Functional Analysis of Promoters, mRNA Cleavage, and mRNA Secondary Structure on esxB-esxA in Mycolicibacterium smegmatis


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Strains and Culture Conditions
2.2. Flow Cytometry
2.3. RNA Extraction
2.4. cDNA Synthesis
2.5. Quantitative PCR
2.6. Rapid Amplification of cDNA Ends (RACE)
2.7. mRNA Half-Life Measurement
2.8. Statistics
3. Results
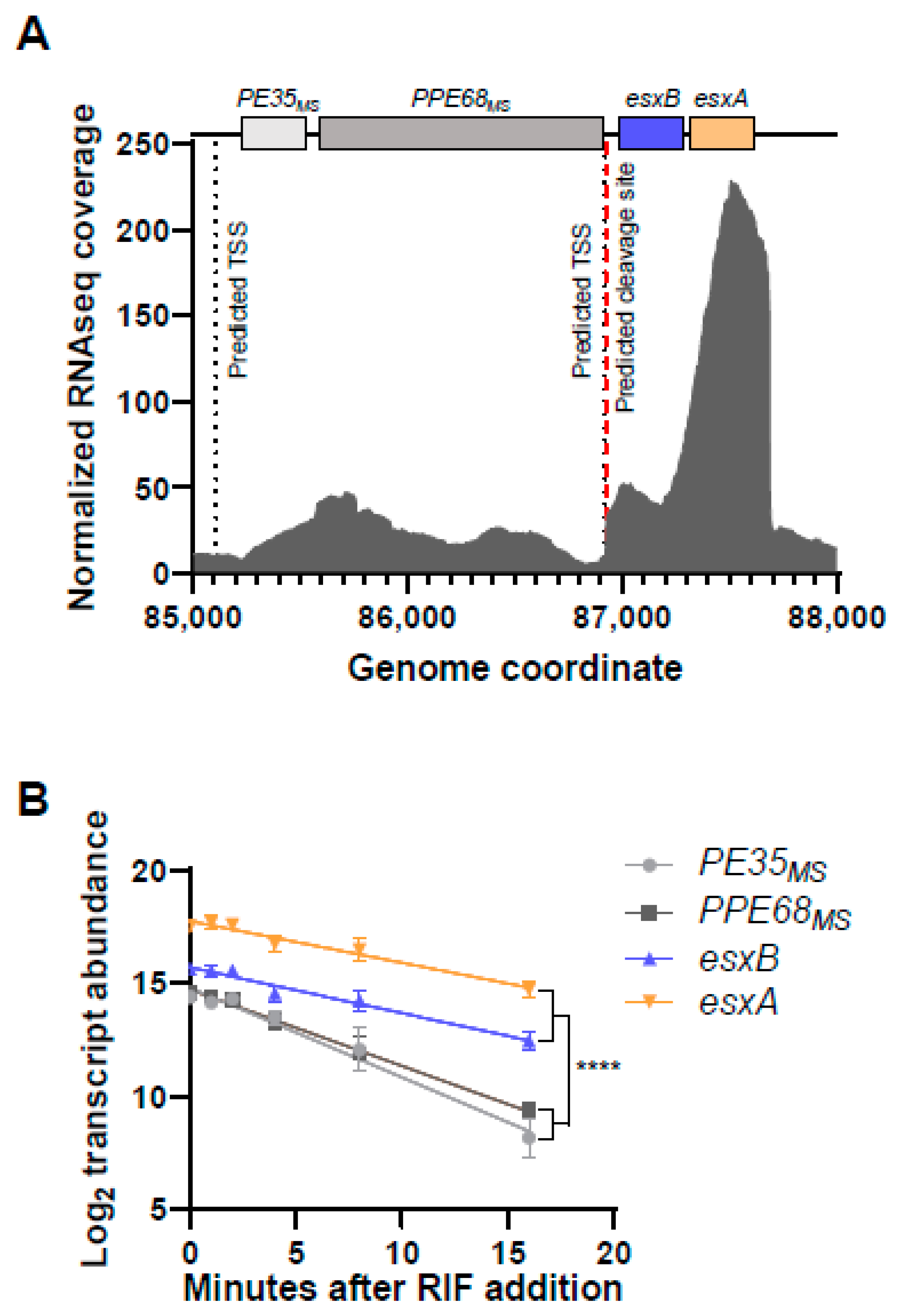
3.1. The PE35ms-PPE68ms-esxB-esxA Locus in M. smegmatis Is Bisected by a Cleavage Site, and the esxB-esxA Portion Is More Abundant and More Stable than the Upstream Portion
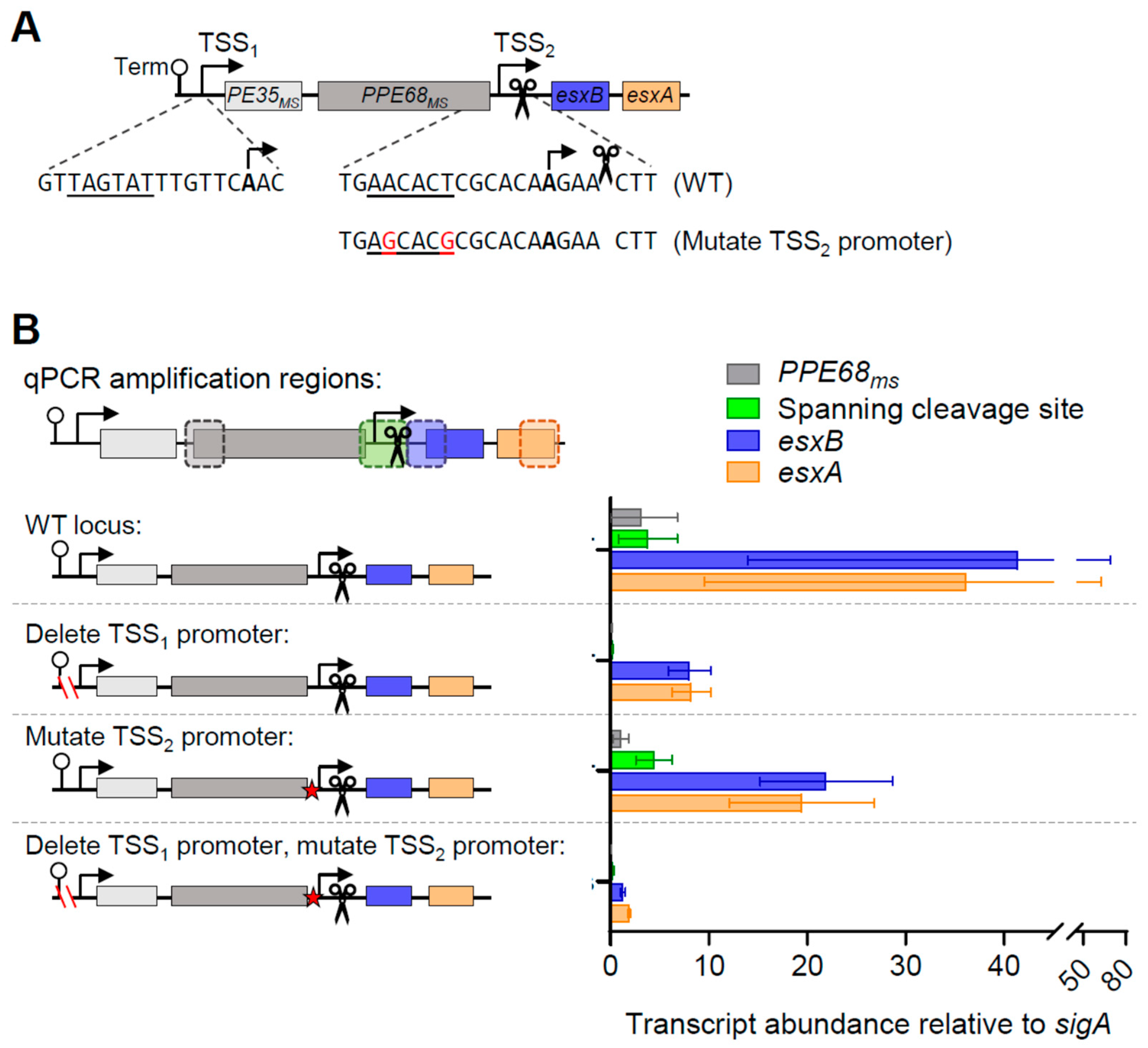
3.2. At Least Two Promoters Contribute to esxB and esxA Expression in M. smegmatis
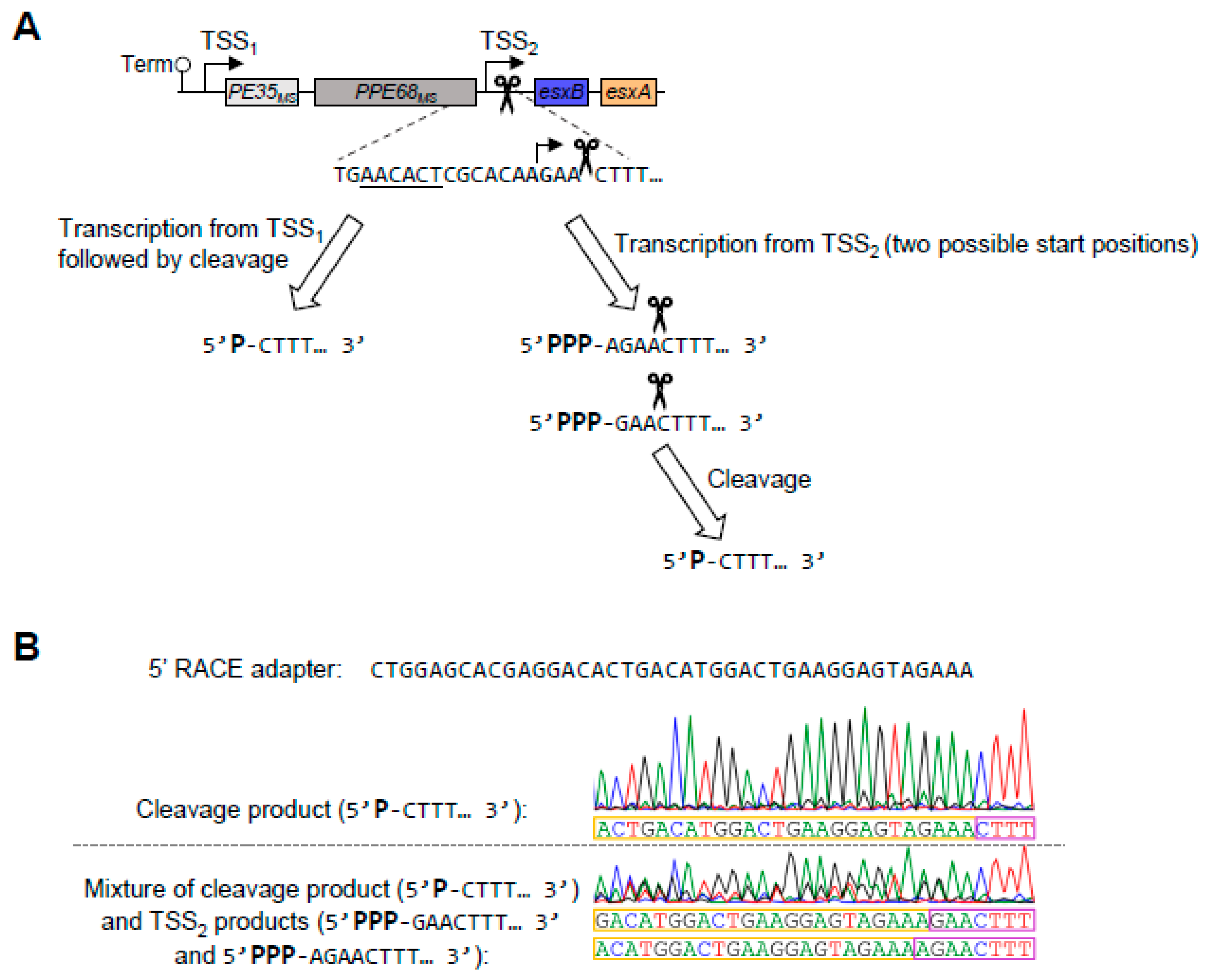
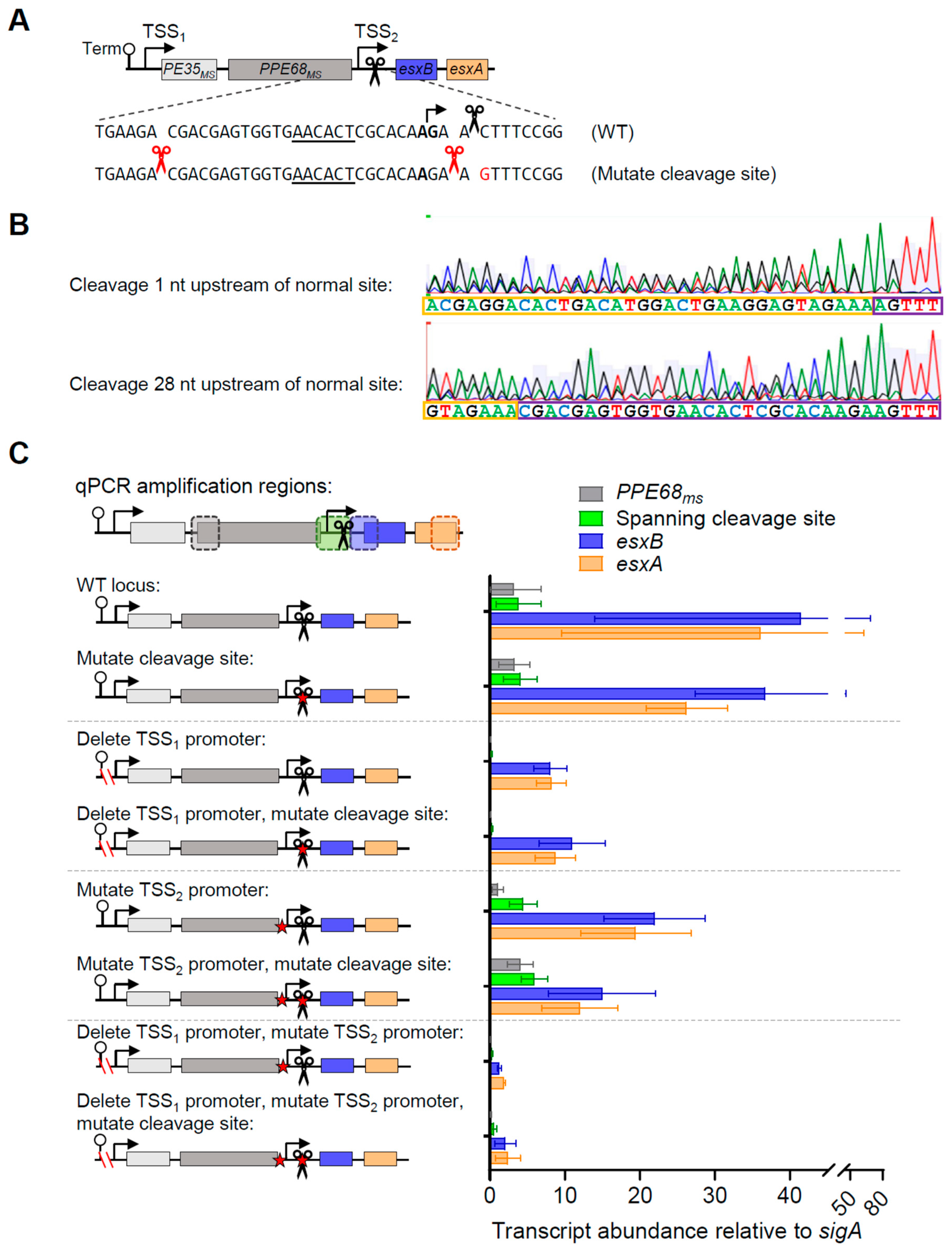
3.3. mRNA Cleavage Can Be Reprogramed by a Single Point Mutation but Is Not Required for Normal Levels of esxB and esxA Expression in M. smegmatis
3.4. The Stability of the M. smegmatis esxB-esxA Transcript Is Not Dependent upon 5′ End Secondary Structure
3.5. Secondary Structure near the 5′ Ends of 5′ UTRs Is Not Generally Correlated with mRNA Stability in M. smegmatis
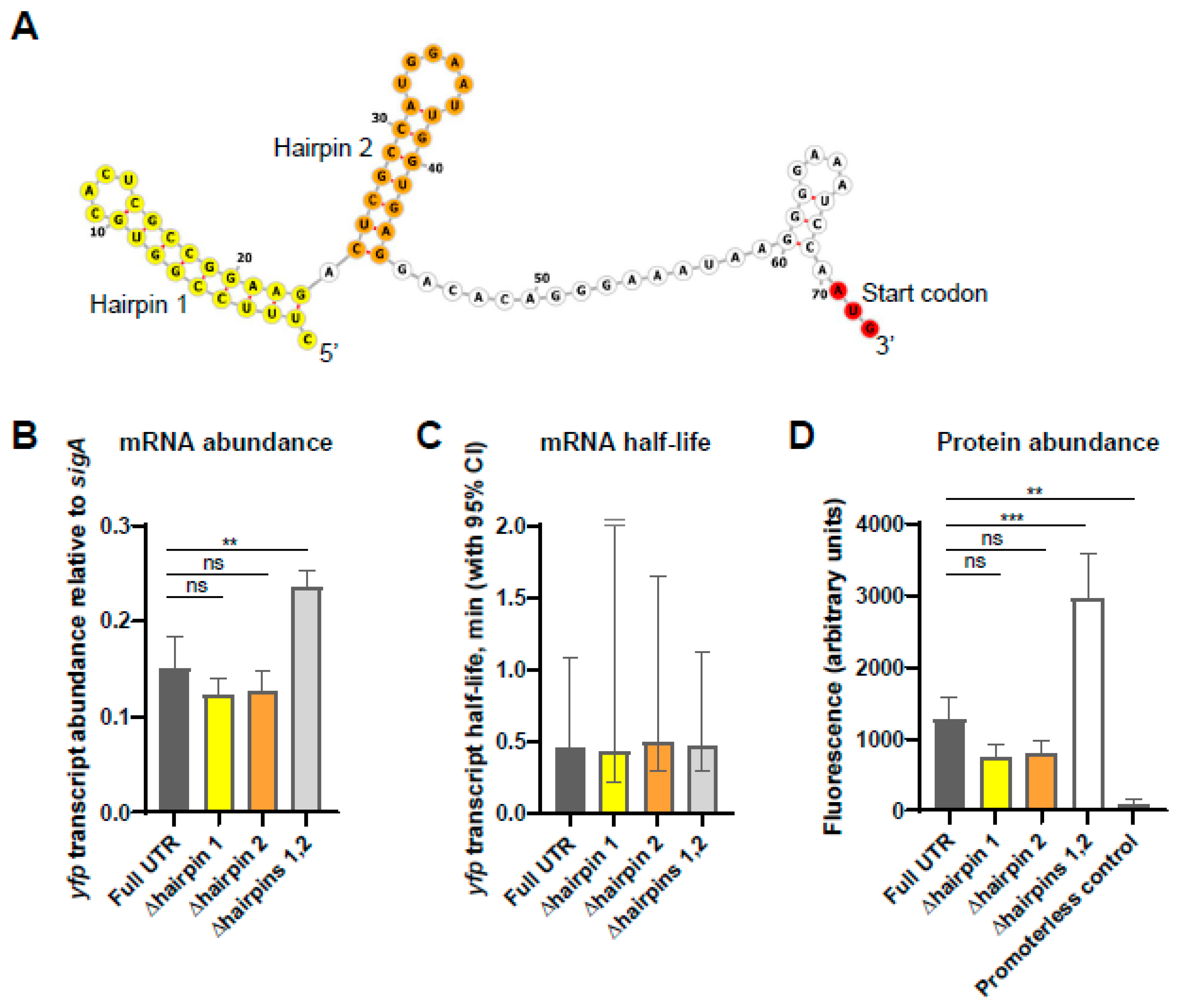

4. Discussion and Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- World Health Organization. Global Tuberculosis Report; WHO: Geneva, Switzerland, 2023. [Google Scholar]
- Bosserman, R.E.; Champion, P.A. Esx Systems and the Mycobacterial Cell Envelope: What’s the Connection? J. Bacteriol. 2017, 199, e00131-17. [Google Scholar] [CrossRef] [PubMed]
- Passos, B.B.S.; Araujo-Pereira, M.; Vinhaes, C.L.; Amaral, E.P.; Andrade, B.B. The role of ESAT-6 in tuberculosis immunopathology. Front. Immunol. 2024, 15, 1383098. [Google Scholar] [CrossRef]
