
Estimating the Current Routes of Transmission in HIV-1 F1 Subtype Infected Persons in Romania: Differences Between Self-Reporting and Phylogenetic Data


Abstract
1. Introduction
2. Materials and Methods
2.1. Study Population
2.2. Cluster Analysis
2.3. Statistical Analysis
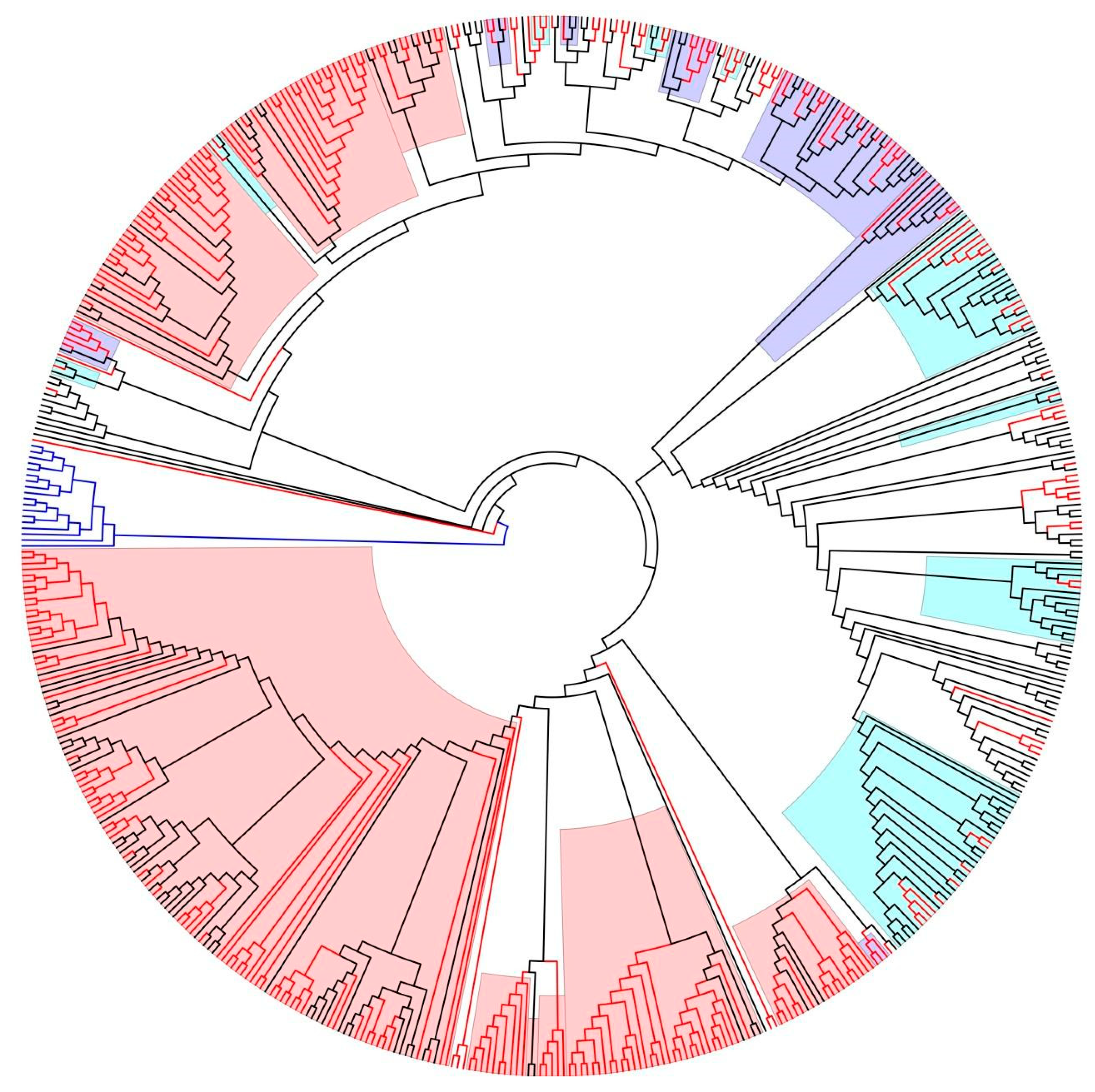
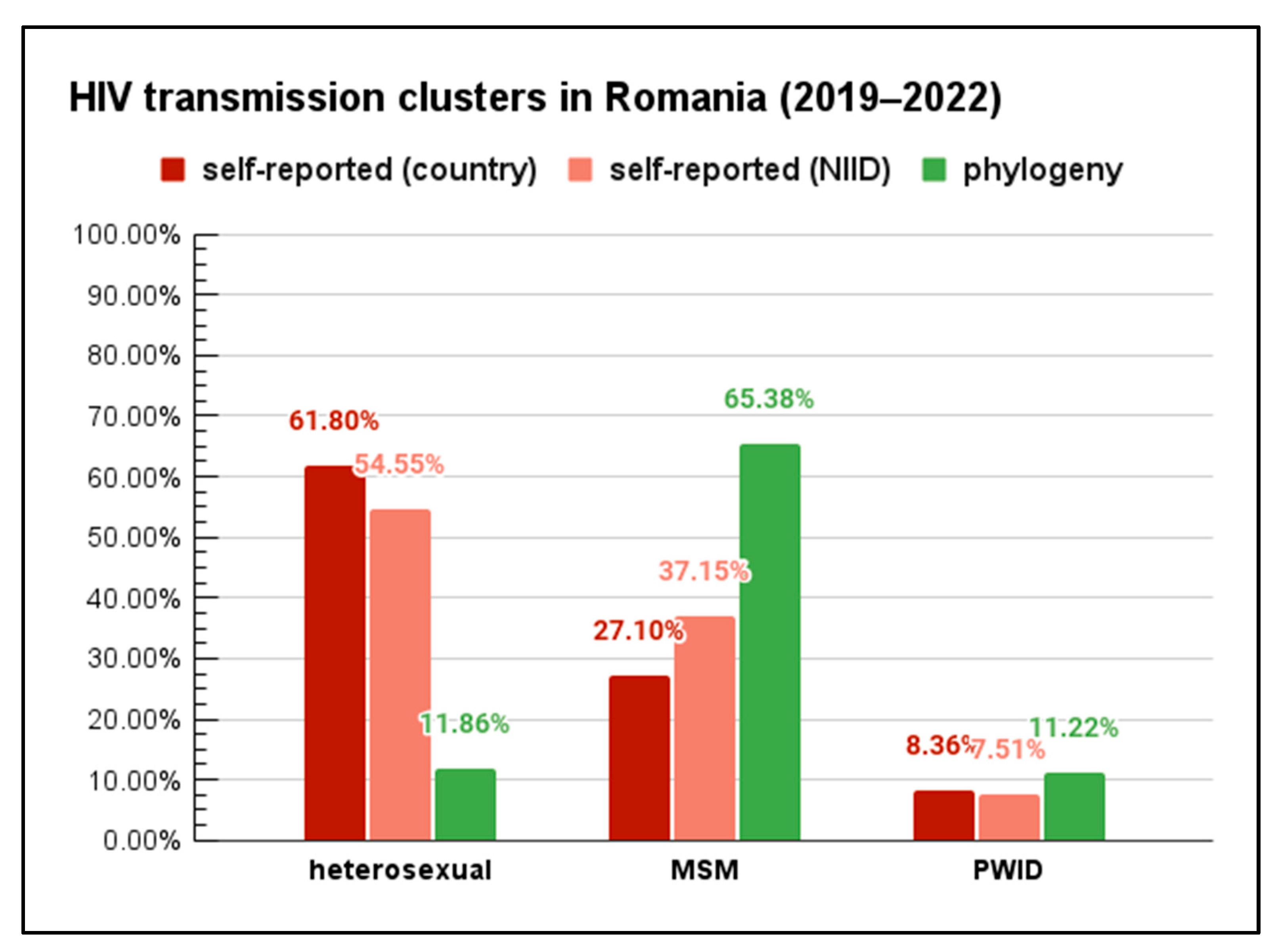
3. Results
- Seven clusters (comprising in total of 62 sequences, 37 from 2019 to 2022) of strains associated with heterosexual transmission;
- Nine clusters (271 in total, of which 204 sequences were from 2019 to 2022) of strains associated with MSM transmission;
- Nine clusters (89 sequences, of which 35 sequences were from 2019 to 2022) of strains affiliated with people who inject drugs (PWID).
