
In Silico Identification of Potential Inhibitors of SARS-CoV-2 Main Protease (Mpro)

 , ,
, ,  ,
,  ,
,  , , , , and
, , , , and 
Abstract
:1. Introduction
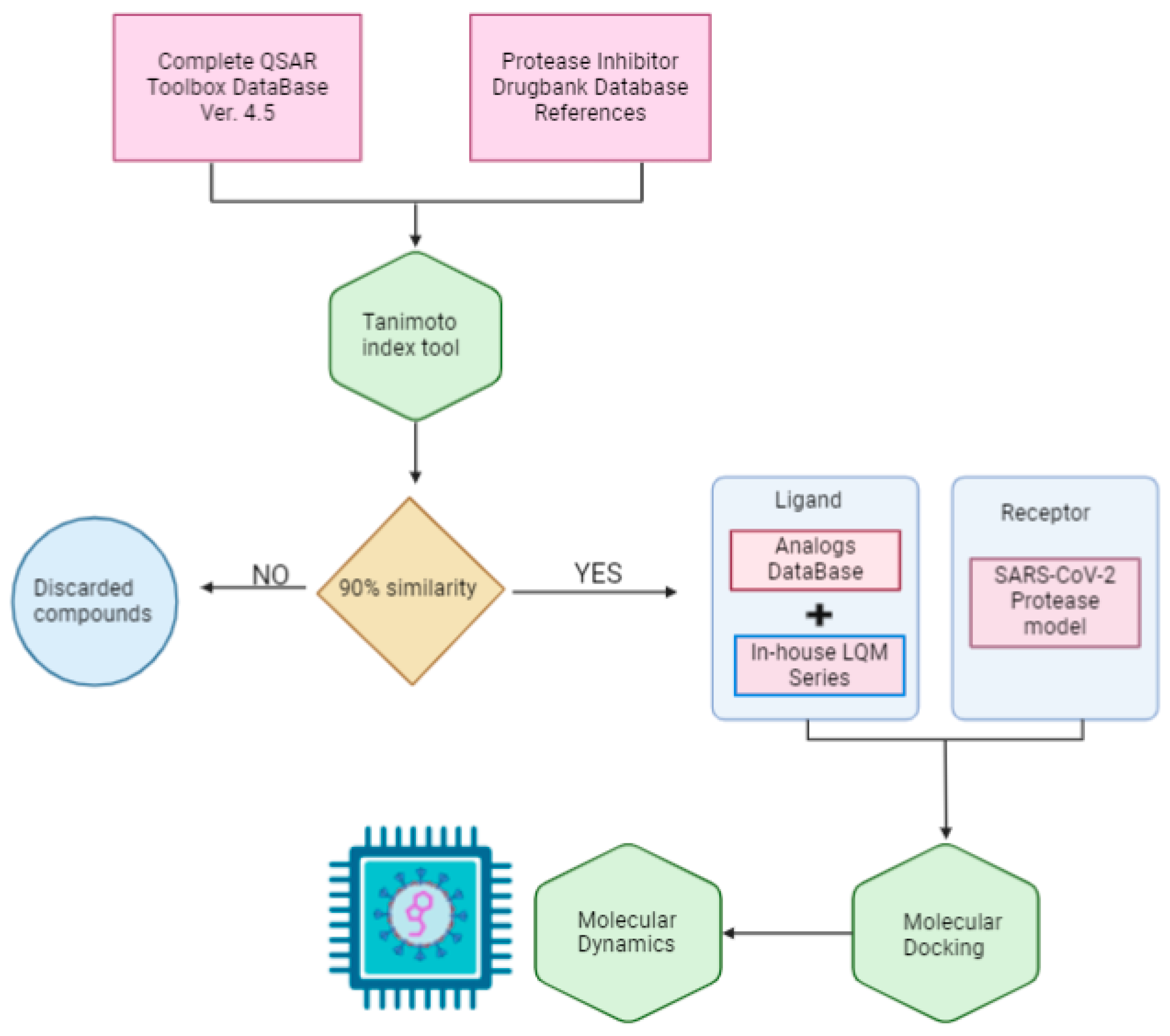
2. Methods
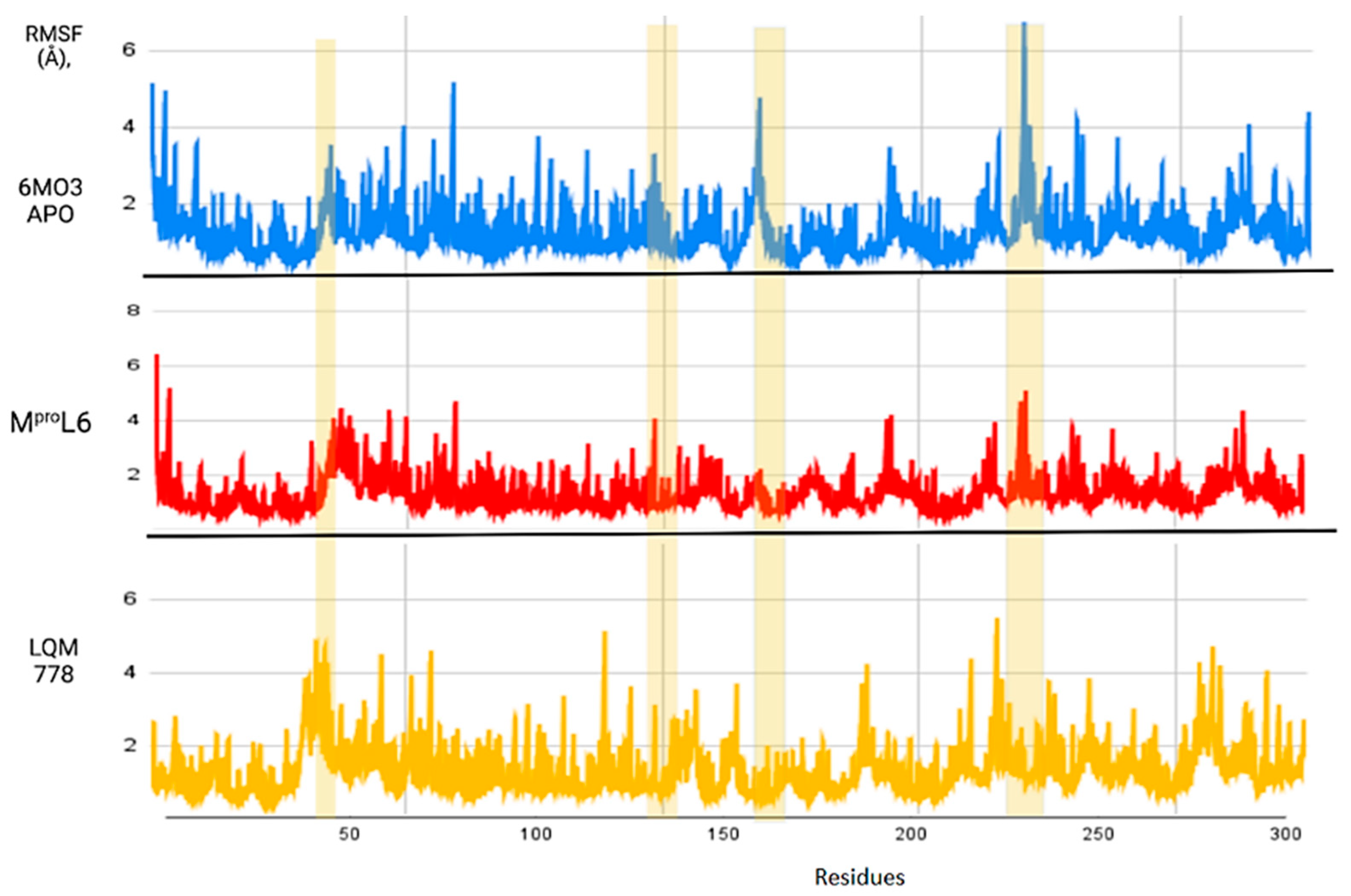
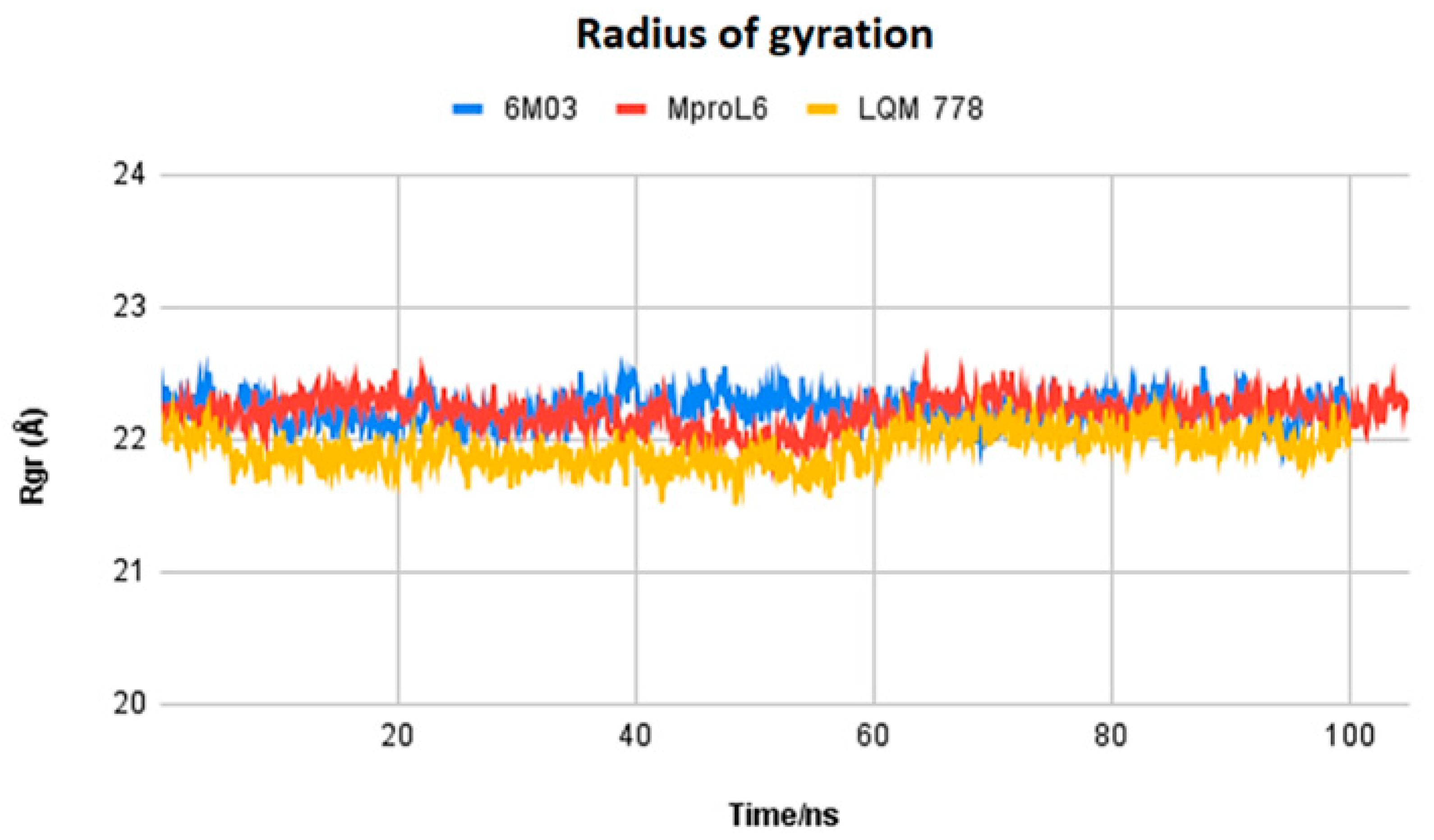
3. Results and Analysis
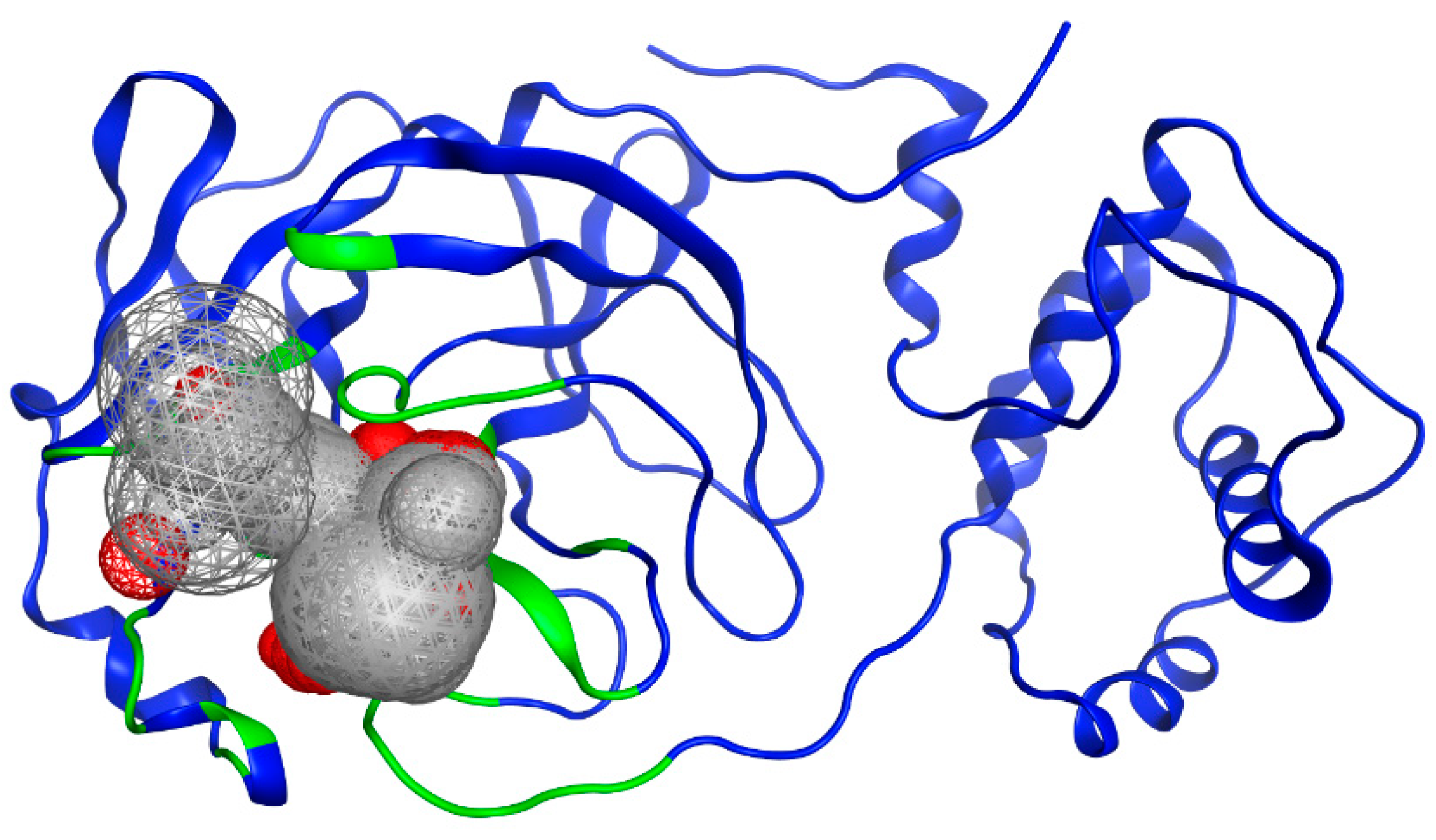
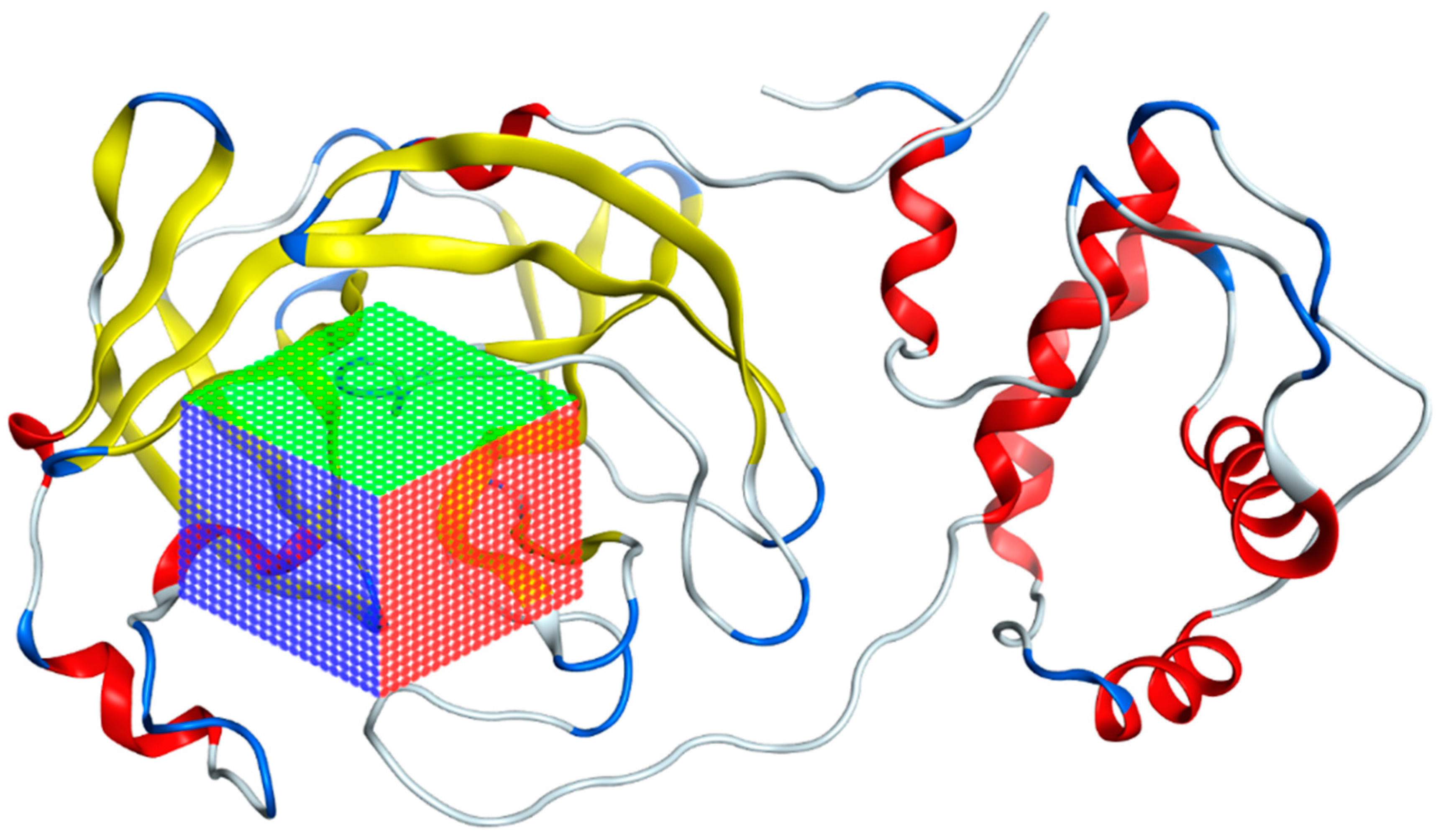
3.1. Mpro Structure Selection
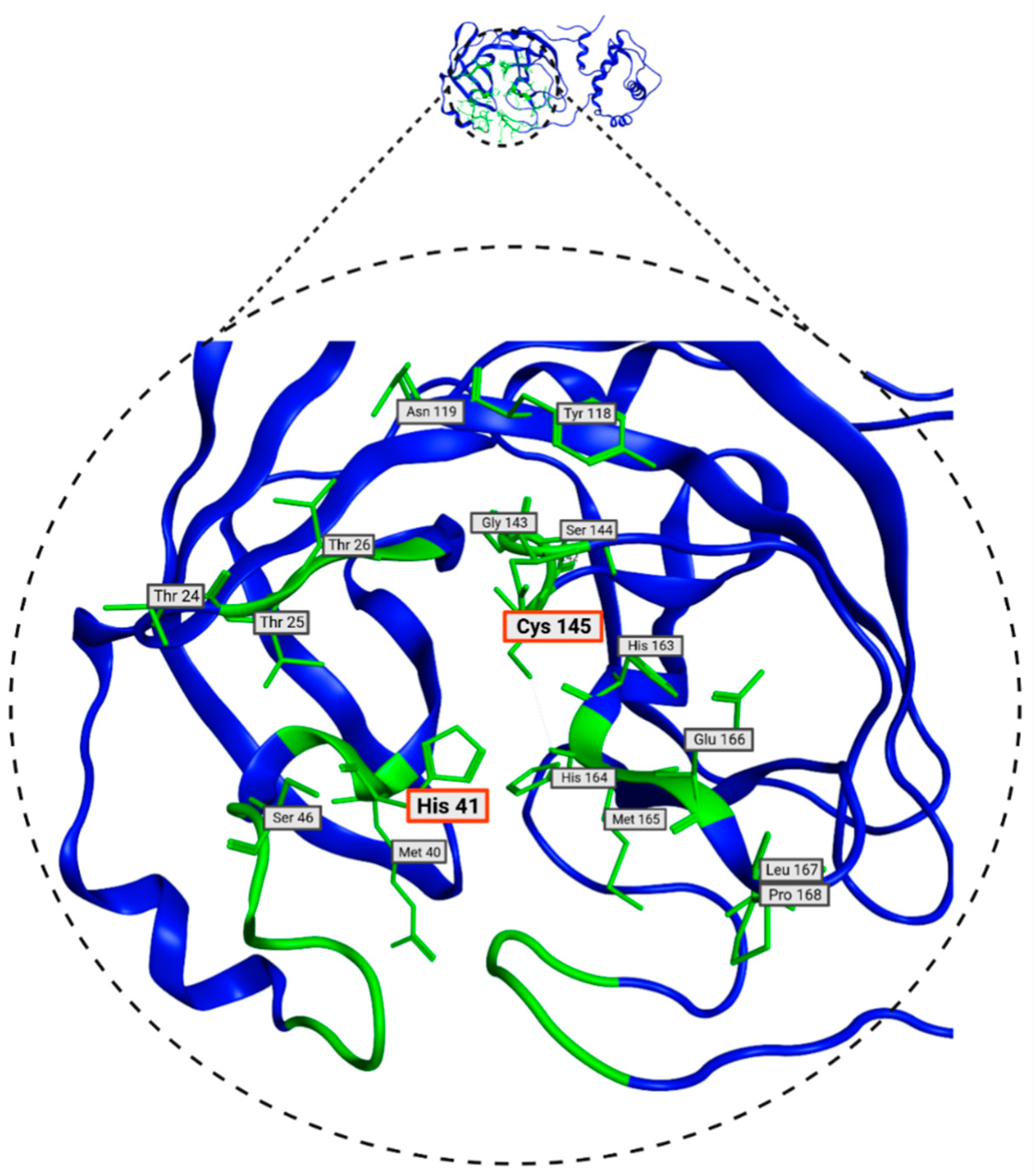
3.2. Selection of Potential Inhibitors of SARS-CoV-2 Mpro
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhu, H.; Wei, L.; Niu, P. The novel coronavirus outbreak in Wuhan, China. Glob. Health Res. Policy 2020, 5, 6. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Li, X.; Li, T.; Zhang, S.; Wang, L.; Wu, X.; Liu, J. The genetic sequence, origin, and diagnosis of SARS-CoV-2. Eur. J. Clin. Microbiol. Infect. Dis. 2020, 39, 1629–1635. [Google Scholar] [CrossRef] [PubMed]
- Cucinotta, D.; Vanelli, M. WHO Declares COVID-19 a Pandemic. Acta Biomed. 2020, 91, 157–160. [Google Scholar] [CrossRef] [PubMed]
- Miyah, Y.; Benjelloun, M.; Lairini, S.; Lahrichi, A. COVID-19 Impact on Public Health, Environment, Human Psychology, Global Socioeconomy, and Education. Sci. World J. 2022, 2022, 5578284. [Google Scholar] [CrossRef]
- W.H.O. Weekly Epidemiological Update on COVID-19. 15 July 2024. Available online: https://www.who.int/publications/m/item/covid-19-epidemiological-update-edition-169 (accessed on 17 July 2024).
- Weiss, S.R.; Leibowitz, J.L. Coronavirus pathogenesis. Adv. Virus Res. 2011, 81, 85–164. [Google Scholar] [CrossRef]
- Dhama, K.; Khan, S.; Tiwari, R.; Sircar, S.; Bhat, S.; Malik, Y.S.; Singh, K.P.; Chaicumpa, W.; Bonilla-Aldana, D.K.; Rodriguez-Morales, A.J. Coronavirus Disease 2019-COVID-19. Clin. Microbiol. Rev. 2020, 33, e00028-20. [Google Scholar] [CrossRef]
- Ding, X.L.; Jia, Q.L.; To, S.F. Human Coronavirus-229E, -OC43, -NL63, and -HKU1 (Coronaviridae). In Encyclopedia of Virology, 4th ed.; Dennis, H.B., Mark, Z., Eds.; Academic Press: Oxford, UK, 2021; pp. 428–440. [Google Scholar] [CrossRef]
- Krishnamoorthy, S.; Swain, B.; Verma, R.S.; Gunthe, S.S. SARS-CoV, MERS-CoV, and 2019-nCoV viruses: An overview of origin, evolution, and genetic variations. Virusdisease 2020, 31, 411–423. [Google Scholar] [CrossRef]
- Lu, R.; Zhao, X.; Li, J.; Niu, P.; Yang, B.; Wu, H.; Wang, W.; Song, H.; Huang, B.; Zhu, N.; et al. Genomic characterisation and epidemiology of 2019 novel Coronavirus: Implications for virus origins and receptor binding. Lancet 2020, 395, 565–574. [Google Scholar] [CrossRef]
- Ravi, V.; Saxena, S.; Panda, P.S. Basic virology of SARS-CoV 2. Indian J. Med. Microbiol. 2022, 40, 182–186. [Google Scholar] [CrossRef]
