.Q.; Barabe, N.D.; Huang, Y.M.; Rayner, G.A.; Christopher, M.E.; Schmaltz, F.L. Pre- and post-exposure protection against Western equine encephalitis virus after single inoculation with adenovirus vector expressing interferon alpha. Virology 2007, 369, 206–213. [Google Scholar] [CrossRef]
- Feld, J.J.; Hoofnagle, J.H. Mechanism of action of interferon and ribavirin in treatment of hepatitis C. Nature 2005, 436, 967–972. [Google Scholar] [CrossRef]
- Pereira, C.R.; Machado, J.; Rodrigues, J.; de Oliveira, N.M.; Criado, M.B.; Greten, H.J. Effectiveness of Acupuncture in Parkinson’s Disease Symptoms-A Systematic Review. Healthcare 2022, 10, 2334. [Google Scholar] [CrossRef] [PubMed]
- Taylor, A.; Herrero, L.J.; Rudd, P.A.; Mahalingam, S. Mouse models of alphavirus-induced inflammatory disease. J. Gen. Virol. 2015, 96 Pt 2, 221–238. [Google Scholar] [CrossRef]
- Walf, A.A.; Frye, C.A. The use of the elevated plus maze as an assay of anxiety-related behavior in rodents. Nat. Protoc. 2007, 2, 322–328. [Google Scholar] [CrossRef]
- Kraeuter, A.K.; Guest, P.C.; Sarnyai, Z. The Y-Maze for Assessment of Spatial Working and Reference Memory in Mice. Methods Mol. Biol. 2019, 1916, 105–111. [Google Scholar] [CrossRef]
- Gilli, F.; Royce, D.B.; Pachner, A.R. Measuring Progressive Neurological Disability in a Mouse Model of Multiple Sclerosis. J. Vis. Exp. 2016, 117, 54616. [Google Scholar] [CrossRef]
- Bey, A.L.; Jiang, Y.H. Overview of mouse models of autism spectrum disorders. Curr. Protoc. Pharmacol. 2014, 66, 5.66.1–5.66.26. [Google Scholar] [CrossRef]

| Animal Model | Viral Strain | Infection Route | % Mortality | Clinical Observations | Neuropathological Features | Refs. | |
|---|---|---|---|---|---|---|---|
| Species | Strain | ||||||
| NHP | Cynomolgus macaque, common marmoset | V105-00210 | AE * | 60–100% | Weight loss, fever, ataxia, nystagmus, lethargy, twitching, tremors, and seizures (lethal) | Meningoencephalitis and vasculitis, neuronal degeneration and necrosis, perivascular cuffs, cellular debris, gliosis, satellitosis, edema, hemorrhage, infiltrating neutrophils, and lymphocytic infiltrates within the hippocampus, amygdala, corpus striatum, thalamus, mesencephalon, and medulla oblongata. | [47,48,49] |
| NHP | Common marmoset | FL939-39 | IN ** | 100% | Weight loss, anorexia, lethargy, hunched posture, somnolence | Encephalitis, mononuclear cell leptomeningitis, perivascular cuffing in cerebral cortex and hippocampus. Microglial activation and neuronal necrosis, viral antigen in cerebral cortex. | [42] |
| NHP | Cynomolgus macaque | FL939-39 | SC *** | 75% | Dehydration, ataxia, intermittent tremors, lethargy, difficulty standing on both feet, abnormal vocalizations, hyperthermia, excessive salivation, recumbency, facial edema, anorexia | Neuronal necrosis and gliosis in the cerebrum, inflammation in the meninges, and perivascular cuffing in the cerebrum and cerebellum. Acute inflammation in the cerebrum, cerebellum, and brain stem neuropil; chronic inflammation in the meninges, choroid plexus, and cerebral neuropil; gliosis in the brain stem; hemorrhage in the cerebrum and brainstem neuropil | [4] |
| Mice | BALB/c, CD-1 | FL939-39, EEEV NA V105 | AE | 80–100% | Weight loss, ataxia, ruffled fur, hunched posture, lethargy, dehydration, head tilt, circling, fixed gaze, paralysis, lateral recumbency, and seizures (rare) | Meningoencephalitis; Neuronal vacuolation, spongiosis, necrosis. Viral antigen in nasal cavity, olfactory bulbs, frontal cortex, hippocampus, thalamus, midbrain, cerebellum, brain stem, spinal cord, and pituitary gland. | [50,51,52,53] |
| Mice | BALB/c | FL939-39 | IN | Not described [50,51] | Viremia, weight loss, ataxia, ruffled fur, hunched posture, lethargy, dehydration, head tilt, circling, fixed gaze, paralysis, lateral recumbency, and seizures (rare) | Neuronal cell death in the hippocampus. Viral antigen in olfactory epithelium, olfactory bulb, neurons in the cerebrum, frontal cortex, and midbrain. | [50,51] |
| Mice | BALB/c, C57BL/6 | FL939-39 | SC | 100% [38], Not described [50,51] | Ruffled fur, hunched posture, lethargy, tremors, lateral recumbency | Neuronal apoptosis, necrosis, spongiosis, meningoencephalitis, vasculitis, thrombosis, and perivascular cuffing. Viral antigen in olfactory epithelium, cortex, hippocampus, caudate putamen, thalamus. | [38,50,51] |
| Host Factor | Function | Impact on EEEV Pathogenesis | Ref. |
|---|---|---|---|
| VLDLR | Entry receptor for EEEV | Increased survival in VLDLR KO mice | [58] |
| miR-142-3p | Hematopoietic cell-specific microRNA | Increased survival following infection with combinatorial mutations of EEEV miR-142-3p | [61] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Woodson, C.M.; Carney, S.K.; Kehn-Hall, K. Neuropathogenesis of Encephalitic Alphaviruses in Non-Human Primate and Mouse Models of Infection. Pathogens 2025, 14, 193. https://doi.org/10.3390/pathogens14020193
Woodson CM, Carney SK, Kehn-Hall K. Neuropathogenesis of Encephalitic Alphaviruses in Non-Human Primate and Mouse Models of Infection. Pathogens. 2025; 14(2):193. https://doi.org/10.3390/pathogens14020193
Chicago/Turabian StyleWoodson, Caitlin M., Shannon K. Carney, and Kylene Kehn-Hall. 2025. "Neuropathogenesis of Encephalitic Alphaviruses in Non-Human Primate and Mouse Models of Infection" Pathogens 14, no. 2: 193. https://doi.org/10.3390/pathogens14020193
APA StyleWoodson, C. M., Carney, S. K., & Kehn-Hall, K. (2025). Neuropathogenesis of Encephalitic Alphaviruses in Non-Human Primate and Mouse Models of Infection. Pathogens, 14(2), 193. https://doi.org/10.3390/pathogens14020193