

Total Infectome Characterization of Respiratory Infections during the 2022–23 COVID-19 Outbreak in China Revealed Extensive Coinfections with Links to SARS-CoV-2 Status, Age, and Disease Severity
 and
and
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample and Clinical Data Collection
2.2. Sample Processing and DNA Extraction
2.3. Metagenomic Sequencing
2.4. Bioinformatic Analysis
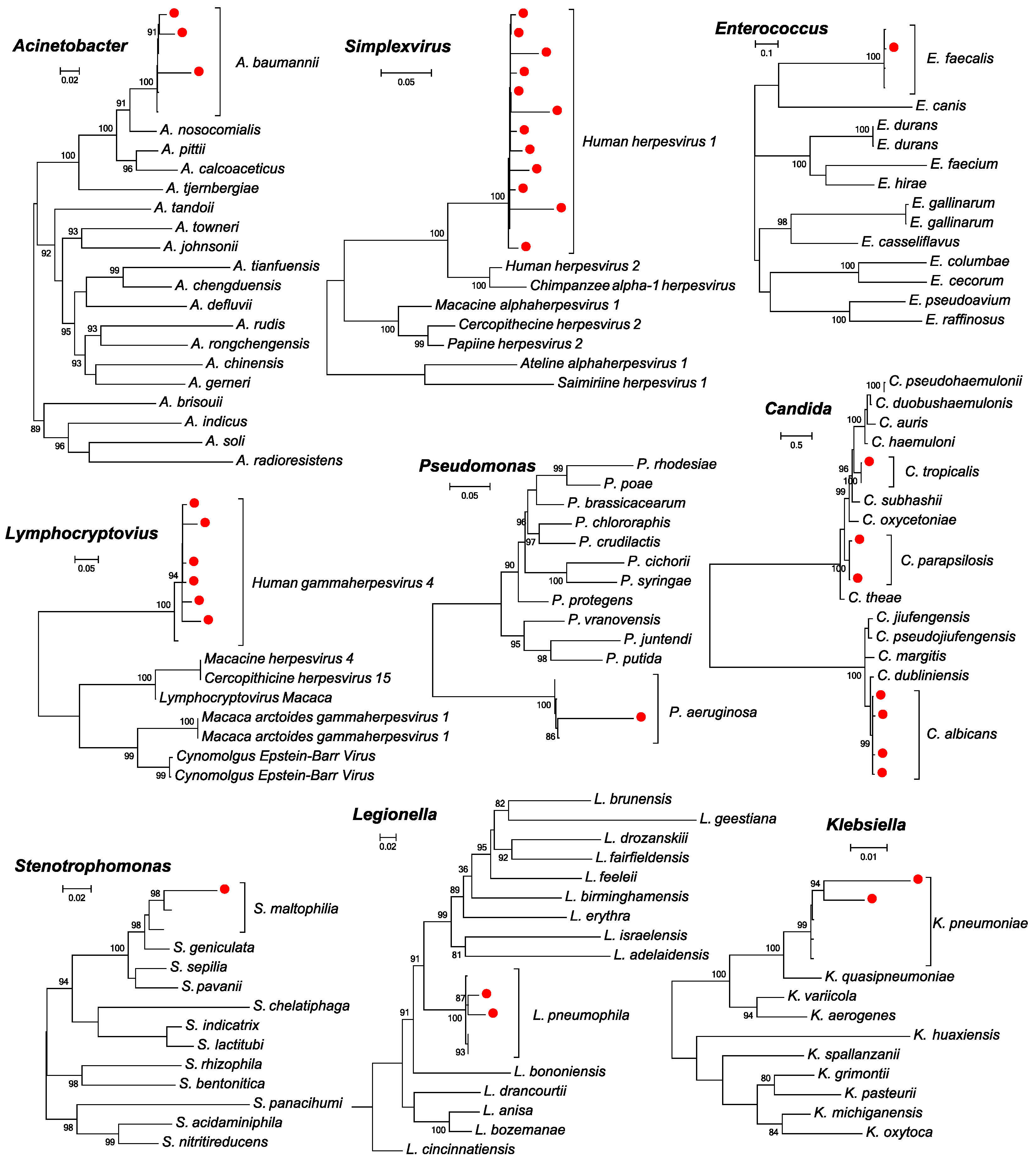
2.5. Evolutionary Analyses
2.6. Statistical Analyses
3. Results
3.1. Participant Recruitment and Clinical Factors
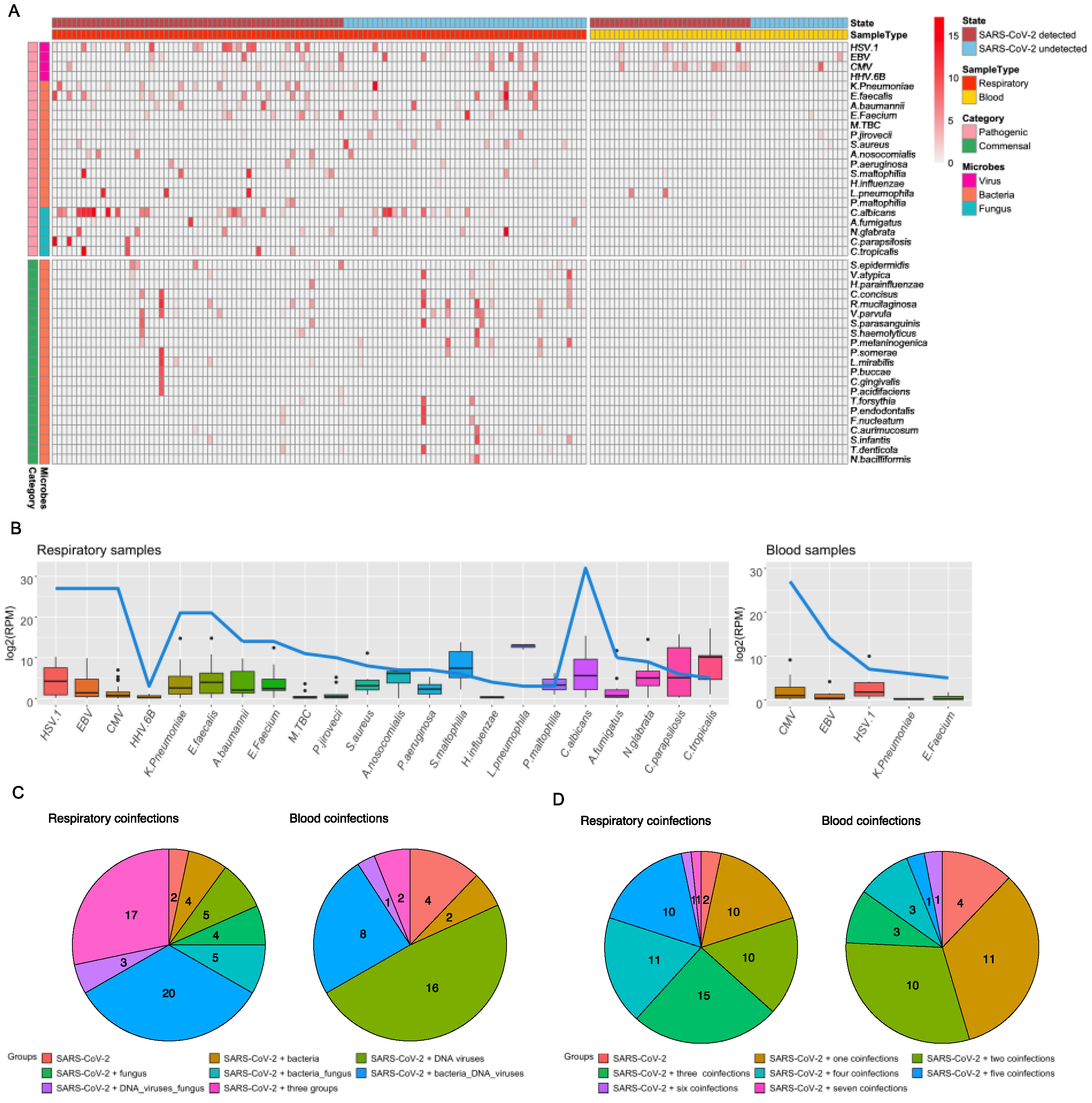
3.2. Total Infectome and Coinfection Characterizations
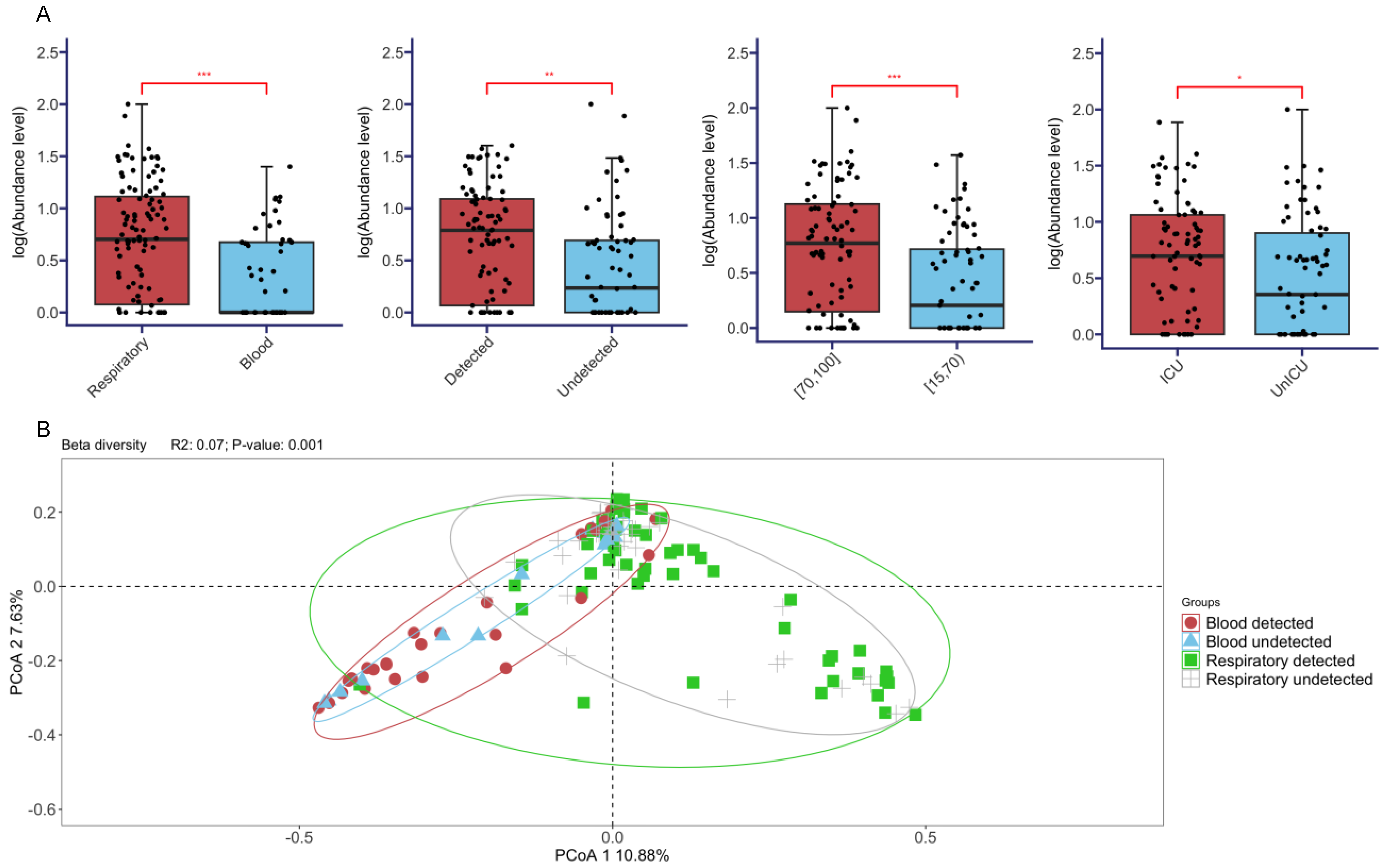
3.3. Comparisons of Diversities across Different Sample Types, SARS-CoV-2 Status, Age, and Disease Severity
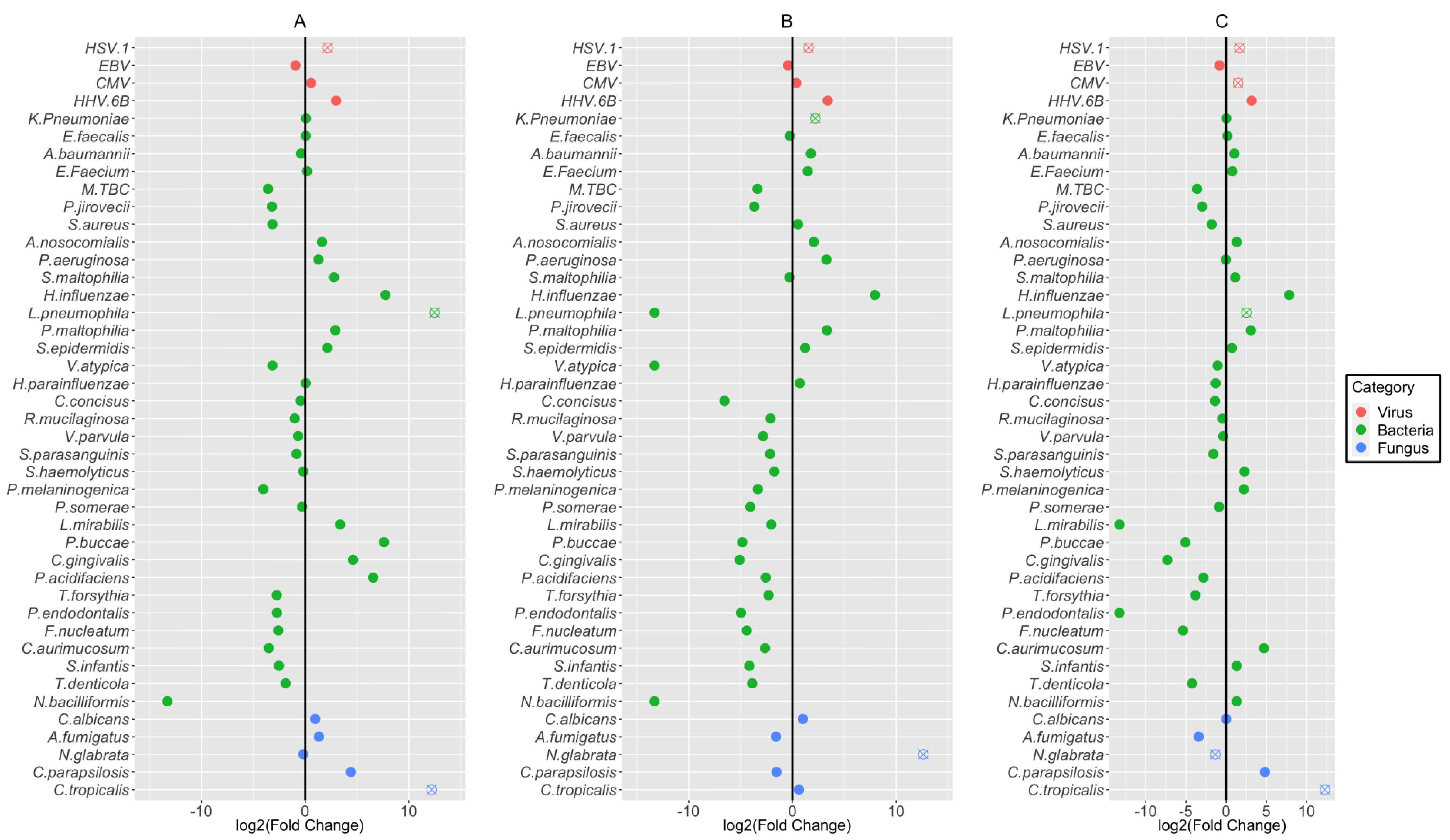
3.4. Association of Coinfecting Pathogens with Detection Status, Age, and Disease Severity
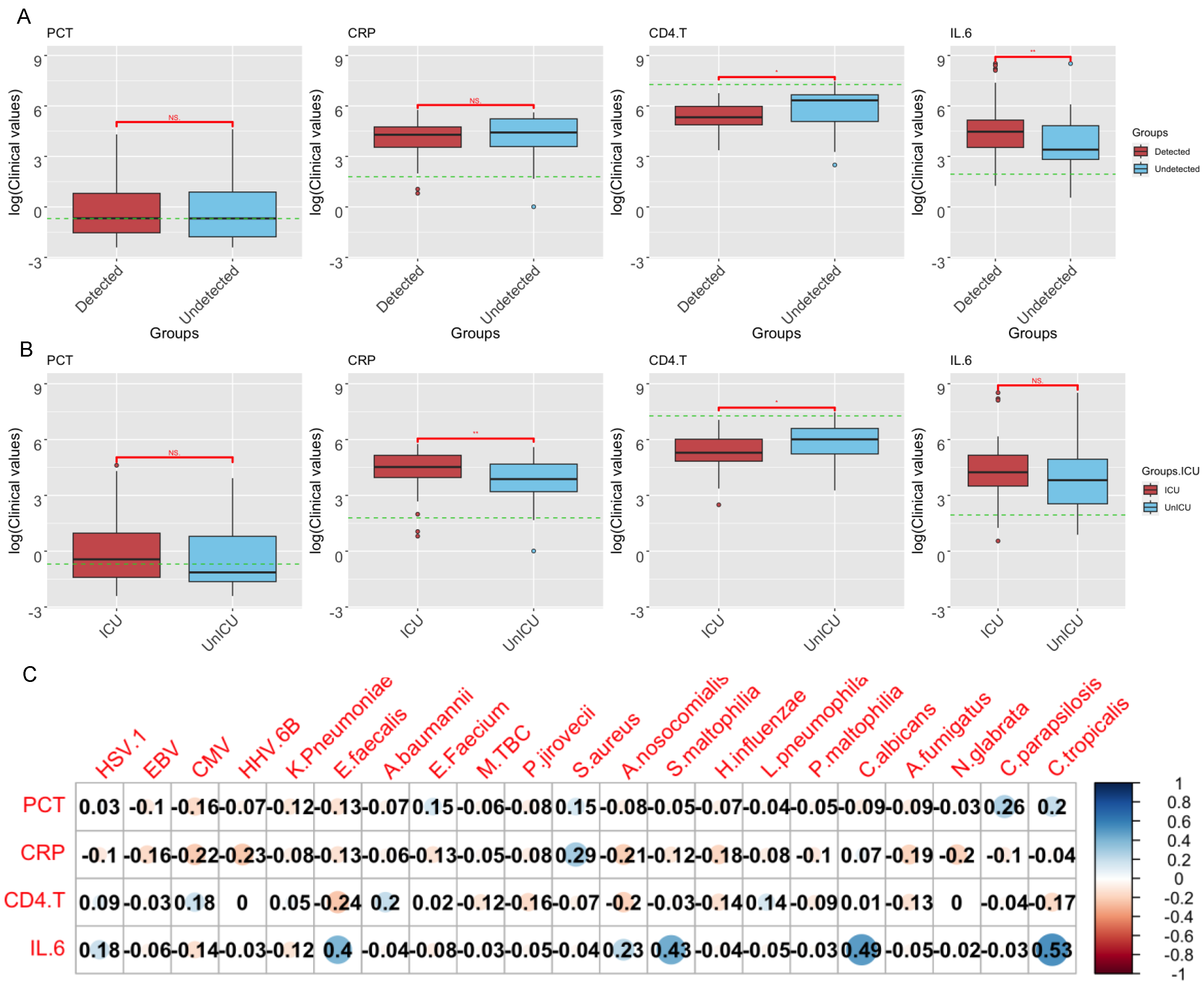
3.5. Correlations of Clinical Indicators and Pathogen Abundance
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Huang, S.; Gao, Z.; Wang, S. China’s COVID-19 reopening measures-warriors and weapons. Lancet 2023, 401, 643–644. [Google Scholar] [CrossRef]
- Huang, J.; Zhao, S.; Chong, K.C.; Zhou, Y.; Lu, W.; Fang, F.; Cheung, P.P.H.; Lai, K.C.; Hui, D.S.; Mok, C.K.P. Infection rate in Guangzhou after easing the zero-COVID policy: Seroprevalence results to ORF8 antigen. Lancet Infect. Dis. 2023, 23, 403–404. [Google Scholar] [CrossRef]
- Lansbury, L.; Lim, B.; Baskaran, V.; Lim, W.S. Co-infections in people with COVID-19: A systematic review and meta-analysis. J. Infect. 2020, 81, 266–275. [Google Scholar] [CrossRef] [PubMed]
- Zhong, H.; Wang, Y.; Shi, Z.; Zhang, L.; Ren, H.; He, W.; Zhang, Z.; Zhu, A.; Zhao, J.; Xiao, F.; et al. Characterization of respiratory microbial dysbiosis in hospitalized COVID-19 patients. Cell Discov. 2021, 7, 23. [Google Scholar] [CrossRef] [PubMed]