- Jungreis, I.; Sealfon, R.; Kellis, M. SARS-CoV-2 gene content and COVID-19 mutation impact by comparing 44 Sarbecovirus genomes. Nat. Commun. 2021, 12, 2642. [Google Scholar] [CrossRef]
- Redondo, N.; Zaldivar-Lopez, S.; Garrido, J.J.; Montoya, M. SARS-CoV-2 Accessory Proteins in Viral Pathogenesis: Knowns and Unknowns. Front. Immunol. 2021, 12, 708264. [Google Scholar] [CrossRef] [PubMed]
- Mariano, G.; Farthing, R.J.; Lale-Farjat, S.L.M.; Bergeron, J.R.C. Structural Characterization of SARS-CoV-2: Where We Are, and Where We Need to Be. Front. Mol. Biosci. 2020, 7, 605236. [Google Scholar] [CrossRef] [PubMed]
- Jackson, C.B.; Farzan, M.; Chen, B.; Choe, H. Mechanisms of SARS-CoV-2 entry into cells. Nat. Rev. Mol. Cell Biol. 2022, 23, 3–20. [Google Scholar] [CrossRef] [PubMed]
- Rastogi, M.; Pandey, N.; Shukla, A.; Singh, S.K. SARS coronavirus 2: From genome to infectome. Respir. Res. 2020, 21, 318. [Google Scholar] [CrossRef]
- Malone, B.; Urakova, N.; Snijder, E.J.; Campbell, E.A. Structures and functions of coronavirus replication-transcription complexes and their relevance for SARS-CoV-2 drug design. Nat. Rev. Mol. Cell Biol. 2022, 23, 21–39. [Google Scholar] [CrossRef]
- Triggle, C.R.; Bansal, D.; Ding, H.; Islam, M.M.; Farag, E.; Hadi, H.A.; Sultan, A.A. A Comprehensive Review of Viral Characteristics, Transmission, Pathophysiology, Immune Response, and Management of SARS-CoV-2 and COVID-19 as a Basis for Controlling the Pandemic. Front. Immunol. 2021, 12, 631139. [Google Scholar] [CrossRef]
- Armstrong, L.A.; Lange, S.M.; Dee Cesare, V.; Matthews, S.P.; Nirujogi, R.S.; Cole, I.; Hope, A.; Cunningham, F.; Toth, R.; Mukherjee, R.; et al. Biochemical characterization of protease activity of Nsp3 from SARS-CoV-2 and its inhibition by nanobodies. PLoS ONE 2021, 16, e0253364. [Google Scholar] [CrossRef]
- Ullrich, S.; Ekanayake, K.B.; Otting, G.; Nitsche, C. Main protease mutants of SARS-CoV-2 variants remain susceptible to nirmatrelvir. Bioorg. Med. Chem. Lett. 2022, 62, 128629. [Google Scholar] [CrossRef]
- Zhang, L.; Lin, D.; Sun, X.; Curth, U.; Drosten, C.; Sauerhering, L.; Becker, S.; Rox, K.; Hilgenfeld, R. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved alpha-ketoamide inhibitors. Science 2020, 368, 409–412. [Google Scholar] [CrossRef]
- Hu, Q.; Xiong, Y.; Zhu, G.H.; Zhang, Y.N.; Zhang, Y.W.; Huang, P.; Ge, G.B. The SARS-CoV-2 main protease (Mpro): Structure, function, and emerging therapies for COVID-19. MedComm 2022, 3, e151. [Google Scholar] [CrossRef]
- Jin, Z.; Du, X.; Xu, Y.; Deng, Y.; Liu, M.; Zhao, Y.; Zhang, B.; Li, X.; Zhang, L.; Peng, C.; et al. Structure of Mpro from SARS-CoV-2 and discovery of its inhibitors. Nature 2020, 582, 289–293. [Google Scholar] [CrossRef] [PubMed]
- Baden, L.R.; El Sahly, H.M.; Essink, B.; Kotloff, K.; Frey, S.; Novak, R.; Diemert, D.; Spector, S.A.; Rouphael, N.; Creech, C.B.; et al. Efficacy and Safety of the mRNA-1273 SARS-CoV-2 Vaccine. N. Engl. J. Med. 2021, 384, 403–416. [Google Scholar] [CrossRef] [PubMed]