- Parbhoo, T.; Mouton, J.M.; Sampson, S.L. Phenotypic adaptation of Mycobacterium tuberculosis to host-associated stressors that induce persister formation. Front. Cell. Infect. Microbiol. 2022, 12, 956607. [Google Scholar] [CrossRef] [PubMed]
- Rivera-Calzada, A.; Famelis, N.; Llorca, O.; Geibel, S. Type VII secretion systems: Structure, functions and transport models. Nat. Rev. Microbiol. 2021, 19, 567–584. [Google Scholar] [CrossRef]
- Coros, A.; Callahan, B.; Battaglioli, E.; Derbyshire, K.M. The specialized secretory apparatus ESX-1 is essential for DNA transfer in Mycobacterium smegmatis. Mol. Microbiol. 2008, 69, 794–808. [Google Scholar] [CrossRef]
- Flint, J.L.; Kowalski, J.C.; Karnati, P.K.; Derbyshire, K.M. The RD1 virulence locus of Mycobacterium tuberculosis regulates DNA transfer in Mycobacterium smegmatis. Proc. Natl. Acad. Sci. USA 2004, 101, 12598–12603. [Google Scholar] [CrossRef]
- Mahairas, G.G.; Sabo, P.J.; Hickey, M.J.; Singh, D.C.; Stover, C.K. Molecular analysis of genetic differences between Mycobacterium bovis BCG and virulent M. bovis. J. Bacteriol. 1996, 178, 1274–1282. [Google Scholar] [CrossRef] [PubMed]
- Behr, M.A.; Wilson, M.A.; Gill, W.P.; Salamon, H.; Schoolnik, G.K.; Rane, S.; Small, P.M. Comparative genomics of BCG vaccines by whole-genome DNA microarray. Science 1999, 284, 1520–1523. [Google Scholar] [CrossRef]
- Fortune, S.M.; Jaeger, A.; Sarracino, D.A.; Chase, M.R.; Sassetti, C.M.; Sherman, D.R.; Bloom, B.R.; Rubin, E.J. Mutually dependent secretion of proteins required for mycobacterial virulence. Proc. Natl. Acad. Sci. USA 2005, 102, 10676–10681. [Google Scholar] [CrossRef]
- Minch, K.J.; Rustad, T.R.; Peterson, E.J.R.; Winkler, J.; Reiss, D.J.; Ma, S.; Hickey, M.; Brabant, W.; Morrison, B.; Turkarslan, S.; et al. The DNA-binding network of Mycobacterium tuberculosis. Nat. Commun. 2015, 6, 5829. [Google Scholar] [CrossRef]
