


Preservation of scRNA-Seq Libraries Using Existing Inactivation Protocols


Abstract
1. Introduction
2. Materials and Methods
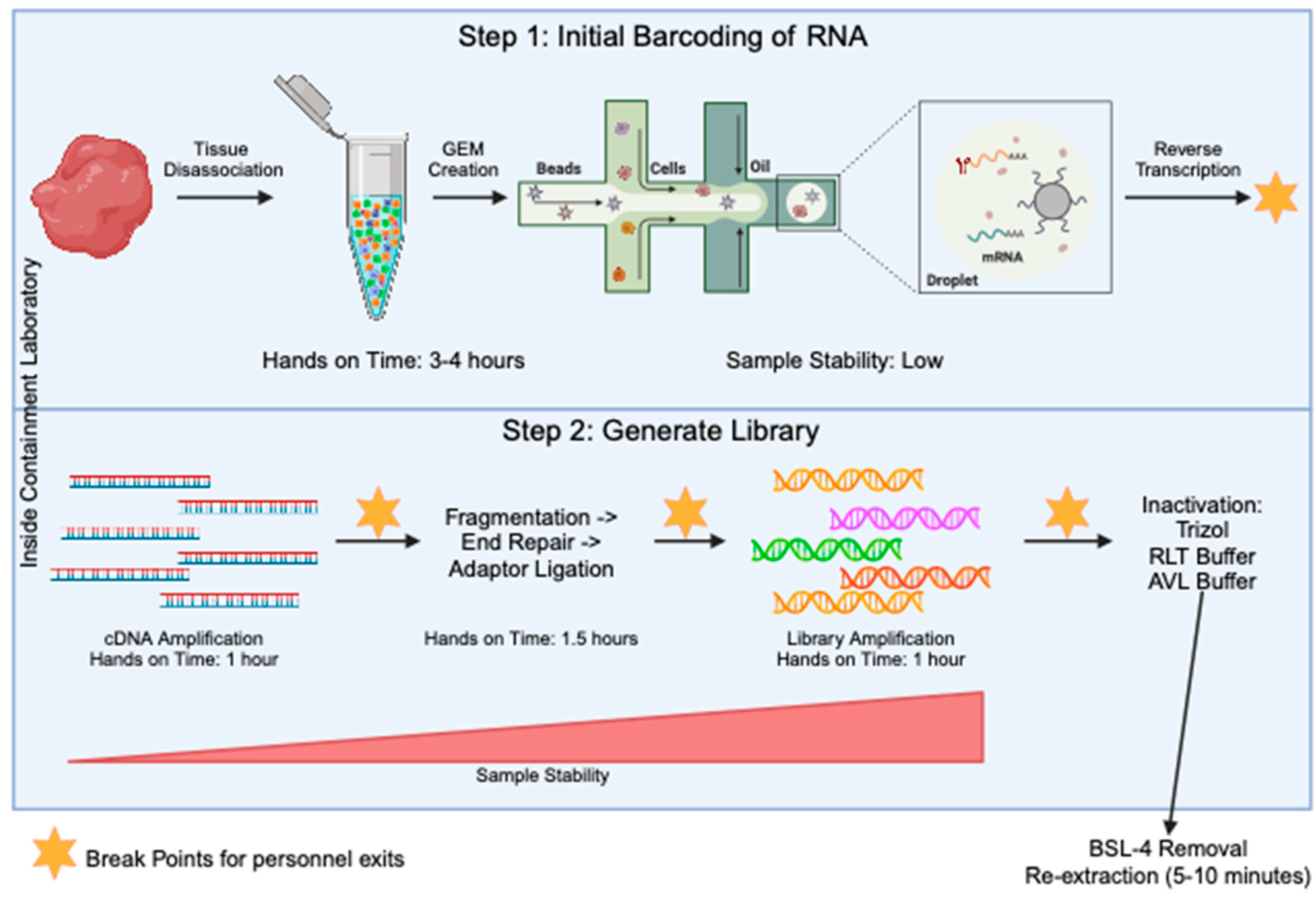
2.1. Single-Cell Sample Prep
2.2. Single-Cell Library Prep