- Polack, F.P.; Thomas, S.J.; Kitchin, N.; Absalon, J.; Gurtman, A.; Lockhart, S.; Perez, J.L.; Perez Marc, G.; Moreira, E.D.; Zerbini, C.; et al. Safety and Efficacy of the BNT162b2 mRNA COVID-19 Vaccine. N. Engl. J. Med. 2020, 383, 2603–2615. [Google Scholar] [CrossRef] [PubMed]
- Hammond, J.; Leister-Tebbe, H.; Gardner, A.; Abreu, P.; Bao, W.; Wisemandle, W.; Baniecki, M.; Hendrick, V.M.; Damle, B.; Simon-Campos, A.; et al. Oral Nirmatrelvir for High-Risk, Nonhospitalized Adults with COVID-19. N. Engl. J. Med. 2022, 386, 1397–1408. [Google Scholar] [CrossRef] [PubMed]
- Beigel, J.H.; Tomashek, K.M.; Dodd, L.E.; Mehta, A.K.; Zingman, B.S.; Kalil, A.C.; Hohmann, E.; Chu, H.Y.; Luetkemeyer, A.; Kline, S.; et al. Remdesivir for the Treatment of COVID-19—Final Report. N. Engl. J. Med. 2020, 383, 1813–1826. [Google Scholar] [CrossRef]
- Jayk Bernal, A.; Gomes da Silva, M.M.; Musungaie, D.B.; Kovalchuk, E.; Gonzalez, A.; Delos Reyes, V.; Martin-Quiros, A.; Caraco, Y.; Williams-Diaz, A.; Brown, M.L.; et al. Molnupiravir for Oral Treatment of COVID-19 in Nonhospitalized Patients. N. Engl. J. Med. 2022, 386, 509–520. [Google Scholar] [CrossRef]
- Sheward, D.J.; Kim, C.; Fischbach, J.; Muschiol, S.; Ehling, R.A.; Bjorkstrom, N.K.; Karlsson Hedestam, G.B.; Reddy, S.T.; Albert, J.; Peacock, T.P.; et al. Evasion of neutralising antibodies by omicron sublineage BA.2.75. Lancet Infect. Dis. 2022, 22, 1421–1422. [Google Scholar] [CrossRef]
- Gandhi, S.; Klein, J.; Robertson, A.; Pena-Hernandez, M.A.; Lin, M.J.; Roychoudhury, P.; Lu, P.; Fournier, J.; Ferguson, D.; Mohamed Bakhash, S.A.; et al. De novo emergence of a remdesivir resistance mutation during treatment of persistent SARS-CoV-2 infection in an immunocompromised patient: A case report. medRxiv 2021. [Google Scholar] [CrossRef]
- Wang, Q.; Guo, Y.; Iketani, S.; Nair, M.S.; Li, Z.; Mohri, H.; Wang, M.; Yu, J.; Bowen, A.D.; Chang, J.Y.; et al. Antibody evasion by SARS-CoV-2 Omicron subvariants BA.2.12.1, BA.4 and BA.5. Nature 2022, 608, 603–608. [Google Scholar] [CrossRef]
- Morgnanesi, D.; Heinrichs, E.J.; Mele, A.R.; Wilkinson, S.; Zhou, S.; Kulp, J.L., 3rd. A computational chemistry perspective on the current status and future direction of hepatitis B antiviral drug discovery. Antiviral. Res. 2015, 123, 204–215. [Google Scholar] [CrossRef]
- Sliwoski, G.; Kothiwale, S.; Meiler, J.; Lowe, E.W., Jr. Computational methods in drug discovery. Pharmacol. Rev. 2014, 66, 334–395. [Google Scholar] [CrossRef] [PubMed]
- Muratov, E.N.; Amaro, R.; Andrade, C.H.; Brown, N.; Ekins, S.; Fourches, D.; Isayev, O.; Kozakov, D.; Medina-Franco, J.L.; Merz, K.M.; et al. A critical overview of computational approaches employed for COVID-19 drug discovery. Chem. Soc. Rev. 2021, 50, 9121–9151. [Google Scholar] [CrossRef] [PubMed]