- Solans, L.; Aguiló, N.; Samper, S.; Pawlik, A.; Frigui, W.; Martín, C.; Brosch, R.; Gonzalo-Asensio, J. A specific polymorphism in Mycobacterium tuberculosis H37Rv causes differential ESAT-6 expression and identifies WhiB6 as a novel ESX-1 component. Infect. Immun. 2014, 82, 3446–3456. [Google Scholar] [CrossRef] [PubMed]
- Cortes, T.; Schubert, O.T.; Rose, G.; Arnvig, K.B.; Comas, I.; Aebersold, R.; Young, D.B. Genome-wide Mapping of Transcriptional Start Sites Defines an Extensive Leaderless Transcriptome in Mycobacterium tuberculosis. Cell Rep. 2013, 5, 1121–1131. [Google Scholar] [CrossRef] [PubMed]
- Shell, S.S.; Wang, J.; Lapierre, P.; Mir, M.; Chase, M.R.; Pyle, M.M.; Gawande, R.; Ahmad, R.; Sarracino, D.A.; Ioerger, T.R.; et al. Leaderless Transcripts and Small Proteins Are Common Features of the Mycobacterial Translational Landscape. PLoS Genet. 2015, 11, e1005641. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Sun, H.; Rapiejko, A.R.; Vargas-Blanco, D.A.; Martini, M.C.; Chase, M.R.; Joubran, S.R.; Davis, A.B.; Dainis, J.P.; Kelly, J.M.; et al. Mycobacterial RNase E cleaves with a distinct sequence preference and controls the degradation rates of most Mycolicibacterium smegmatis mRNAs. J. Biol. Chem. 2023, 299, 105312. [Google Scholar] [CrossRef]
- Dinan, A.M.; Tong, P.; Lohan, A.J.; Conlon, K.M.; Miranda-CasoLuengo, A.A.; Malone, K.M.; Gordon, S.V.; Loftus, B.J. Relaxed Selection Drives a Noisy Noncoding Transcriptome in Members of the Mycobacterium tuberculosis Complex. mBio 2014, 5, e01169-14. [Google Scholar] [CrossRef]