- Clementi, N.; Ghosh, S.; De Santis, M.; Castelli, M.; Criscuolo, E.; Zanoni, I.; Clementi, M.; Mancini, N. Viral Respiratory Pathogens and Lung Injury. Clin. Microbiol. Rev. 2021, 34, e00103-20. [Google Scholar] [CrossRef] [PubMed]
- Xu, R.; Lu, R.; Zhang, T.; Wu, Q.; Cai, W.; Han, X.; Wan, Z.; Jin, X.; Zhang, Z.; Zhang, C. Temporal association between human upper respiratory and gut bacterial microbiomes during the course of COVID-19 in adults. Commun. Biol. 2021, 4, 240. [Google Scholar] [CrossRef] [PubMed]
- De Castilhos, J.; Zamir, E.; Hippchen, T.; Rohrbach, R.; Schmidt, S.; Hengler, S.; Schumacher, H.; Neubauer, M.; Kunz, S.; Muller-Esch, T.; et al. Severe Dysbiosis and Specific Haemophilus and Neisseria Signatures as Hallmarks of the Oropharyngeal Microbiome in Critically Ill Coronavirus Disease 2019 (COVID-19) Patients. Clin. Infect. Dis. 2022, 75, e1063–e1071. [Google Scholar] [CrossRef] [PubMed]
- Ren, Z.; Wang, H.; Cui, G.; Lu, H.; Wang, L.; Luo, H.; Chen, X.; Ren, H.; Sun, R.; Liu, W.; et al. Alterations in the human oral and gut microbiomes and lipidomics in COVID-19. Gut 2021, 70, 1253–1265. [Google Scholar] [CrossRef]
- Swets, M.C.; Russell, C.D.; Harrison, E.M.; Docherty, A.B.; Lone, N.; Girvan, M.; Hardwick, H.E.; Investigators, I.C.; Visser, L.G.; Openshaw, P.J.M.; et al. SARS-CoV-2 co-infection with influenza viruses, respiratory syncytial virus, or adenoviruses. Lancet 2022, 399, 1463–1464. [Google Scholar] [CrossRef]
- Alhumaid, S.; Alabdulqader, M.; Al Dossary, N.; Al Alawi, Z.; Alnaim, A.A.; Al Mutared, K.M.; Al Noaim, K.; Al Ghamdi, M.A.; Albahrani, S.J.; Alahmari, A.A.; et al. Global Coinfections with Bacteria, Fungi, and Respiratory Viruses in Children with SARS-CoV-2: A Systematic Review and Meta-Analysis. Trop. Med. Infect. Dis. 2022, 7, 380. [Google Scholar] [CrossRef]