- Fischer, A.; Sellner, M.; Neranjan, S.; Smiesko, M.; Lill, M.A. Potential Inhibitors for Novel Coronavirus Protease Identified by Virtual Screening of 606 Million Compounds. Int. J. Mol. Sci. 2020, 21, 3626. [Google Scholar] [CrossRef] [PubMed]
- Harrison, C. Coronavirus puts drug repurposing on the fast track. Nat. Biotechnol. 2020, 38, 379–381. [Google Scholar] [CrossRef]
- Dimitrov, S.D.; Diderich, R.; Sobanski, T.; Pavlov, T.S.; Chankov, G.V.; Chapkanov, A.S.; Karakolev, Y.H.; Temelkov, S.G.; Vasilev, R.A.; Gerova, K.D.; et al. QSAR Toolbox—Workflow and major functionalities. SAR QSAR Environ. Res. 2016, 27, 203–219. [Google Scholar] [CrossRef]
- QSAR Toolbox. Available online: https://qsartoolbox.org (accessed on 2 October 2021).
- Anastasiu, D.C.; Karypis, G. Efficient identification of Tanimoto nearest neighbors. Int. J. Data Sci. Anal. 2017, 4, 153–172. [Google Scholar] [CrossRef]
- Citarella, A.; Scala, A.; Piperno, A.; Micale, N. SARS-CoV-2 M(pro): A Potential Target for Peptidomimetics and Small-Molecule Inhibitors. Biomolecules 2021, 11, 607. [Google Scholar] [CrossRef]
- Chen, Y.W.; Yiu, C.B.; Wong, K.Y. Prediction of the SARS-CoV-2 (2019-nCoV) 3C-like protease (3CLpro) structure: Virtual screening reveals velpatasvir, ledipasvir, and other drug repurposing candidates. F1000Research 2020, 9, 129. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef]
- Chemical Computing Group, ULC. Molecular Operating Environment (MOE) 2022.02; Chemical Computing Group, ULC: Montreal, QC, Canada, 2022. [Google Scholar]
- Mengist, H.M.; Dilnessa, T.; Jin, T. Structural Basis of Potential Inhibitors Targeting SARS-CoV-2 Main Protease. Front. Chem. 2021, 9, 622898. [Google Scholar] [CrossRef]
- Volkamer, A.; Kuhn, D.; Grombacher, T.; Rippmann, F.; Rarey, M. Combining global and local measures for structure-based druggability predictions. J. Chem. Inf. Model. 2012, 52, 360–372. [Google Scholar] [CrossRef] [PubMed]
- Owens, J. Determining druggability. Nat. Rev. Drug Discov. 2007, 6, 187. [Google Scholar] [CrossRef]
- Michel, M.; Visnes, T.; Homan, E.J.; Seashore-Ludlow, B.; Hedenstrom, M.; Wiita, E.; Vallin, K.; Paulin, C.B.J.; Zhang, J.; Wallner, O.; et al. Computational and Experimental Druggability Assessment of Human DNA Glycosylases. ACS Omega 2019, 4, 11642–11656. [Google Scholar] [CrossRef] [PubMed]
- Patel, C.N.; Jani, S.P.; Prasanth Kumar, S.; Modi, K.M.; Kumar, Y. Computational investigation of natural compounds as potential main protease (Mpro) inhibitors for SARS-CoV-2 virus. Comput. Biol. Med. 2022, 151, 106318. [Google Scholar] [CrossRef]
- CoVariants SARS-CoV-Mutations and Variants of Interest. Available online: https://covariants.org/ (accessed on 31 October 2023).