- Berthet, F.X.; Rasmussen, P.B.; Rosenkrands, I.; Andersen, P.; Gicquel, B. A Mycobacterium tuberculosis operon encoding ESAT = 6 and a novel low-molecular-mass culture filtrate protein (CFP-10). Microbiology 1998, 144, 3195–3203. [Google Scholar] [CrossRef]
- Gray, T.A.; Krywy, J.A.; Harold, J.; Palumbo, M.J.; Derbyshire, K.M. Distributive conjugal transfer in mycobacteria generates progeny with meiotic-like genome-wide mosaicism, allowing mapping of a mating identity locus. PLoS Biol. 2013, 11, e1001602. [Google Scholar] [CrossRef]
- Gray, T.A.; Clark, R.R.; Boucher, N.; Lapierre, P.; Smith, C.; Derbyshire, K.M. Intercellular communication and conjugation are mediated by ESX secretion systems in mycobacteria. Science 2016, 354, 347–350. [Google Scholar] [CrossRef]
- Martini, M.C.; Zhou, Y.; Sun, H.; Shell, S.S. Defining the Transcriptional and Post-transcriptional Landscapes of Mycobacterium smegmatis in Aerobic Growth and Hypoxia. Front. Microbiol. 2019, 10, 591. [Google Scholar] [CrossRef]
- Czyz, A.; Mooney, R.A.; Iaconi, A.; Landick, R. Mycobacterial RNA Polymerase Requires a U-Tract at Intrinsic Terminators and Is Aided by NusG at Suboptimal Terminators. mBio 2014, 5, e00931. [Google Scholar] [CrossRef]
- Ehrt, S.; Guo, X.V.; Hickey, C.M.; Ryou, M.; Monteleone, M.; Riley, L.W.; Schnappinger, D. Controlling gene expression in mycobacteria with anhydrotetracycline and Tet repressor. Nucleic Acids Res. 2005, 33, e21. [Google Scholar] [CrossRef]
- Nguyen, T.G.; Vargas-Blanco, D.A.; Roberts, L.A.; Shell, S.S. The Impact of Leadered and Leaderless Gene Structures on Translation Efficiency, Transcript Stability, and Predicted Transcription Rates in Mycobacterium smegmatis. J. Bacteriol. 2020, 202, e00746-19. [Google Scholar] [CrossRef] [PubMed]