- Liu, Y.; Ling, L.; Wong, S.H.; Wang, M.H.; Fitzgerald, J.R.; Zou, X.; Fang, S.; Liu, X.; Wang, X.; Hu, W.; et al. Outcomes of respiratory viral-bacterial co-infection in adult hospitalized patients. eClinicalMedicine 2021, 37, 100955. [Google Scholar] [CrossRef] [PubMed]
- Aykac, K.; Ozsurekci, Y.; Cura Yayla, B.C.; Evren, K.; Lacinel Gurlevik, S.; Oygar, P.D.; Yucel, M.; Karakoc, A.E.; Alp, A.; Cengiz, A.B.; et al. Pneumococcal carriage in children with COVID-19. Hum. Vaccines Immunother. 2021, 17, 1628–1634. [Google Scholar] [CrossRef] [PubMed]
- Fan, J.; Li, X.; Gao, Y.; Zhou, J.; Wang, S.; Huang, B.; Wu, J.; Cao, Q.; Chen, Y.; Wang, Z.; et al. The lung tissue microbiota features of 20 deceased patients with COVID-19. J. Infect. 2020, 81, e64–e67. [Google Scholar] [CrossRef] [PubMed]
- Altinkaya Cavus, M.; Sav, H. Opportunistic Candida Infections in Critical COVID-19 Patients. Pol. J. Microbiol. 2022, 71, 411–419. [Google Scholar] [CrossRef]
- Choudhary, R.; Webber, B.J.; Womack, L.S.; Dupont, H.K.; Chiu, S.K.; Wanga, V.; Gerdes, M.E.; Hsu, S.; Shi, D.S.; Dulski, T.M.; et al. Factors Associated With Severe Illness in Patients Aged <21 Years Hospitalized for COVID-19. Hosp. Pediatr. 2022, 12, 760–783. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, C.J.; Pho, M.T.; Pitrak, D.; Ridgway, J.P.; Pettit, N.N. Community-acquired Coinfection in Coronavirus Disease 2019: A Retrospective Observational Experience. Clin. Infect. Dis. 2021, 72, 1450–1452. [Google Scholar] [CrossRef]
- Fan, H.; Zhou, L.; Lv, J.; Yang, S.; Chen, G.; Liu, X.; Han, C.; Tan, X.; Qian, S.; Wu, Z.; et al. Bacterial coinfections contribute to severe COVID-19 in winter. Cell Res. 2023, 33, 562–564. [Google Scholar] [CrossRef]