2.3. Sample Inactivation and Re-Extraction
2.4. Single-Cell Library Data Processing
2.5. Statistic
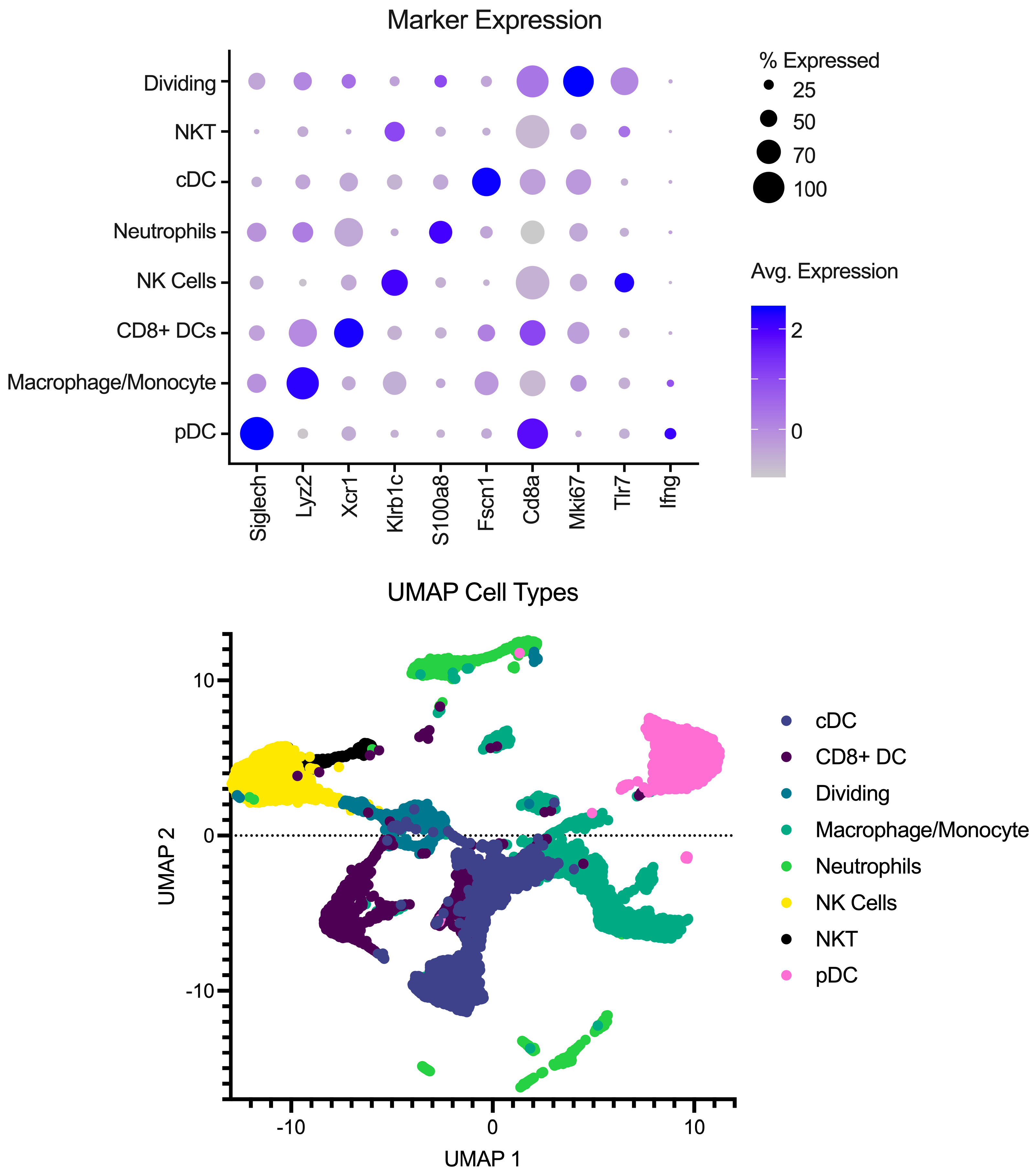
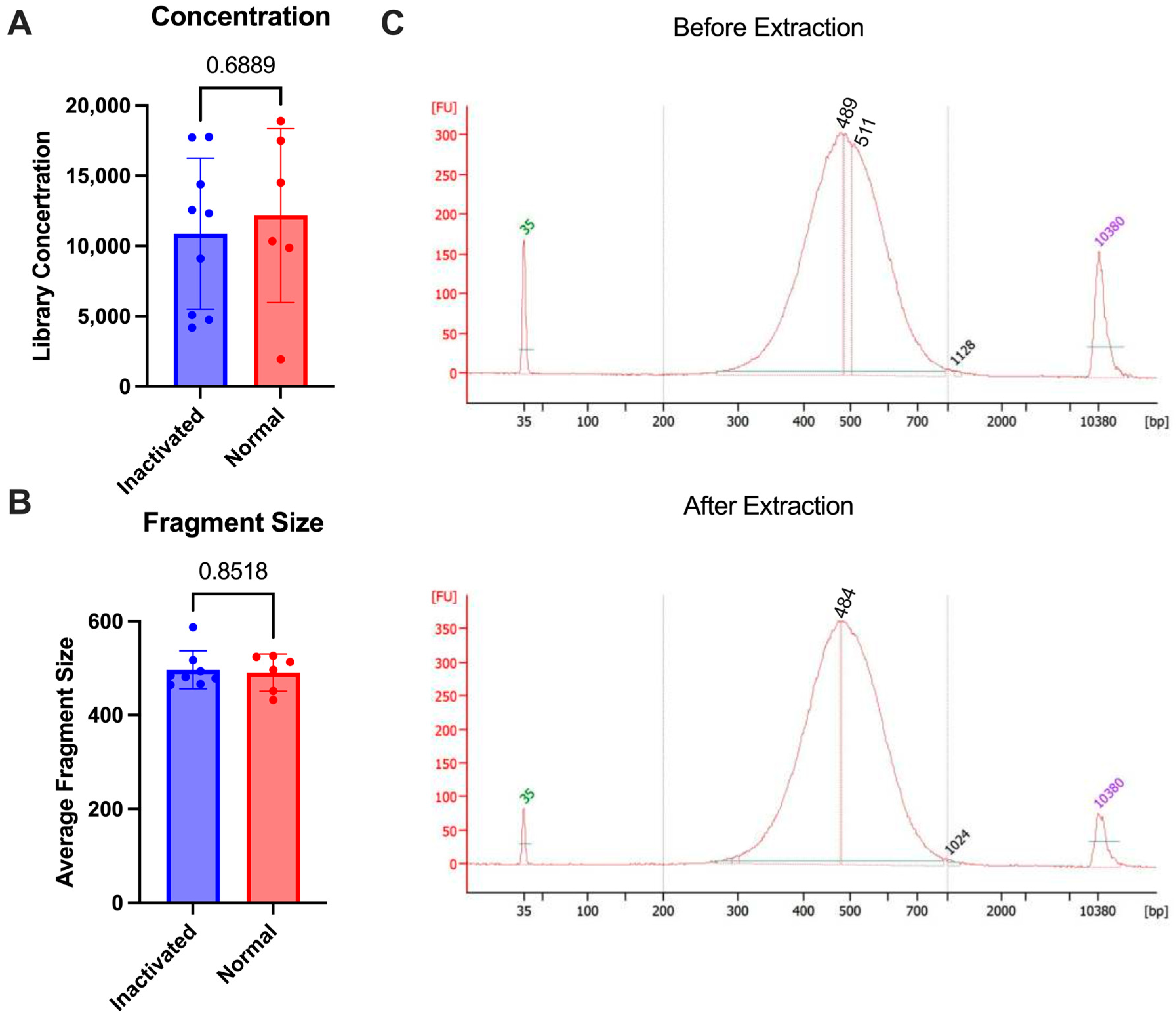
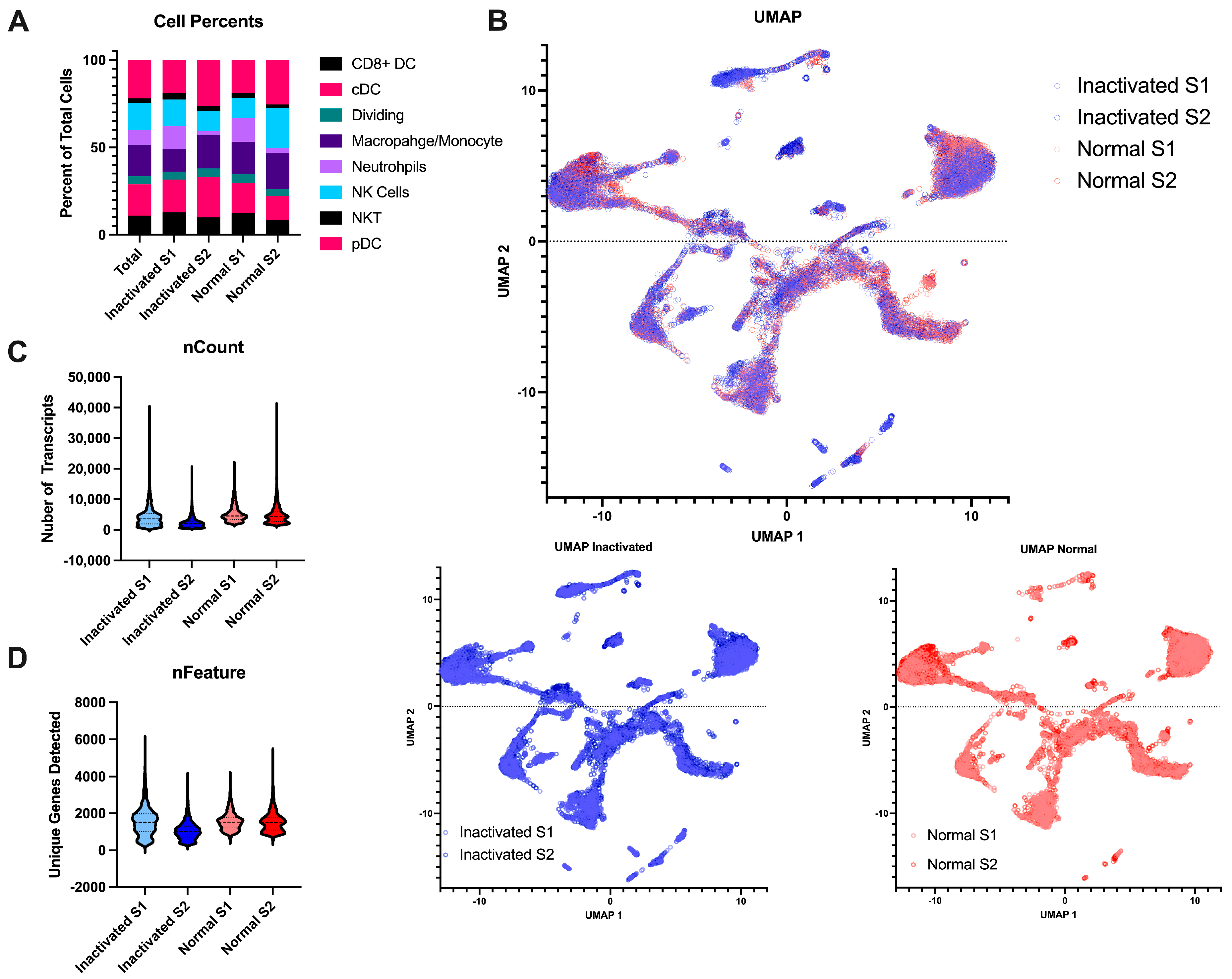
3. Results
3.1. Sample Stablilty and Inactivation Procedure
3.2. Preservation of Sample Quality Following Inactivation
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Speranza, E. Understanding virus–host interactions in tissues. Nat. Microbiol. 2023, 8, 1397–1407. [Google Scholar] [CrossRef] [PubMed]
- Meechan, P.J.; Potts, J. Biosafety in Microbiological and Biomedical Laboratories; No. (CDC) 300859; HHS Publication: Atlanta, GA, USA, 2020.