- Meinken, C.; Blencke, H.-M.; Ludwig, H.; Stülke, J. Expression of the glycolytic gapA operon in Bacillus subtilis: Differential syntheses of proteins encoded by the operon. Microbiology 2003, 149, 751–761. [Google Scholar] [CrossRef] [PubMed]
- Bouvet, P.; Belasco, J.G. Control of RNase E-mediated RNA degradation by 5′-terminal base pairing in E. coli. Nature 1992, 360, 488–491. [Google Scholar] [CrossRef] [PubMed]
- Emory, S.A.; Bouvet, P.; Belasco, J.G. A 5′-terminal stem-loop structure can stabilize mRNA in Escherichia coli. Genes Dev. 1992, 6, 135–148. [Google Scholar] [CrossRef] [PubMed]
- Heck, C.; Rothfuchs, R.; Jäger, A.; Rauhut, R.; Klug, G. Effect of the pufQ-pufB intercistronic region on puf mRNA stability in Rhodobacter capsulatus. Mol. Microbiol. 1996, 20, 1165–1178. [Google Scholar] [CrossRef]
- Hambraeus, G.; Karhumaa, K.; Rutberg, B. A 5′ stem-loop and ribosome binding but not translation are important for the stability of Bacillus subtilis aprE leader mRNA. Microbiology 2002, 148, 1795–1803. [Google Scholar] [CrossRef]
- Amilon, K.R.; Letley, D.P.; Winter, J.A.; Robinson, K.; Atherton, J.C. Expression of the Helicobacter pylori virulence factor vacuolating cytotoxin A (vacA) is influenced by a potential stem-loop structure in the 5′ untranslated region of the transcript. Mol. Microbiol. 2015, 98, 831–846. [Google Scholar] [CrossRef]
- Carrier, T.A.; Keasling, J.D. Library of synthetic 5′ secondary structures to manipulate mRNA stability in Escherichia coli. Biotechnol. Prog. 1999, 15, 58–64. [Google Scholar] [CrossRef]
- Ching, C.; Gozzi, K.; Heinemann, B.; Chai, Y.; Godoy, V.G. RNA-Mediated cis Regulation in Acinetobacter baumannii Modulates Stress-Induced Phenotypic Variation. J. Bacteriol. 2017, 199, e00799-16. [Google Scholar] [CrossRef]
- Sharp, J.S.; Bechhofer, D.H. Effect of 5′-proximal elements on decay of a model mRNA in Bacillus subtilis. Mol. Microbiol. 2005, 57, 484–495. [Google Scholar] [CrossRef] [PubMed]