- The Lancet Regional Health-Western Pacific. The end of zero-COVID-19 policy is not the end of COVID-19 for China. Lancet Reg. Health West. Pac. 2023, 30, 100702. [Google Scholar] [CrossRef]
- Ioannidis, J.P.A.; Zonta, F.; Levitt, M. What Really Happened During the Massive SARS-CoV-2 Omicron Wave in China? JAMA Intern. Med. 2023, 183, 633–634. [Google Scholar] [CrossRef]
- Burki, T. Moving away from zero COVID in China. Lancet Respir. Med. 2023, 11, 132. [Google Scholar] [CrossRef]
- Shi, M.; Zhao, S.; Yu, B.; Wu, W.C.; Hu, Y.; Tian, J.H.; Yin, W.; Ni, F.; Hu, H.L.; Geng, S.; et al. Total infectome characterization of respiratory infections in pre-COVID-19 Wuhan, China. PLoS Pathog. 2022, 18, e1010259. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Yin, Y.; Gao, H.; Guo, Y.; Dong, Z.; Wang, X.; Zhang, Y.; Yang, S.; Peng, Q.; Liu, Y.; et al. Clinical Utility of In-house Metagenomic Next-generation Sequencing for the Diagnosis of Lower Respiratory Tract Infections and Analysis of the Host Immune Response. Clin. Infect. Dis. 2020, 71, S416–S426. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Lu, Z.; Bao, Y.; Yang, Y.; de Groot, R.; Dai, W.; de Jonge, M.I.; Zheng, Y. Clinical diagnostic application of metagenomic next-generation sequencing in children with severe nonresponding pneumonia. PLoS ONE 2020, 15, e0232610. [Google Scholar] [CrossRef] [PubMed]
- Li, C.X.; Li, W.; Zhou, J.; Zhang, B.; Feng, Y.; Xu, C.P.; Lu, Y.Y.; Holmes, E.C.; Shi, M. High resolution metagenomic characterization of complex infectomes in paediatric acute respiratory infection. Sci. Rep. 2020, 10, 3963. [Google Scholar] [CrossRef] [PubMed]
- Thi Kha Tu, N.; Thi Thu Hong, N.; Thi Han Ny, N.; My Phuc, T.; Thi Thanh Tam, P.; Doorn, H.R.V.; Dang Trung Nghia, H.; Thao Huong, D.; An Han, D.; Thi Thu Ha, L.; et al. The Virome of Acute Respiratory Diseases in Individuals at Risk of Zoonotic Infections. Viruses 2020, 12, 960. [Google Scholar] [CrossRef] [PubMed]