- Prioritizing Diseases for Research and Development in Emergency Contexts. 2021. Available online: https://www.who.int/activities/prioritizing-diseases-for-research-and-development-in-emergency-contexts/prioritizing-diseases-for-research-and-development-in-emergency-contexts (accessed on 28 November 2023).
- HHS and USDA Select Agents and Toxins. 2023. Available online: https://www.selectagents.gov/sat/list.htm (accessed on 28 November 2023).
- CDC. Guidance on the Inactivation or Removal of Select Agents and Toxins for Future Use; 7 CFR Part 331, 9 CFR Part 121.3, 42 CFR Part 73.3; CDC: Atlanta, GA, USA, 2018.
- Haddock, E.; Feldmann, F.; Shupert, W.L.; Feldmann, H. Inactivation of SARS-CoV-2 Laboratory Specimens. Am. J. Trop. Med. Hyg. 2021, 104, 2195–2198. [Google Scholar] [CrossRef]
- Feldmann, F.; Shupert, W.L.; Haddock, E.; Twardoski, B.; Feldmann, H. Gamma Irradiation as an Effective Method for Inactivation of Emerging Viral Pathogens. Am. J. Trop. Med. Hyg. 2019, 100, 1275–1277. [Google Scholar] [CrossRef] [PubMed]
- Haddock, E.; Feldmann, F.; Feldmann, H. Effective Chemical Inactivation of Ebola Virus. Emerg. Infect. Dis. 2016, 22, 1292–1294. [Google Scholar] [CrossRef] [PubMed]
- Triant, D.A.; Whitehead, A. Simultaneous Extraction of High-Quality RNA and DNA from Small Tissue Samples. J. Hered. 2008, 100, 246–250. [Google Scholar] [CrossRef]
- Hao, Y.; Hao, S.; Andersen-Nissen, E.; Mauck, W.M.; Zheng, S.; Butler, A.; Lee, M.J.; Wilk, A.J.; Darby, C.; Zager, M.; et al. Integrated analysis of multimodal single-cell data. Cell 2021, 184, 3573–3587.e29. [Google Scholar] [CrossRef]
- Speranza, E.; Williamson, B.N.; Feldmann, F.; Sturdevant, G.L.; Pérez-Pérez, L.; Meade-White, K.; Smith, B.J.; Lovaglio, J.; Martens, C.; Munster, V.J.; et al. Single-cell RNA sequencing reveals SARS-CoV-2 infection dynamics in lungs of African green monkeys. Sci. Transl. Med. 2021, 13, eabe8146. [Google Scholar] [CrossRef]
- Roberts, L.M.; Schwarz, B.; Speranza, E.; Leighton, I.; Wehrly, T.; Best, S.; Bosio, C.M. Pulmonary infection induces persistent, pathogen-specific lipidomic changes influencing trained immunity. iScience 2021, 24, 103025. [Google Scholar] [CrossRef]
- Speranza, E.; Purushotham, J.N.; Port, J.R.; Schwarz, B.; Flagg, M.; Williamson, B.N.; Feldmann, F.; Singh, M.; Pérez-Pérez, L.; Sturdevant, G.L.; et al. Age-related differences in immune dynamics during SARS-CoV-2 infection in rhesus macaques. Life Sci. Alliance 2022, 5, e202101314. [Google Scholar] [CrossRef]