- Dierynck, I.; Van Marck, H.; Van Ginderen, M.; Jonckers, T.H.; Nalam, M.N.; Schiffer, C.A.; Raoof, A.; Kraus, G.; Picchio, G. TMC310911, a novel human immunodeficiency virus type 1 protease inhibitor, shows in vitro an improved resistance profile and higher genetic barrier to resistance compared with current protease inhibitors. Antimicrob. Agents Chemother. 2011, 55, 5723–5731. [Google Scholar] [CrossRef]
- Kaldor, S.W.; Kalish, V.J.; Davies, J.F., 2nd; Shetty, B.V.; Fritz, J.E.; Appelt, K.; Burgess, J.A.; Campanale, K.M.; Chirgadze, N.Y.; Clawson, D.K.; et al. Viracept (nelfinavir mesylate, AG1343): A potent, orally bioavailable inhibitor of HIV-1 protease. J. Med. Chem. 1997, 40, 3979–3985. [Google Scholar] [CrossRef]
- Islam, R.; Parves, M.R.; Paul, A.S.; Uddin, N.; Rahman, M.S.; Mamun, A.A.; Hossain, M.N.; Ali, M.A.; Halim, M.A. A molecular modeling approach to identify effective antiviral phytochemicals against the main protease of SARS-CoV-2. J. Biomol. Struct. Dyn. 2021, 39, 3213–3224. [Google Scholar] [CrossRef]
- Sheahan, T.P.; Sims, A.C.; Leist, S.R.; Schafer, A.; Won, J.; Brown, A.J.; Montgomery, S.A.; Hogg, A.; Babusis, D.; Clarke, M.O.; et al. Comparative therapeutic efficacy of remdesivir and combination lopinavir, ritonavir, and interferon beta against MERS-CoV. Nat. Commun. 2020, 11, 222. [Google Scholar] [CrossRef]
- Zanasi, A.; Mazzolini, M.; Kantar, A. A reappraisal of the mucoactive activity and clinical efficacy of bromhexine. Multidiscip. Respir. Med. 2017, 12, 7. [Google Scholar] [CrossRef]
- Hull, M.W.; Montaner, J.S. Ritonavir-boosted protease inhibitors in HIV therapy. Ann. Med. 2011, 43, 375–388. [Google Scholar] [CrossRef]
- Kiser, J.J.; Flexner, C. Direct-acting antiviral agents for hepatitis C virus infection. Annu. Rev. Pharmacol. Toxicol. 2013, 53, 427–449. [Google Scholar] [CrossRef] [PubMed]
- Kupferschmidt, H.H.; Fattinger, K.E.; Ha, H.R.; Follath, F.; Krahenbuhl, S. Grapefruit juice enhances the bioavailability of the HIV protease inhibitor saquinavir in man. Br. J. Clin. Pharmacol. 1998, 45, 355–359. [Google Scholar] [CrossRef] [PubMed]
- Kitamura, K.; Tomita, K. Proteolytic activation of the epithelial sodium channel and therapeutic application of a serine protease inhibitor for the treatment of salt-sensitive hypertension. Clin. Exp. Nephrol. 2012, 16, 44–48. [Google Scholar] [CrossRef] [PubMed]
- Raboisson, P.; de Kock, H.; Rosenquist, A.; Nilsson, M.; Salvador-Oden, L.; Lin, T.I.; Roue, N.; Ivanov, V.; Wahling, H.; Wickstrom, K.; et al. Structure-activity relationship study on a novel series of cyclopentane-containing macrocyclic inhibitors of the hepatitis C virus NS3/4A protease leading to the discovery of TMC435350. Bioorg. Med. Chem. Lett. 2008, 18, 4853–4858. [Google Scholar] [CrossRef] [PubMed]
- Bafna, K.; White, K.; Harish, B.; Rosales, R.; Ramelot, T.A.; Acton, T.B.; Moreno, E.; Kehrer, T.; Miorin, L.; Royer, C.A.; et al. Hepatitis C virus drugs that inhibit SARS-CoV-2 papain-like protease synergize with remdesivir to suppress viral replication in cell culture. Cell Rep. 2021, 35, 109133. [Google Scholar] [CrossRef]
- Alamri, M.A.; Altharawi, A.; Alabbas, A.B.; Alossaimi, M.A.; Alqahtani, S.M. Structure-based virtual screening and molecular dynamics of phytochemicals derived from Saudi medicinal plants to identify potential COVID-19 therapeutics. Arab J. Chem. 2020, 13, 7224–7234. [Google Scholar] [CrossRef]
- Lu, H. Drug treatment options for the 2019-new coronavirus (2019-nCoV). Biosci. Trends 2020, 14, 69–71. [Google Scholar] [CrossRef]
- Purohit, R.; Sethumadhavan, R. Structural basis for the resilience of Darunavir (TMC114) resistance major flap mutations of HIV-1 protease. Interdiscip. Sci. 2009, 1, 320–328. [Google Scholar] [CrossRef]
- Doyon, L.; Tremblay, S.; Bourgon, L.; Wardrop, E.; Cordingley, M.G. Selection and characterization of HIV-1 showing reduced susceptibility to the non-peptidic protease inhibitor tipranavir. Antiviral. Res. 2005, 68, 27–35. [Google Scholar] [CrossRef]
- Ye, G.; Wang, X.; Tong, X.; Shi, Y.; Fu, Z.F.; Peng, G. Structural Basis for Inhibiting Porcine Epidemic Diarrhea Virus Replication with the 3C-Like Protease Inhibitor GC376. Viruses 2020, 12, 240. [Google Scholar] [CrossRef]
- Kiselev, O.I.; Deeva, E.G.; Mel’nikova, T.I.; Kozeletskaia, K.N.; Kiselev, A.S.; Rusinov, V.L.; Charushin, V.N.; Chupakhin, O.N. A new antiviral drug Triazavirin: Results of phase II clinical trial. Vopr. Virusol. 2012, 57, 9–12. [Google Scholar] [PubMed]
- Khatib, M.; Pieraccini, G.; Innocenti, M.; Melani, F.; Mulinacci, N. An insight on the alkaloid content of Capparis spinosa L. root by HPLC-DAD-MS, MS/MS and 1H qNMR. J. Pharm. Biomed. Anal. 2016, 123, 53–62. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, A.; Siddiqui, S.; Ahmad, R.; Mehrotra, S.; Ahmad, B.; Srivastava, A.N. Exploring nature’s bounty: Identification of Withania somnifera as a promising source of therapeutic agents against COVID-19 by virtual screening and in silico evaluation. J. Biomol. Struct. Dyn. 2022, 40, 1858–1908. [Google Scholar] [CrossRef] [PubMed]