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pimentel, V.; Pineda-Peña, A.; Sebastião, C.S.; de Paula, J.L.; Ahagon, C.M.; Pingarilho, M.; Martins, M.R.O.; Coelho, L.P.O.; Matsuda, E.M.; Alves, D.; et al. Dynamics and features of transmission clusters of HIV-1 subtypes in the state of São Paulo, Brazil. Front. Public Health 2024, 12, 1384512. [Google Scholar] [CrossRef]
- Junqueira, D.M.; de Matos Almeida, S.E. HIV-1 subtype B: Traces of a pandemic. Virology 2016, 495, 173–184. [Google Scholar] [CrossRef]
- Zhan, X.-Y.; Zha, G.-F.; He, Y. Evolutionary dissection of monkeypox virus: Positive Darwinian selection drives the adaptation of virus-host interaction proteins. Front. Cell. Infect. Microbiol. 2022, 12, 1083234. [Google Scholar] [CrossRef]
- Paraschiv, S.; Otelea, D.; Batan, I.; Baicus, C.; Magiorkinis, G.; Paraskevis, D. Molecular typing of the recently expanding subtype B HIV-1 epidemic in Romania: Evidence for local spread among MSMs in Bucharest area. Infect. Genet. Evol. 2012, 12, 1052–1057. [Google Scholar] [CrossRef]
- Department for Monitoring and Evaluation of HIV/AIDS Infection in Romania CNLAS. Available online: https://www.cnlas.ro/index.php (accessed on 7 October 2024).
- Apetrei, C.; Necula, A.; Holm-Hansen, C.; Loussert-Ajaka, I.; Pandrea, I.; Cozmei, C.; Streinu-Cercel, A.; Pascu, F.R.; Negut, E.; Molnar, G.; et al. HIV-1 diversity in Romania. AIDS 1998, 12, 1079–1085. [Google Scholar] [CrossRef]
- Niculescu, I.; Paraschiv, S.; Paraskevis, D.; Abagiu, A.; Batan, I.; Banica, L.; Otelea, D. Recent HIV-1 Outbreak Among Intravenous Drug Users in Romania: Evidence for Cocirculation of CRF14_BG and Subtype F1 Strains. AIDS Res. Hum. Retroviruses 2015, 31, 488–495. [Google Scholar] [CrossRef]
- Paraskevis, D.; Paraschiv, S.; Sypsa, V.; Nikolopoulos, G.; Tsiara, C.; Magiorkinis, G.; Psichogiou, M.; Flampouris, A.; Mardarescu, M.; Niculescu, I.; et al. Enhanced HIV-1 surveillance using molecular epidemiology to study and monitor HIV-1 outbreaks among intravenous drug users (IDUs) in Athens and Bucharest. Infect. Genet. Evol. 2015, 35, 109–121. [Google Scholar] [CrossRef]
- CNLAS Date Statistice. Available online: https://www.cnlas.ro/index.php/date-statistice (accessed on 30 September 2024).
- Frentz, D.; van de Vijver, D.; Abecasis, A.; Albert, J.; Hamouda, O.; Jørgensen, L.; Kücherer, C.; Struck, D.; Schmit, J.-C.; Vercauteren, J.; et al. Patterns of transmitted HIV drug resistance in Europe vary by risk group. PLoS ONE 2014, 9, e94495. [Google Scholar] [CrossRef]
- Ragonnet-Cronin, M.; Hodcroft, E.; Hué, S.; Fearnhill, E.; Delpech, V.; Brown, A.J.L.; Lycett, S. HIV Drug Resistance Database. Automated analysis of phylogenetic clusters. BMC Bioinform. 2013, 14, 317. [Google Scholar] [CrossRef]
- De Oliveira, T.; Deforche, K.; Cassol, S.; Salminen, M.; Paraskevis, D.; Seebregts, C.; Snoeck, J.; van Rensburg, E.J.; Wensing, A.M.J.; van de Vijver, D.A.; et al. An automated genotyping system for analysis of HIV-1 and other microbial sequences. Bioinformatics 2005, 21, 3797–3800. [Google Scholar] [CrossRef]
- Struck, D.; Lawyer, G.; Ternes, A.M.; Schmit, J.C.; Bercoff, D.P. COMET: Adaptive context-based modelling for ultrafast HIV-1 subtype identification. Nucleic Acids Res. 2014, 42, e144. [Google Scholar] [CrossRef] [PubMed]
- Los Alamos National Laboratory—HIV Sequence Database. Available online: https://www.hiv.lanl.gov/content/sequence/HIV/mainpage.html (accessed on 2 October 2024).