- Long, Y.; Zhang, Y.; Gong, Y.; Sun, R.; Su, L.; Lin, X.; Shen, A.; Zhou, J.; Caiji, Z.; Wang, X.; et al. Diagnosis of Sepsis with Cell-free DNA by Next-Generation Sequencing Technology in ICU Patients. Arch. Med. Res. 2016, 47, 365–371. [Google Scholar] [CrossRef]
- Jeon, Y.J.; Zhou, Y.; Li, Y.; Guo, Q.; Chen, J.; Quan, S.; Zhang, A.; Zheng, H.; Zhu, X.; Lin, J.; et al. The feasibility study of non-invasive fetal trisomy 18 and 21 detection with semiconductor sequencing platform. PLoS ONE 2014, 9, e110240. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef]
- Katoh, K.; Toh, H. Recent developments in the MAFFT multiple sequence alignment program. Brief Bioinform. 2008, 9, 286–298. [Google Scholar] [CrossRef]
- Capella-Gutierrez, S.; Silla-Martinez, J.M.; Gabaldon, T. trimAl: A tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 2009, 25, 1972–1973. [Google Scholar] [CrossRef]
- Guindon, S.; Dufayard, J.F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New algorithms and methods to estimate maximum-likelihood phylogenies: Assessing the performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef] [PubMed]
- Merenstein, C.; Liang, G.; Whiteside, S.A.; Cobian-Guemes, A.G.; Merlino, M.S.; Taylor, L.J.; Glascock, A.; Bittinger, K.; Tanes, C.; Graham-Wooten, J.; et al. Signatures of COVID-19 Severity and Immune Response in the Respiratory Tract Microbiome. mBio 2021, 12, e0177721. [Google Scholar] [CrossRef] [PubMed]
- Bussolati, E.; Cultrera, R.; Quaranta, A.; Cricca, V.; Marangoni, E.; La Rosa, R.; Bertacchini, S.; Bellonzi, A.; Ragazzi, R.; Volta, C.A.; et al. Effect of the Pandemic Outbreak on ICU-Associated Infections and Antibiotic Prescription Trends in Non-COVID19 Acute Respiratory Failure Patients. J. Clin. Med. 2022, 11, 7080. [Google Scholar] [CrossRef] [PubMed]