- Arnold, T.E.; Yu, J.; Belasco, J.G. mRNA stabilization by the ompA 5′ untranslated region: Two protective elements hinder distinct pathways for mRNA degradation. RNA 1998, 4, 319–330. [Google Scholar] [PubMed]
- Sun, H.; Vargas-Blanco, D.A.; Zhou, Y.; Masiello, C.S.; Kelly, J.M.; Moy, J.K.; Korkin, D.; Shell, S.S. Diverse intrinsic properties shape transcript stability and stabilization in Mycolicibacterium smegmatis. NAR Genom. Bioinform. 2024, 6, lqae147. [Google Scholar] [CrossRef] [PubMed]
- Raghavan, S.; Manzanillo, P.; Chan, K.; Dovey, C.; Cox, J.S. Secreted transcription factor controls Mycobacterium tuberculosis virulence. Nature 2008, 454, 717–721. [Google Scholar] [CrossRef]
- Anil Kumar, V.; Goyal, R.; Bansal, R.; Singh, N.; Sevalkar, R.R.; Kumar, A.; Sarkar, D. EspR-dependent ESAT-6 secretion of Mycobacterium tuberculosis requires the presence of virulence regulator PhoP. J. Biol. Chem. 2016, 291, 19018–19030. [Google Scholar] [CrossRef]
- Bosserman, R.E.; Nguyen, T.T.; Sanchez, K.G.; Chirakos, A.E.; Ferrell, M.J.; Thompson, C.R.; Champion, M.M.; Abramovitch, R.B.; Champion, P.A. WhiB6 regulation of ESX-1 gene expression is controlled by a negative feedback loop in Mycobacterium marinum. Proc. Natl. Acad. Sci. USA 2017, 114, E10772–E10781. [Google Scholar] [CrossRef]
- Abdallah, A.M.; Weerdenburg, E.M.; Guan, Q.; Ummels, R.; Borggreve, S.; Adroub, S.A.; Malas, T.B.; Naeem, R.; Zhang, H.; Otto, T.D.; et al. Integrated transcriptomic and proteomic analysis of pathogenic mycobacteria and their esx-1 mutants reveal secretion-dependent regulation of ESX-1 substrates and WhiB6 as a transcriptional regulator. PLoS ONE 2019, 14, e0211003. [Google Scholar] [CrossRef]