- Tao, Y.; Qu, H.; Wang, S.; Yan, F.; Wang, C.; Zhang, M. Computational assessment of herbal medicine-derived compounds as potential inhibitors of SARS-CoV-2 main protease. J. Biomol. Struct. Dyn. 2023, 41, 9602–9613. [Google Scholar] [CrossRef]
- Zhang, L.; Lin, D.; Kusov, Y.; Nian, Y.; Ma, Q.; Wang, J.; von Brunn, A.; Leyssen, P.; Lanko, K.; Neyts, J.; et al. alpha-Ketoamides as Broad-Spectrum Inhibitors of Coronavirus and Enterovirus Replication: Structure-Based Design, Synthesis, and Activity Assessment. J. Med. Chem. 2020, 63, 4562–4578. [Google Scholar] [CrossRef]
- Sharma, G.; Kumar, N.; Sharma, C.S.; Mishra, S.S. In silico guided screening of active components of C. lanceolata as 3-chymotrypsin-like protease inhibitors of novel coronavirus. 3 Biotech 2023, 13, 324. [Google Scholar] [CrossRef]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Kruger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280 e278. [Google Scholar] [CrossRef]
- Yamamoto, M.; Matsuyama, S.; Li, X.; Takeda, M.; Kawaguchi, Y.; Inoue, J.I.; Matsuda, Z. Identification of Nafamostat as a Potent Inhibitor of Middle East Respiratory Syndrome Coronavirus S Protein-Mediated Membrane Fusion Using the Split-Protein-Based Cell-Cell Fusion Assay. Antimicrob. Agents Chemother. 2016, 60, 6532–6539. [Google Scholar] [CrossRef]
- Eberhardt, J.; Santos-Martins, D.; Tillack, A.F.; Forli, S. AutoDock Vina 1.2.0: New Docking Methods, Expanded Force Field, and Python Bindings. J. Chem. Inf. Model. 2021, 61, 3891–3898. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef]
- Antonopoulou, I.; Sapountzaki, E.; Rova, U.; Christakopoulos, P. Inhibition of the main protease of SARS-CoV-2 (Mpro) by repurposing/designing drug-like substances and utilizing nature’s toolbox of bioactive compounds. Comput. Struct. Biotechnol. J. 2022, 20, 1306–1344. [Google Scholar] [CrossRef] [PubMed]
- Dallakyan, S.; Olson, A.J. Small-molecule library screening by docking with PyRx. Methods Mol. Biol. 2015, 1263, 243–250. [Google Scholar] [CrossRef] [PubMed]
- Phillips, J.C.; Hardy, D.J.; Maia, J.D.C.; Stone, J.E.; Ribeiro, J.V.; Bernardi, R.C.; Buch, R.; Fiorin, G.; Henin, J.; Jiang, W.; et al. Scalable molecular dynamics on CPU and GPU architectures with NAMD. J. Chem. Phys. 2020, 153, 044130. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein | Volume (Å3) | Surface (Å2) | Drug Score (Druggability) |
|---|---|---|---|
| 7TOB | 673.33 | 790.00 | 0.81 |
| 5R81 | 623.55 | 648.20 | 0.71 |
| 5R7Y | 618.69 | 718.99 | 0.77 |
| 5R83 | 623.80 | 701.32 | 0.77 |
| 5R7Z | 623.89 | 669.87 | 0.78 |
| 5R82 | 628.54 | 764.97 | 0.72 |
| 5R84 | 588.10 | 717.52 | 0.74 |
| 6LU7 | 398.59 | 629.05 | 0.73 |
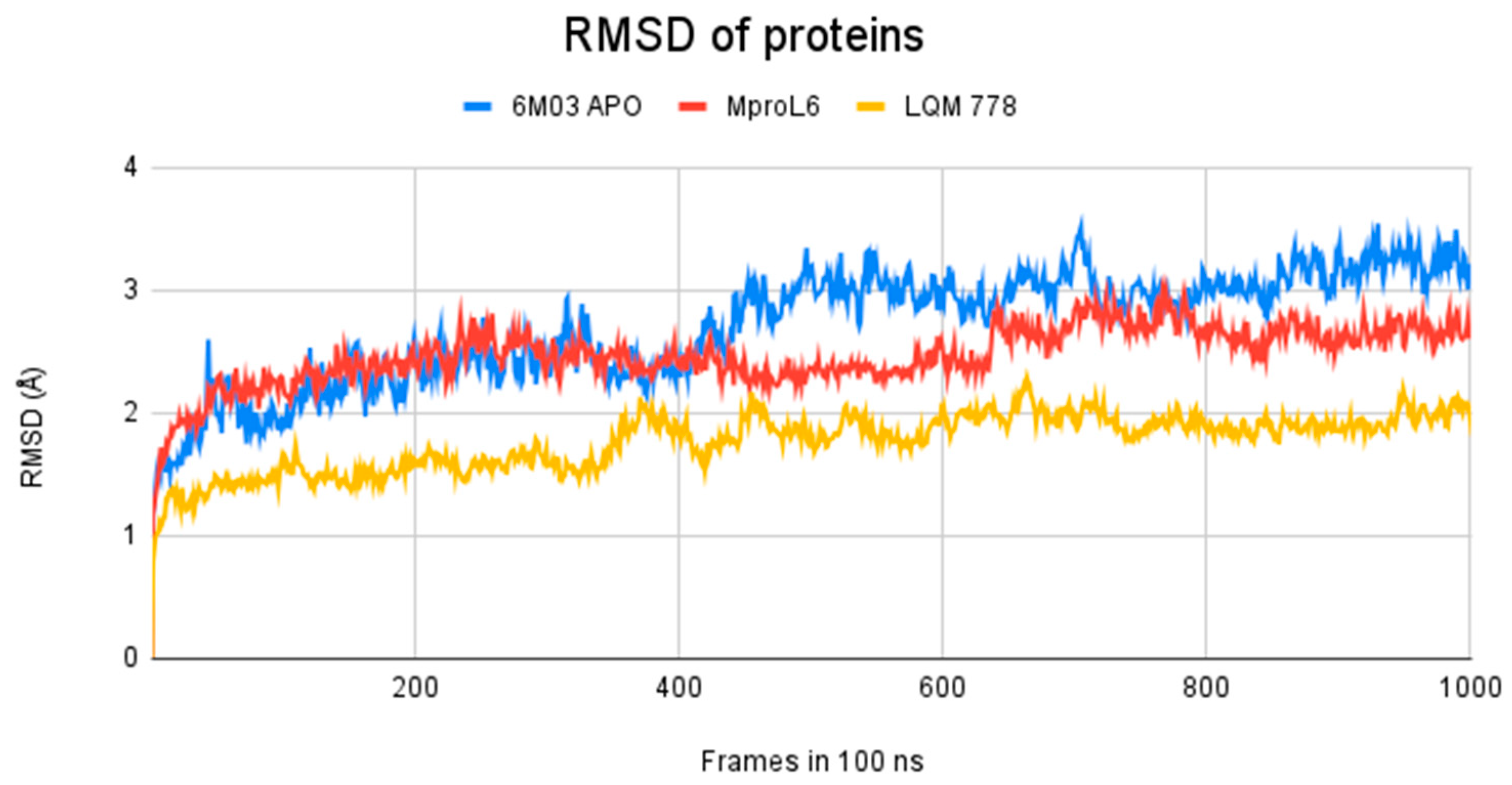
| 6M03 | 523.87 | 747.87 | 0.78 |
| 6Y2F | 600.64 | 672.26 | 0.70 |
| 6Y2G.A | 672.29 | 756.12 | 0.80 |
| 6Y2G.B | 660.16 | 670.12 | 0.73 |
| 6Y8E | 589.38 | 775.76 | 0.79 |
| 6YB7 | 529.98 | 682.30 | 0.76 |
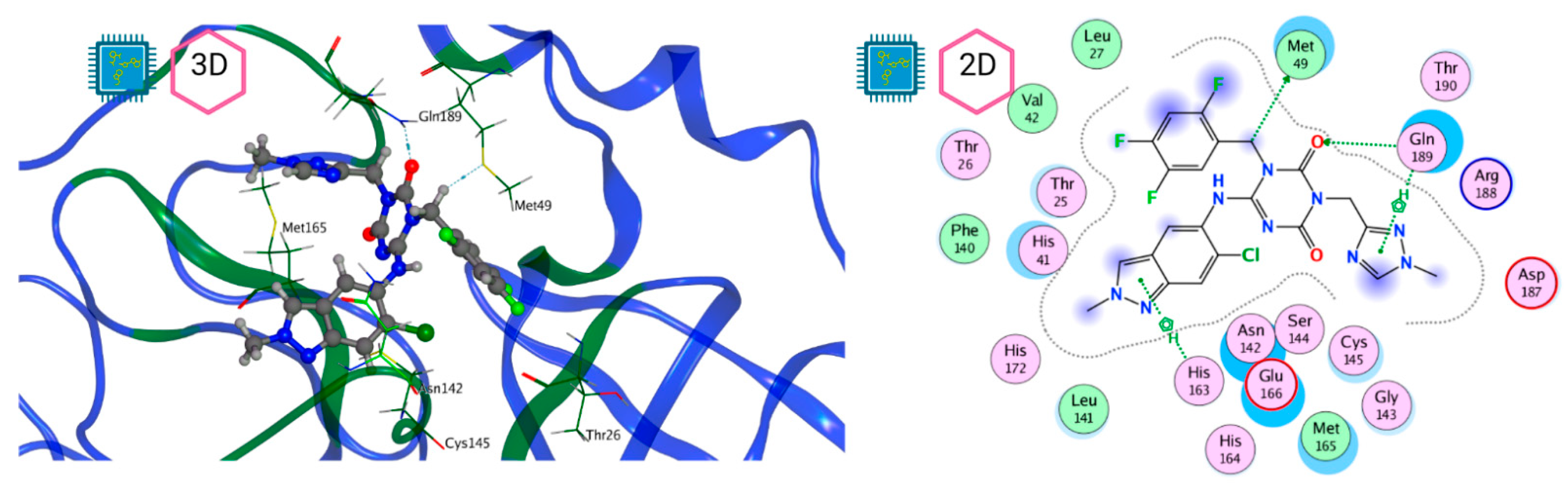
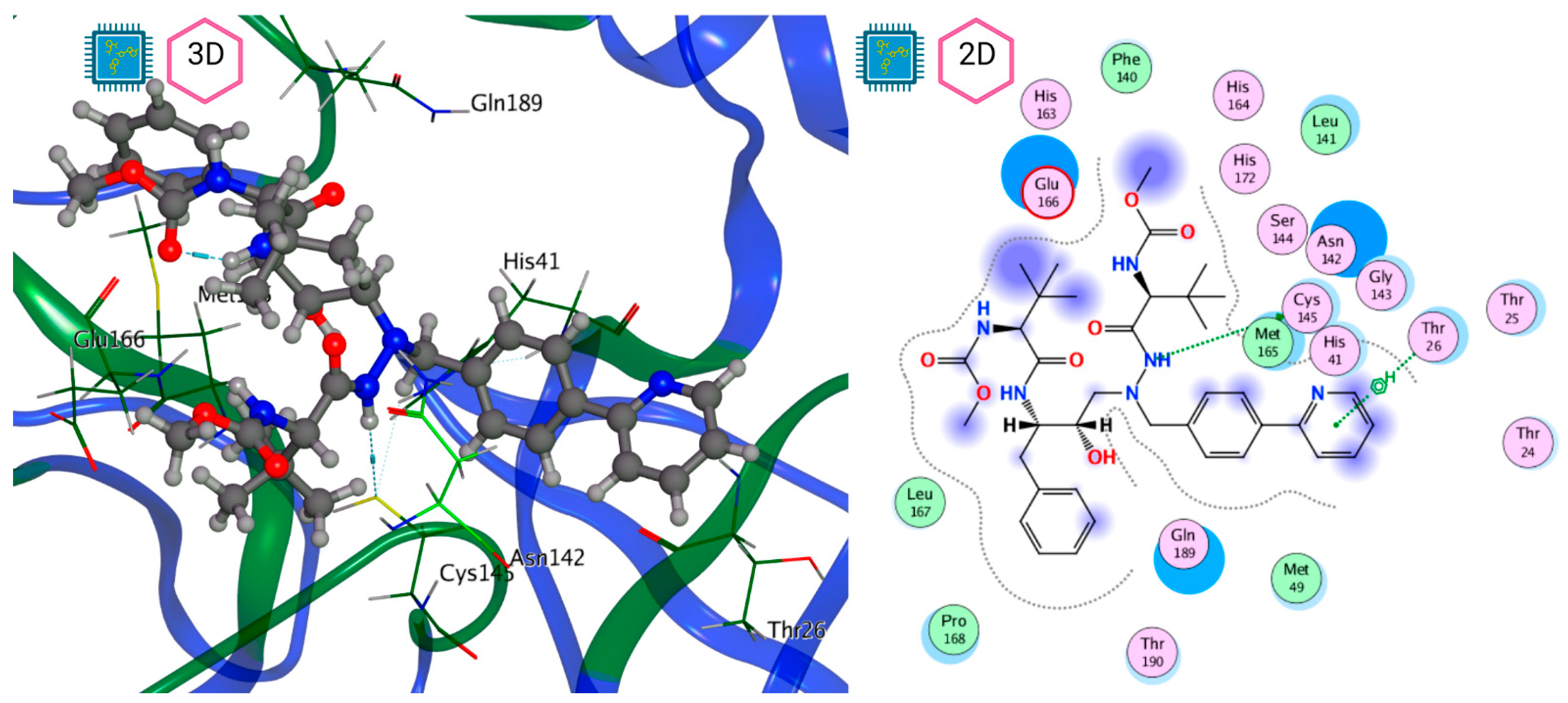
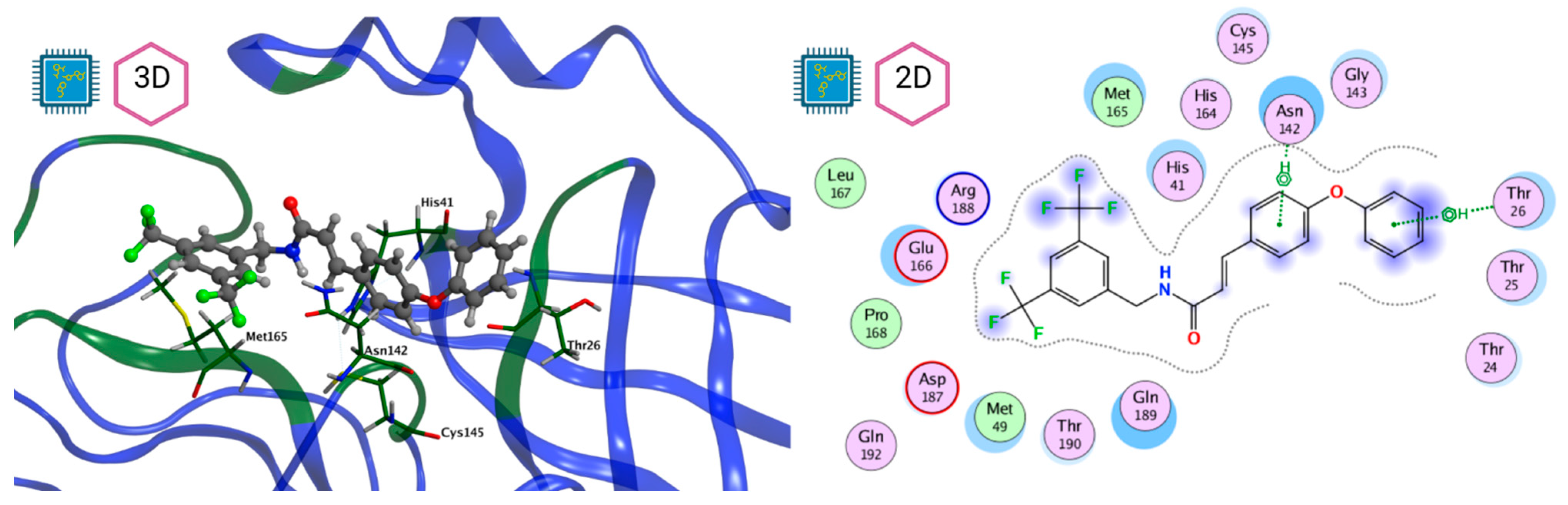
| Thr26, Leu27, His41, Ser46, Met49, Tyr118, Asn119, Phe140, Leu141, Asn142, Gly143, Ser144, Cys145, His163, His164, Met165, Glu166, Leu167, Pro168, His172, Gln189 |
| Protease Inhibitor | Number of Analogs | Activity | Target/Uniprot ID | References |
|---|---|---|---|---|
| ASC09 (TMC-310911) | 4 | Protease inhibitor (PI) with activity against a variety of HIV-1 strains including multi-PI-resistant strains | Gag-Pol polyprotein/P03366 HIV-1 protease/O90777 | Dierynck et al. (2011) [50] |
| Nelfinavir | 16 | Used in the treatment of HIV infection, inhibits viral proteinase enzyme which prevents cleavage of the gag-pol polyprotein, resulting in noninfectious, immature viral particles. | HIV-1 protease/O90777 | Kaldor et al. (1997) [51] |
| Baicalein | 276 | Baicalein is under investigation in clinical trials in the treatment of healthy adults with influenza fever. | Lactoylglutathione lyase/Q04760 Tumor necrosis factor/P01375 Xanthine dehydrogenase-oxidase/P47989 L-selectin/P14151 Prolyl endopeptidase/P48147 Polyunsaturated fatty acid 5-lipoxygenase/P09917 | Islam et al. (2021) [52] |
| Remdesivir | 3 | Nucleoside analog used to treat RNA virus infections by inhibiting the RNA polymerase (RdRp) enzyme complex for genomic replication. | Replicase polyprotein 1ab/SARS-CoV: P0C6X7 SARS-CoV-2: P0DTD1 RNA-directed RNA polymerase L/Q05318 | Sheahan et al. (2020) [53] |
| Bromhexine | 305 | Mucolytic agent, derived from the Adhatoda vasica plant; used for a variety of respiratory conditions associated with increased mucus secretion. | Transmembrane protease serine 2/O15393 Angiotensin-converting enzyme 2/Q9BYF1 | Zanasi et al. (2017) [54] |