- Sulaiman, I.; Chung, M.; Angel, L.; Tsay, J.J.; Wu, B.G.; Yeung, S.T.; Krolikowski, K.; Li, Y.; Duerr, R.; Schluger, R.; et al. Microbial signatures in the lower airways of mechanically ventilated COVID-19 patients associated with poor clinical outcome. Nat. Microbiol. 2021, 6, 1245–1258. [Google Scholar] [CrossRef]
- Dunning, J.; Blankley, S.; Hoang, L.T.; Cox, M.; Graham, C.M.; James, P.L.; Bloom, C.I.; Chaussabel, D.; Banchereau, J.; Brett, S.J.; et al. Progression of whole-blood transcriptional signatures from interferon-induced to neutrophil-associated patterns in severe influenza. Nat. Immunol. 2018, 19, 625–635. [Google Scholar] [CrossRef] [PubMed]
- Bermejo-Martin, J.F.; Garcia-Mateo, N.; Motos, A.; Resino, S.; Tamayo, L.; Ryan Murua, P.; Bustamante-Munguira, E.; Gallego Curto, E.; Ubeda-Iglesias, A.; de la Torre, M.D.C.; et al. Effect of viral storm in patients admitted to intensive care units with severe COVID-19 in Spain: A multicentre, prospective, cohort study. Lancet Microbe 2023, 4, e431–e441. [Google Scholar] [CrossRef] [PubMed]
- Chong, S.; Kim, T.S.; Cho, E.Y. Herpes simplex virus pneumonia: High-resolution CT findings. Br. J. Radiol. 2010, 83, 585–589. [Google Scholar] [CrossRef]
- Luyt, C.E.; Combes, A.; Deback, C.; Aubriot-Lorton, M.H.; Nieszkowska, A.; Trouillet, J.L.; Capron, F.; Agut, H.; Gibert, C.; Chastre, J. Herpes simplex virus lung infection in patients undergoing prolonged mechanical ventilation. Am. J. Respir. Crit. Care Med. 2007, 175, 935–942. [Google Scholar] [CrossRef]
- Russell, C.D.; Fairfield, C.J.; Drake, T.M.; Turtle, L.; Seaton, R.A.; Wootton, D.G.; Sigfrid, L.; Harrison, E.M.; Docherty, A.B.; de Silva, T.I.; et al. Co-infections, secondary infections, and antimicrobial use in patients hospitalised with COVID-19 during the first pandemic wave from the ISARIC WHO CCP-UK study: A multicentre, prospective cohort study. Lancet Microbe 2021, 2, e354–e365. [Google Scholar] [CrossRef]