- Sanchez, K.G.; Ferrell, M.J.; Chirakos, A.E.; Nicholson, K.R.; Abramovitch, R.B.; Champion, M.M.; Champion, P.A. EspM Is a Conserved Transcription Factor That Regulates Gene Expression in Response to the ESX-1 System. mBio 2020, 11, e02807-19. [Google Scholar] [CrossRef]
- Nicholson, K.R.; Cronin, R.M.; Prest, R.J.; Menon, A.R.; Yang, Y.; Jennisch, M.K.; Champion, M.M.; Tobin, D.M.; Champion, P.A. The antagonistic transcription factors, EspM and EspN, regulate the ESX-1 secretion system in M. marinum. mBio 2024, 15, e0335723. [Google Scholar] [CrossRef]
- Kaberdin, V.R.; Walsh, A.P.; Jakobsen, T.; McDowall, K.J.; von Gabain, A. Enhanced cleavage of RNA mediated by an interaction between substrates and the arginine-rich domain of E. coli ribonuclease E. J. Mol. Biol. 2000, 301, 257–264. [Google Scholar] [CrossRef]
- Mackie, G.A. Ribonuclease E is a 5′-end-dependent endonuclease. Nature 1998, 395, 720–723. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Belasco, J.G. Catalytic activation of multimeric RNase E and RNase G by 5′-monophosphorylated RNA. Proc. Natl. Acad. Sci. USA 2004, 101, 9211–9216. [Google Scholar] [CrossRef] [PubMed]
- Zeller, M.-E.; Csanadi, A.; Miczak, A.; Rose, T.; Bizebard, T.; Kaberdin, V.R. Quaternary structure and biochemical properties of mycobacterial RNase E/G. Biochem. J. 2007, 403, 207. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Diwa, A.; Belasco, J.G. Regions of RNase E important for 5′-end-dependent RNA cleavage and autoregulated synthesis. J. Bacteriol. 2000, 182, 2468–2475. [Google Scholar] [CrossRef]
- Bandyra, K.J.; Wandzik, J.M.; Luisi, B.F. Substrate Recognition and Autoinhibition in the Central Ribonuclease RNase E. Mol. Cell 2018, 72, 275–285.E4. [Google Scholar] [CrossRef]