- Sayers, E.W.; Bolton, E.E.; Brister, J.R.; Canese, K.; Chan, J.; Comeau, D.C.; Connor, R.; Funk, K.; Kelly, C.; Kim, S.; et al. Database resources of the national center for biotechnology information. Nucleic Acids Res. 2022, 50, D20–D26. [Google Scholar] [CrossRef] [PubMed]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree 2—approximately maximum-likelihood trees for large alignments. PLoS ONE 2010, 5, e9490. [Google Scholar] [CrossRef] [PubMed]
- Rambaut, A. FigTree. 2018. Available online: http://tree.bio.ed.ac.uk (accessed on 2 October 2024).
- Bouckaert, R.; Vaughan, T.G.; Barido-Sottani, J.; Duchêne, S.; Fourment, M.; Gavryushkina, A.; Heled, J.; Jones, G.; Kühnert, D.; De Maio, N.; et al. BEAST 2.5: An advanced software platform for Bayesian evolutionary analysis. PLoS Comput. Biol. 2019, 15, e1006650. [Google Scholar] [CrossRef] [PubMed]
- Drummond, A.J.; Rambaut, A.; Shapiro, B.; Pybus, O.G. Bayesian coalescent inference of past population dynamics from molecular sequences. Mol. Biol. Evol. 2005, 22, 1185–1192. [Google Scholar] [CrossRef] [PubMed]
- Drummond, A.J.; Ho, S.Y.W.; Phillips, M.J.; Rambaut, A. Relaxed phylogenetics and dating with confidence. PLoS Biol. 2006, 4, e88. [Google Scholar] [CrossRef]
- Rambaut, A.; Drummond, A.J.; Xie, D.; Baele, G.; Suchard, M.A. Posterior summarization in Bayesian phylogenetics using Tracer 1.7. Syst. Biol. 2018, 67, 901–904. [Google Scholar] [CrossRef]
- Drummond, A.J.; Nicholls, G.K.; Rodrigo, A.G.; Solomon, W. Estimating mutation parameters, population history and genealogy simultaneously from temporally spaced sequence data. Genetics 2002, 161, 1307–1320. [Google Scholar] [CrossRef]
- HIV-Global. Available online: https://www.who.int/health-topics/hiv-aids/#tab=tab_1 (accessed on 2 October 2024).
- HIV and AIDS. Available online: https://www.who.int/news-room/fact-sheets/detail/hiv-aids (accessed on 2 October 2024).
- Paraschiv, S.; Banica, L.; Nicolae, I.; Niculescu, I.; Abagiu, A.; Jipa, R.; Pineda-Peña, A.-C.; Pingarilho, M.; Neaga, E.; Theys, K.; et al. Epidemic dispersion of HIV and HCV in a population of co-infected Romanian injecting drug users. PLoS ONE 2017, 12, e0185866. [Google Scholar] [CrossRef]
- Planinić, A.; Begovac, J.; Rokić, F.; Šimičić, P.; Oroz, M.; Jakovac, K.; Vugrek, O.; Zidovec-Lepej, S. Characterization of Human Immunodeficiency Virus-1 Transmission Clusters and Transmitted Drug-Resistant Mutations in Croatia from 2019 to 2022. Viruses 2023, 15, 2408. [Google Scholar] [CrossRef]
- Poon, A.F.Y. Impacts and shortcomings of genetic clustering methods for infectious disease outbreaks. Virus Evol. 2016, 2, vew031. [Google Scholar] [CrossRef] [PubMed]
- Junqueira, D.M.; Sibisi, Z.; Wilkinson, E.; de Oliveira, T. Factors influencing HIV-1 phylogenetic clustering. Curr. Opin. HIV AIDS 2019, 14, 161–172. [Google Scholar] [CrossRef] [PubMed]
- Magalis, B.R.; Riva, A.; Marini, S.; Salemi, M.; Prosperi, M. Novel insights on unraveling dynamics of transmission clusters in outbreaks using phylogeny-based methods. Infect. Genet. Evol. 2024, 124, 105661. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Zhao, B.; An, M.; Zhong, P.; Shang, H. Molecular network-based intervention brings us closer to ending the HIV pandemic. Front. Med. 2020, 14, 136–148. [Google Scholar] [CrossRef]