| Ritonavir | 61 | HIV protease inhibitor used in combination with other antiviral agents for the treatment of HIV infection. | Gag-Pol polyprotein/P03366 | Hull et al. (2011) [55] |
| Boceprevir | 77 | NS3/4A protease inhibitor for hepatitis C virus, used in combination with other medications to treat chronic hepatitis C genotype 1 infection. It is not indicated for use as monotherapy. | Genome polyprotein/P26664 | Kiser et al. (2013) [56] |
| Saquinavir | 56 | HIV protease inhibitor used in combination with other antiretroviral agents for the treatment of HIV-1 in patients with advanced immunodeficiency. | Gag-Pol polyprotein/P03366 | Kupferschmidt et al. (1998) [57] |
| Camostat | 554 | Serine protease inhibitor approved in Japan for the treatment of chronic pancreatitis. | Metal ion binding/P07477 | Kitamura et al. (2012) [58] |
| Simeprevir | 2 | Direct-acting antiviral agent that inhibits the HCV NS3/4A protease; used to treat chronic hepatitis C virus (HCV) infection in adults with HCV genotype 1 or 4. | Genome polyprotein/P26664 | Raboisson et al. (2008) [59] Bafna et al. (2011) [60] |
| Cyanidin 3-glucoside | 56 | Anthocyanidin phytochemical and metabolite found in several plants such as angiosperms; also produced by Saccharomyces cerevisiae. | No information about a specific protease. The ability to inhibit the activity of cyclooxygenase enzymes has been demonstrated. | Islam et al. (2021) [52] |
| Vaniprevir | 2 | In clinical trials for the treatment and diagnosis of Hepatitis C, including chronic forms and genotype 1 infections. | Gag-Pol polyprotein/P03366 | Bafna et al. (2011) [60] |
| Chrysphanol 8′(6-galloylgluside | 16 | Anthraquinone derivative isolated from rhubarb; it has an inhibitory effect on platelet aggregation induced by collagen and thrombin. | No information about a specific protease. | Alamri et al. (2020) [61] |
| Umifenovir | 193 | Dual-function antiviral and host-targeting agent used for the treatment and prevention of influenza and other respiratory viruses. | No information about a specific protease. | Lu et al. (2020) [62] |
| Darunavir | 62 | HIV protease inhibitor employed in treating HIV infection, particularly in patients with a history of previous antiretroviral therapies. | Aspartic-type endopeptidase activity UniProt: Q72874 | Purohit et al. (2009) [63] |
| Tipranavir | 26 | Protease inhibitor used to treat HIV-1 that is resistant to multiple other protease inhibitors. | Aspartic-type endopeptidase activity UniProt: Q72874 | Doyon et al. (2005) [64] |
| Gc376 | 155 | Direct-acting antiviral for coronaviruses such as MERS-CoV, feline, ferret, and mink. | Replicase polyprotein 1ab SARS CoV II: P0DTD1 Feline: Uniprot Q98VG9 | Ye et al. (2020) [65] |
| Triazavirin | 33 | Influenza A and B infections | Triazavirin is a guanosine nucleotide analog that inhibits RNA synthesis. | Kiselev et al. (2012) [66] |
| Isocodonocarpine | 110 | Phytochemical identified in silico for its binding to SARS-CoV-2 papain. | No information about a specific protease | Khatib et al. (2016) [67] |
| Withanolide A | 19 | Phytochemical that exhibits stronger binding with Mpro compared to hydroxychloroquine. | No information about a specific protease | Srivastava et al. (2022) [68] |
| Iso-mulbel-rochromene | 13 | Phytochemical inhibitor identified for inhibition of 3CLpro protease. | No information about a specific protease | Tao et al. (2023) [69] |
| α-Ketoamide- 11r | 83 | Peptidomimetic compound designed for antiviral activity against Mpro coronavirus and the enterovirus 3C protease. | Replicase polyprotein 1ab/P0C6X7 | Islam et al. (2021) [52] Zhang et al. (2020) [70] |
| Lopinavir | 101 | HIV-1 protease inhibitor used in combination with ritonavir for the treatment of HIV infection. | Aspartic-type endopeptidase activity/Q72874 | Sheahan et al. (2020) [53] |
| x77 | 174 | Standard inhibitor identified for the inhibition of the 3CLpro protease | No information about a specific protease | Sharma et al. (2023) [71] |
| Nafamostat | 168 | Used in trials studying the prevention of liver transplantation and postreperfusion syndrome. Anticoagulant therapy for patients undergoing continuous renal replacement therapy. | Tumor necrosis factor/P01375 Prothrombin/P00734 Coagulation factor X/P00742 Serine protease 1/P07477 Kallikrein-1/P06870 Intercellular adhesion molecule 1/P05362 | Hoffmann et al. (2020) [72] Yamamoto et al. (2016) [73] |
| Total analogs | 2565 |
| Ligand | Structure | AutoDock Vina Score (kcal/mol) | MOE Score (kcal/mol) | Residues of the Mpro That Interact with the Ligands |
|---|---|---|---|---|
| Ensitrelvir (S-217622) | 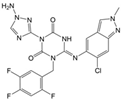 | −7.3 | −8.57 | Met49, Cys145, His163, Gln189 |