- Espinosa, V.; Dutta, O.; McElrath, C.; Du, P.; Chang, Y.J.; Cicciarelli, B.; Pitler, A.; Whitehead, I.; Obar, J.J.; Durbin, J.E.; et al. Type III interferon is a critical regulator of innate antifungal immunity. Sci. Immunol. 2017, 2, eaan5357. [Google Scholar] [CrossRef]
- Llorens-Rico, V.; Gregory, A.C.; Van Weyenbergh, J.; Jansen, S.; Van Buyten, T.; Qian, J.; Braz, M.; Menezes, S.M.; Van Mol, P.; Vanderbeke, L.; et al. Clinical practices underlie COVID-19 patient respiratory microbiome composition and its interactions with the host. Nat. Commun. 2021, 12, 6243. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient Characteristics | SARS-CoV-2 Undetected (n = 57) | SARS-CoV-2 Detected (n = 59) |
|---|---|---|
| Age | 60.2 (15~91) | 70.7 (31~91) |
| Gender [Number (%)] Male Female | 38 (66.7) 19 (33.3) | 48 (81.4) 11 (18.6) |
| Outcomes [Number (%)] Died Discharged Transferred Undischarged NA | 5 (8.8) 36 (63.1) 8 (14.0) 3 (5.3) 5 (8.8) | 14 (23.7) 27 (45.8) 13 (22.0) 4 (6.8) 1 (1.7) |
| Severity [Number (%)] ICU UnICU | 17 (29.8) 40 (70.2) | 32 (54.2) 27 (45.8) |
| Symptoms [Number (%)] Chest.Pain Cough Cough up Phlegm (CuP) Diarrhoea Fever Loss of Appetite (LoA) Vomiting | 9 (15.8) 42 (73.7) 36 (63.2) 7 (12.3) 38 (66.7) 15 (26.3) 8 (14.0) | 10 (16.9) 53 (89.8) 51 (86.4) 18 (30.5) 48 (81.4) 26 (44.1) 9 (15.3) |
| Comorbidities [Number (%)] Coronary Artery Disease (CAD) Chronic Lung Disease (CLD) Diabetes Hyperlipidemia Hypertension Other infectious disease (OID) Tumor | 16 (28.1) 4 (7.0) 14 (24.6) 0 (0) 18 (31.6) 4 (7.0) 10 (17.5) | 31 (52.5) 7 (11.9) 16 (21.7) 1 (1.7) 32 (54.2) 4 (6.8) 14 (23.7) |