- Paraskevis, D.; Nikolopoulos, G.; Tsiara, C.; Paraskeva, D.; Antoniadou, A.; Lazanas, M.; Gargalianos, P.; Psychogiou, M.; Malliori, M.; Kremastinou, J.; et al. HIV-1 outbreak among injecting drug users in Greece, 2011: A preliminary report. Euro Surveill. 2011, 16, 19962. [Google Scholar] [CrossRef]
- Kostaki, E.-G.; Nikolopoulos, G.K.; Pavlitina, E.; Williams, L.; Magiorkinis, G.; Schneider, J.; Skaathun, B.; Morgan, E.; Psichogiou, M.; Daikos, G.L.; et al. Molecular Analysis of Human Immunodeficiency Virus Type 1 (HIV-1)-Infected Individuals in a Network-Based Intervention (Transmission Reduction Intervention Project): Phylogenetics Identify HIV-1-Infected Individuals With Social Links. J. Infect. Dis. 2018, 218, 707–715. [Google Scholar] [CrossRef]
- Ou, C.Y.; Ciesielski, C.A.; Myers, G.; Bandea, C.I.; Luo, C.C.; Korber, B.T.; Mullins, J.I.; Schochetman, G.; Berkelman, R.L.; Economou, A.N. Molecular epidemiology of HIV transmission in a dental practice. Science 1992, 256, 1165–1171. [Google Scholar] [CrossRef]
- Enfield, K.B.; Sharapov, U.; Hall, K.K.; Leiner, J.; Berg, C.L.; Xia, G.; Thompson, N.D.; Ganova-Raeva, L.; Sifri, C.D. Transmission of hepatitis B virus from an orthopedic surgeon with a high viral load. Clin. Infect. Dis. 2013, 56, 218–224. [Google Scholar] [CrossRef]
- Otani, M.; Shiino, T.; Kondo, M.; Hachiya, A.; Nishizawa, M.; Kikuchi, T.; Matano, T.; Japanese Drug Resistance HIV-1 Surveillance Network. Phylodynamic analysis reveals changing transmission dynamics of HIV-1 CRF01_AE in Japan from heterosexuals to men who have sex with men. Int. J. Infect. Dis. 2021, 108, 397–405. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
| Cluster ID | Total Sequences (N) | Newly Diagnosed (N) | Sequences Most Often/Exclusively Encountered in |
|---|---|---|---|
| 1 | 29 | 16 | heterosexuals |
| 2 | 8 | 5 | heterosexuals |
| 3 | 6 | 5 | heterosexuals |
| 4 | 9 | 4 | heterosexuals |
| 5 | 4 | 3 | heterosexuals |
| 6 | 3 | 3 | heterosexuals |
| 7 | 3 | 1 | heterosexuals |
| 8 | 122 | 79 | MSM |
| 9 | 34 | 31 | MSM |
| 10 | 36 | 29 | MSM |
| 11 | 28 | 24 | MSM |
| 12 | 19 | 17 | MSM |
| 13 | 10 | 10 | MSM |
| 14 | 14 | 8 | MSM |
| 15 | 5 | 5 | MSM |
| 16 | 3 | 1 | MSM |
| 17 | 23 | 12 | PWID |
| 18 | 33 | 10 | PWID |
| 19 | 3 | 3 | PWID |
| 20 | 14 | 2 | PWID |
| 21 | 4 | 2 | PWID |
| 22 | 3 | 2 | PWID |
| 23 | 3 | 2 | PWID |
| 24 | 3 | 1 | PWID |
| 25 | 3 | 1 | PWID |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hohan, R.; Paraschiv, S.; Nicolae, I.; Oțelea, D. Estimating the Current Routes of Transmission in HIV-1 F1 Subtype Infected Persons in Romania: Differences Between Self-Reporting and Phylogenetic Data. Pathogens 2024, 13, 960. https://doi.org/10.3390/pathogens13110960
Hohan R, Paraschiv S, Nicolae I, Oțelea D. Estimating the Current Routes of Transmission in HIV-1 F1 Subtype Infected Persons in Romania: Differences Between Self-Reporting and Phylogenetic Data. Pathogens. 2024; 13(11):960. https://doi.org/10.3390/pathogens13110960
Chicago/Turabian StyleHohan, Robert, Simona Paraschiv, Ionelia Nicolae, and Dan Oțelea. 2024. "Estimating the Current Routes of Transmission in HIV-1 F1 Subtype Infected Persons in Romania: Differences Between Self-Reporting and Phylogenetic Data" Pathogens 13, no. 11: 960. https://doi.org/10.3390/pathogens13110960
APA StyleHohan, R., Paraschiv, S., Nicolae, I., & Oțelea, D. (2024). Estimating the Current Routes of Transmission in HIV-1 F1 Subtype Infected Persons in Romania: Differences Between Self-Reporting and Phylogenetic Data. Pathogens, 13(11), 960. https://doi.org/10.3390/pathogens13110960