| Atazanavir (BMS-232632) | 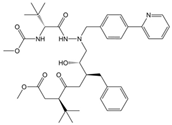 | −6.3 | −9.48 | Thr26, Asn142, Cys145, Glu166 |
| MproL6 | 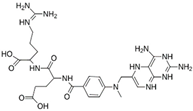 | −7.8 | −8.12 | Asn142, Thr25, Glu166, Cys145 |
| MproL13 | 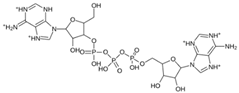 | −7.4 | −7.22 | Asn142, Glu166, Cys145, Thr25, Met49 |
| MproL16 | 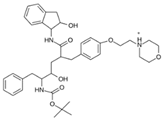 | −6.8 | −7.81 | Asn142, Glu166, Thr25 |
| MproL4 | 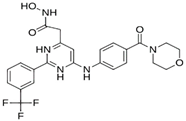 | −7.5 | −6.84 | Asn142, Thr25, Glu166, Gln143 |
| MproL11 | 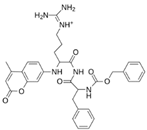 | −6.7 | −7.47 | Asn142, Glu166, Cys145, His41, Thr25 |
| MproL21 | 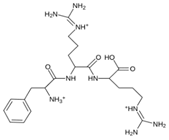 | −6.8 | −7.3 | Asn142, Glu166, Cys145, Thr26, Met49 |
| MproL5 |  | −7.2 | −6.81 | Gly143, Thr25, Glu166, Cys145 |
| MproL7 | 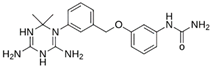 | −7.4 | −6.56 | Thr25, Asn142, Glu166, Met165 |
| MproL8 | 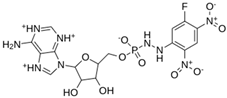 | −7 | −6.92 | Asn142, Cys145, Glu166, Met165, Thr25 |
| MproL17 |  | −6.4 | −7.11 | Asn142, Cys145, Glu166, Thr25 |
| MproL15 | 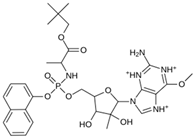 | −6.6 | −6.75 | Asn142, Cys145, Thr25 |
| MproL22 | 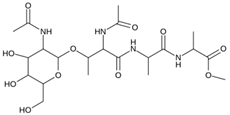 | −6.1 | −7.24 | Asn142, Cys145, Glu166, Thr25, Met49 |
| MproL18 | 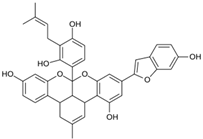 | −6.4 | −6.55 | Asn142, Ser46, Thr25, Glu166 |
| MproL2 | 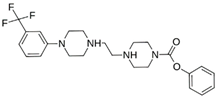 | −6.2 | −6.65 | Asn142, Cys145, Thr25, His41, Met49, Glu166 |
| MproL10 | 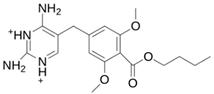 | −6.3 | −6.5 | Asn142, Glu166, Thr25, Met165 |
| MproL14 | 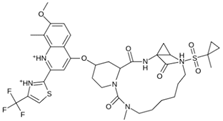 | −5.6 | −7.05 | Asn142, Thr25, Glu166, Cys145 |
| MproL20 | 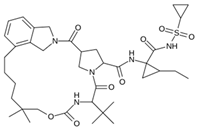 | −5.5 | −7.13 | Asn142, Thr25, Glu166, Met49, Cys145 |
| MproL19 | 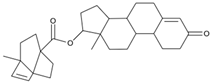 | −6.1 | −6.3 | Asn142, Thr25, Glu166 |
| MproL12 | 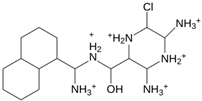 | −6.8 | −4.91 | Asn142, Glu166, Cys145, Met49, His41, Thr25 |
| MproL1 | 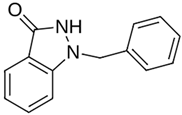 | −5.6 | −5.29 | Asn142, Cys145, Gly143 |
| MproL9 | 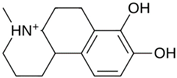 | −5.7 | −4.92 | Asn142, Leu141, His41, Glu166 |
| MproL3 | 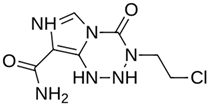 | −5.1 | −5.17 | Cys145, Met165, Glu166, Arg188, Gln189 |
| LQM 778 | 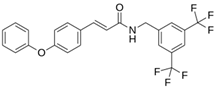 | −8.7 | −7.2 | His41, Cys145, Met165 |
| LQM 769 | 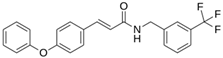 | −8.3 | −6.8 | Asn142, Cys145, Glu166 |
| LQM 794 | 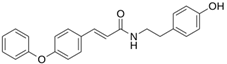 | −7.2 | −7.2 | Thr25, Asn142, Glu 166, Met165 |
| LQM 764 | 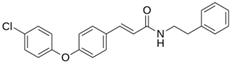 | −6.9 | −6 | Asn142, Cys145, Glu166 |
| LQM 796 | 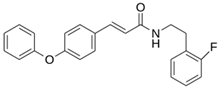 | −6.9 | −5.8 | Cys145, Thr25, His41 |
| LQM 797 | 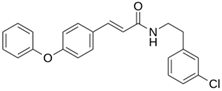 | −6.9 | −5.9 | Asn142, Glu166, Met165 |
| LQM 795 |  | −6.9 | −6.2 | His41, Met49, Glu169 |
| LQM 721 | 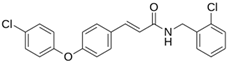 | −6.7 | −5.2 | Glu169, Met49 |
| LQM 755 |  | −6.6 | −5.6 | Asn142, Glu166, Thr25 |
| LQM 798 | 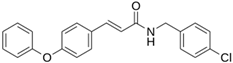 | −6.4 | −5.3 | Ser46, Thr25, His41 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hernández-Serda, M.A.; Vázquez-Valadez, V.H.; Aguirre-Vidal, P.; Markarian, N.M.; Medina-Franco, J.L.; Cardenas-Granados, L.A.; Alarcón-López, A.Y.; Martínez-Soriano, P.A.; Velázquez-Sánchez, A.M.; Falfán-Valencia, R.E.; et al. In Silico Identification of Potential Inhibitors of SARS-CoV-2 Main Protease (Mpro). Pathogens 2024, 13, 887. https://doi.org/10.3390/pathogens13100887
Hernández-Serda MA, Vázquez-Valadez VH, Aguirre-Vidal P, Markarian NM, Medina-Franco JL, Cardenas-Granados LA, Alarcón-López AY, Martínez-Soriano PA, Velázquez-Sánchez AM, Falfán-Valencia RE, et al. In Silico Identification of Potential Inhibitors of SARS-CoV-2 Main Protease (Mpro). Pathogens. 2024; 13(10):887. https://doi.org/10.3390/pathogens13100887
Chicago/Turabian StyleHernández-Serda, Manuel Alejandro, Víctor H. Vázquez-Valadez, Pablo Aguirre-Vidal, Nathan M. Markarian, José L. Medina-Franco, Luis Alfonso Cardenas-Granados, Aldo Yoshio Alarcón-López, Pablo A. Martínez-Soriano, Ana María Velázquez-Sánchez, Rodolfo E. Falfán-Valencia, and et al. 2024. "In Silico Identification of Potential Inhibitors of SARS-CoV-2 Main Protease (Mpro)" Pathogens 13, no. 10: 887. https://doi.org/10.3390/pathogens13100887
APA StyleHernández-Serda, M. A., Vázquez-Valadez, V. H., Aguirre-Vidal, P., Markarian, N. M., Medina-Franco, J. L., Cardenas-Granados, L. A., Alarcón-López, A. Y., Martínez-Soriano, P. A., Velázquez-Sánchez, A. M., Falfán-Valencia, R. E., Angeles, E., & Abrahamyan, L. (2024). In Silico Identification of Potential Inhibitors of SARS-CoV-2 Main Protease (Mpro). Pathogens, 13(10), 887. https://doi.org/10.3390/pathogens13100887