| Treatment [Number (%)] Antibiotic therapy (ABT) Antiviral therapy (AVT) ECMO Hormone therapy (HormT) Immunoglobulin therapy (IgT) Invasive ventilation (IV) Non-Invasive ventilation (NIV) | 44 (77.2) 11 (19.3) 1 (1.75) 20 (35.09) 8 (14.04) 13 (22.81) 15 (26.32) | 50 (84.7) 49 (83.1) 0 (0) 42 (71.2) 18 (30.5) 39 (66.1) 23 (40.0) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, Z.; Cai, K.; Liao, Y.; Wu, W.-C.; Xing, L.; Hu, M.; Ren, J.; Zhang, J.; Zhu, X.; Yuan, K.; et al. Total Infectome Characterization of Respiratory Infections during the 2022–23 COVID-19 Outbreak in China Revealed Extensive Coinfections with Links to SARS-CoV-2 Status, Age, and Disease Severity. Pathogens 2024, 13, 216. https://doi.org/10.3390/pathogens13030216
Yang Z, Cai K, Liao Y, Wu W-C, Xing L, Hu M, Ren J, Zhang J, Zhu X, Yuan K, et al. Total Infectome Characterization of Respiratory Infections during the 2022–23 COVID-19 Outbreak in China Revealed Extensive Coinfections with Links to SARS-CoV-2 Status, Age, and Disease Severity. Pathogens. 2024; 13(3):216. https://doi.org/10.3390/pathogens13030216
Chicago/Turabian StyleYang, Zhongzhou, Kanru Cai, Yuqi Liao, Wei-Chen Wu, Li Xing, Minxuan Hu, Jiali Ren, Jieyun Zhang, Xiuyun Zhu, Ke Yuan, and et al. 2024. "Total Infectome Characterization of Respiratory Infections during the 2022–23 COVID-19 Outbreak in China Revealed Extensive Coinfections with Links to SARS-CoV-2 Status, Age, and Disease Severity" Pathogens 13, no. 3: 216. https://doi.org/10.3390/pathogens13030216
APA StyleYang, Z., Cai, K., Liao, Y., Wu, W.-C., Xing, L., Hu, M., Ren, J., Zhang, J., Zhu, X., Yuan, K., Wang, S., Huang, H., Yang, C., Zhang, M., Shi, M., & Lu, H. (2024). Total Infectome Characterization of Respiratory Infections during the 2022–23 COVID-19 Outbreak in China Revealed Extensive Coinfections with Links to SARS-CoV-2 Status, Age, and Disease Severity. Pathogens, 13(3), 216. https://doi.org/10.3390/pathogens13030216