- Heng, T.S.; Painter, M.W.; The Immunological Genome Project Consortium; Elpek, K.; Lukacs-Kornek, V.; Mauermann, N.; Turley, S.J.; Koller, D.; Kim, F.S.; Wagers, A.J.; et al. The Immunological Genome Project: Networks of gene expression in immune cells. Nat. Immunol. 2008, 9, 1091–1094. [Google Scholar] [CrossRef]
- Rosenberg, A.B.; Roco, C.M.; Muscat, R.A.; Kuchina, A.; Sample, P.; Yao, Z.; Graybuck, L.T.; Peeler, D.J.; Mukherjee, S.; Chen, W.; et al. Single-cell profiling of the developing mouse brain and spinal cord with split-pool barcoding. Science 2018, 360, 176–182. [Google Scholar] [CrossRef]
- Clark, I.C.; Fontanez, K.M.; Meltzer, R.H.; Xue, Y.; Hayford, C.; May-Zhang, A.; D’Amato, C.; Osman, A.; Zhang, J.Q.; Hettige, P.; et al. Microfluidics-free single-cell genomics with templated emulsification. Nat. Biotechnol. 2023, 41, 1557–1566. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Wang, R.; Zhou, Y.; Fei, L.; Sun, H.; Lai, S.; Saadatpour, A.; Zhou, Z.; Chen, H.; Ye, F.; et al. Mapping the Mouse Cell Atlas by Microwell-Seq. Cell 2018, 173, 1307. [Google Scholar] [CrossRef] [PubMed]
- Gierahn, T.M.; Wadsworth, M.H.; Hughes, T.K.; Bryson, B.D.; Butler, A.; Satija, R.; Fortune, S.; Love, J.C.; Shalek, A.K. Seq-Well: Portable, low-cost RNA sequencing of single cells at high throughput. Nat. Methods 2017, 14, 395–398. [Google Scholar] [CrossRef]
- Blow, J.A.; Dohm, D.J.; Negley, D.L.; Mores, C.N. Virus inactivation by nucleic acid extraction reagents. J. Virol. Methods 2004, 119, 195–198. [Google Scholar] [CrossRef]
- Avelin, V.; Sissonen, S.; Julkunen, I.; Oesterlund, P. Inactivation efficacy of H5N1 avian influenza virus by commonly used sample preparation reagents for safe laboratory practices. J. Virol. Methods 2022, 304, 114527. [Google Scholar] [CrossRef]
- Stoeckius, M.; Hafemeister, C.; Stephenson, W.; Houck-Loomis, B.; Chattopadhyay, P.K.; Swerdlow, H.; Satija, R.; Smibert, P. Simultaneous epitope and transcriptome measurement in single cells. Nat. Methods 2017, 14, 865–868. [Google Scholar] [CrossRef]
- Buenrostro, J.D.; Wu, B.; Litzenburger, U.M.; Ruff, D.; Gonzales, M.L.; Snyder, M.P.; Chang, H.Y.; Greenleaf, W.J. Single-cell chromatin accessibility reveals principles of regulatory variation. Nature 2015, 523, 486–490. [Google Scholar] [CrossRef]
- Grosselin, K.; Durand, A.; Marsolier, J.; Poitou, A.; Marangoni, E.; Nemati, F.; Dahmani, A.; Lameiras, S.; Reyal, F.; Frenoy, O. High-throughput single-cell ChIP-seq identifies heterogeneity of chromatin states in breast cancer. Nat. Genet. 2019, 51, 1060–1066. [Google Scholar] [CrossRef]
- Hume, A.J.; Olejnik, J.; White, M.R.; Huang, J.; Turcinovic, J.; Heiden, B.; Bawa, P.S.; Williams, C.J.; Gorham, N.G.; Alekseyev, Y.O.; et al. Heat Inactivation of Nipah Virus for Downstream Single-Cell RNA Sequencing Does Not Interfere with Sample Quality. Pathogens 2024, 13, 62. [Google Scholar] [CrossRef] [PubMed]