- Mathy, N.; Bénard, L.; Pellegrini, O.; Daou, R.; Wen, T. 5′-to-3′ Exoribonuclease Activity in Bacteria: Role of RNase J1 in rRNA Maturation and 5′ Stability of mRNA. Cell 2007, 129, 681–692. [Google Scholar] [CrossRef]
- Li de la Sierra-Gallay, I.; Zig, L.; Jamalli, A.; Putzer, H. Structural insights into the dual activity of RNase J. Nat. Struct. Mol. Biol. 2008, 15, 206–212. [Google Scholar] [CrossRef]
- Martini, M.C.; Hicks, N.D.; Xiao, J.; Alonso, M.N.; Barbier, T.; Sixsmith, J.; Fortune, S.M.; Shell, S.S. Loss of RNase J leads to multi-drug tolerance and accumulation of highly structured mRNA fragments in Mycobacterium tuberculosis. PLoS Pathog. 2022, 18, e1010705. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Peters, R.G.; Kelly, J.M.; Bibeau, S.; Zhou, Y.; Shell, S.S. Functional Analysis of Promoters, mRNA Cleavage, and mRNA Secondary Structure on esxB-esxA in Mycolicibacterium smegmatis. Pathogens 2024, 13, 1041. https://doi.org/10.3390/pathogens13121041
Peters RG, Kelly JM, Bibeau S, Zhou Y, Shell SS. Functional Analysis of Promoters, mRNA Cleavage, and mRNA Secondary Structure on esxB-esxA in Mycolicibacterium smegmatis. Pathogens. 2024; 13(12):1041. https://doi.org/10.3390/pathogens13121041
Chicago/Turabian StylePeters, Ryan G., Jessica M. Kelly, Sarah Bibeau, Ying Zhou, and Scarlet S. Shell. 2024. "Functional Analysis of Promoters, mRNA Cleavage, and mRNA Secondary Structure on esxB-esxA in Mycolicibacterium smegmatis" Pathogens 13, no. 12: 1041. https://doi.org/10.3390/pathogens13121041
APA StylePeters, R. G., Kelly, J. M., Bibeau, S., Zhou, Y., & Shell, S. S. (2024). Functional Analysis of Promoters, mRNA Cleavage, and mRNA Secondary Structure on esxB-esxA in Mycolicibacterium smegmatis. Pathogens, 13(12), 1041. https://doi.org/10.3390/pathogens13121041