- Kotliar, D.; Lin, A.E.; Logue, J.; Hughes, T.K.; Khoury, N.M.; Raju, S.S.; Wadsworth, M.H.; Chen, H.; Kurtz, J.R.; Dighero-Kemp, B. Single-Cell Profiling of Ebola Virus Disease In Vivo Reveals Viral and Host Dynamics. Cell 2020, 183, 1383–1401.e19. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| p_val | avg_log2FC | pct.1 | pct.2 | p_val_adj | |
|---|---|---|---|---|---|
| Jchain | 0 | 1.625376 | 0.266 | 0.528 | 0 |
| S100a9 | 7.85 × 10−141 | 1.146031 | 0.281 | 0.25 | 1.57 × 10−137 |
| Msrb1 | 9.21 × 10−140 | 1.057364 | 0.77 | 0.991 | 1.84 × 10−136 |
| Cebpb | 6.42 × 10−110 | 1.066698 | 0.775 | 0.981 | 1.28 × 10−106 |
| Ccl21a | 8.91 × 10−97 | 1.070559 | 0.239 | 0.314 | 1.78 × 10−93 |
| S100a8 | 3.22 × 10−94 | 1.03576 | 0.289 | 0.294 | 6.44 × 10−91 |
| Ptgs2 | 9.34 × 10−74 | 1.368172 | 0.448 | 0.573 | 1.87 × 10−70 |
| Mzb1 | 4.30 × 10−72 | 1.290382 | 0.827 | 0.936 | 8.60 × 10−69 |
| Slpi | 3.91 × 10−57 | 1.379325 | 0.463 | 0.382 | 7.82 × 10−54 |
| Cpa3 | 1.34 × 10−54 | 1.205746 | 0.286 | 0.36 | 2.67 × 10−51 |
| Il1r2 | 1.58 × 10−51 | 1.16859 | 0.519 | 0.684 | 3.16 × 10−48 |
| Dcpp1 | 1.75 × 10−31 | 1.194771 | 0.718 | 0.996 | 3.51 × 10−28 |
| Clu | 4.14 × 10−26 | 1.074544 | 0.329 | 0.356 | 8.27 × 10−23 |
| Mcpt4 | 4.05 × 10−12 | 1.913468 | 0.396 | 0.424 | 8.09 × 10−9 |
| Ifitm1 | 2.04 × 10−11 | 1.125462 | 0.394 | 0.338 | 4.07 × 10−8 |
| Gsn | 4.04 × 10−10 | 1.079454 | 0.771 | 0.66 | 8.08 × 10−7 |
| Cma1 | 1.32 × 10−7 | 1.404933 | 0.443 | 0.42 | 2.65 × 10−4 |
| Il1b | 4.18 × 10−7 | 1.562076 | 0.552 | 0.593 | 8.36 × 10−4 |
| Cxcl2 | 5.47 × 10−6 | 2.385164 | 0.329 | 0.384 | 1.09 × 10−2 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sturdevant, G.L.; Meade-White, K.D.; Best, S.M.; Speranza, E. Preservation of scRNA-Seq Libraries Using Existing Inactivation Protocols. Pathogens 2024, 13, 167. https://doi.org/10.3390/pathogens13020167
Sturdevant GL, Meade-White KD, Best SM, Speranza E. Preservation of scRNA-Seq Libraries Using Existing Inactivation Protocols. Pathogens. 2024; 13(2):167. https://doi.org/10.3390/pathogens13020167
Chicago/Turabian StyleSturdevant, Gail L., Kimberly D. Meade-White, Sonja M. Best, and Emily Speranza. 2024. "Preservation of scRNA-Seq Libraries Using Existing Inactivation Protocols" Pathogens 13, no. 2: 167. https://doi.org/10.3390/pathogens13020167
APA StyleSturdevant, G. L., Meade-White, K. D., Best, S. M., & Speranza, E. (2024). Preservation of scRNA-Seq Libraries Using Existing Inactivation Protocols. Pathogens, 13(2), 167. https://doi.org/10.3390/pathogens13020167