1. Introduction
Avian influenza A viruses (AIVs) belong to the
Orthomyxoviridae virus family and have a single-stranded, negative-sense RNA genome [
1]. AIVs can be categorised into high- and low-pathogenic viruses (HPAIVs and LPAIVs, respectively), depending on their disease severity [
2]. HPAIVs have up to 100% mortality rates and infect chickens systematically, whereas LPAIVs are associated with milder symptoms and lower mortality rates and are limited to respiratory and intestinal tracts [
3]. This difference in pathogenicity is determined by the haemagglutinin (HA) protein, based on which proteases recognise the proteolytic cleavage site sequence [
1]. The H9N2 subtype was first identified in China in the early 1990s and is now considered to be endemic in poultry throughout Asia, Europe, North Africa and the Middle East [
4,
5,
6,
7]. Despite the H9N2 subtype generally having a mortality rate of less than 20%, outbreaks are associated with decreased egg production and clinical symptoms, as well as an increased likelihood of secondary infections with potentially higher mortality rates [
8,
9]. It is also considered zoonotic, as it can be transferred to humans and can also be transferred to other mammals, such as pigs [
8,
10]. Therefore, being able to treat and control the spread of H9N2 AIV in poultry populations is important for preventing economic losses and protecting public health.
There is growing evidence that the commensal bacteria present at the body sites of chickens play an important role in shaping the host’s defences against viral infection [
11]. Variations in the gut microbiome composition have been shown to be caused by viral infection, as well as environmental factors like housing, diet and hygiene [
12,
13]. The relationship between the gut microbiota and AIV in the chicken model has been observed in numerous studies [
14,
15,
16,
17]. These studies have shown that H9N2 AIV infection is associated with the dysregulation of the gut microbiota, as well as changes in innate immune gene expression levels. Yitbarek et al. (2018) showed that the depletion of the gut microbiota by a cocktail of antibiotics leads to a significant downregulation of the interleukin-22 (IL-22) [
16]. IL-22 is an important cytokine in the gastrointestinal tract, as it plays a role in maintaining homeostatic levels and host defences against microbes, particularly in barrier repair [
18]. However, when the gut microbiota is restored using probiotics or faecal matter transplants (FMTs), the levels of IL-22 were restored to levels similar to chickens that had not undergone depletion of their gut microbiota [
16], thus demonstrating the key part that the gut microbiota plays in viral pathogenesis. We have previously shown reduced colon microbiota alpha diversity in the acute period of AIV infection (days 2–3) in both Rhode Island Red and VALO chicken lines which did not reach the same level as in uninfected chickens by day 10 post infection, which may further influence chicken health following recovery from infection [
19].
Compared to the gut microbiota, the respiratory microbiota has been less well studied. It has been shown that the composition of the respiratory microbiota in chickens is also altered by host-related and environmental factors, such as age, temperature and farm location, but little is known about the relationship between the respiratory microbiome and viral infection in chickens [
20,
21,
22]. LPAIVs have been shown to have a tropism for the respiratory tract and replicate there extensively [
11,
23,
24]. Therefore, it is important to understand how the microbiota in the respiratory tract of chickens is altered following influenza A virus infection, as this may have an impact on the innate immune responses elicited there and the resulting susceptibility to infection. Understanding these changes in the respiratory microbiota could provide information useful for the development of disease prevention and treatment strategies in the future.
In this study, Rhode Island Red (RIR) chickens were used to assess temporal changes in the chicken upper respiratory tract (URT) microbiota following H9N2 AIV infection. Additionally, the study aimed to assess how the healthy chicken respiratory microbiota changes during maturation up to 28 days of age.
2. Materials and Methods
2.1. Experiment Design
In this study, two in vivo animal experiments were performed. In Experiment 1, we used 35 specific pathogen-free (SPF) Rhode Island Red (RIR) chickens to assess the effect of H9N2 AIV infection on upper respiratory tract (URT) microbiota. These birds were reared together until two weeks of age when they were randomly separated into two groups: the control (
n = 20) and H9N2-AI-infected groups (
n = 15). At three weeks of age, the infected birds received 100 µL of 10
5 plaque-forming units (pfu/mL) recombinant H9N2 A/chicken/Pakistan/UDL01/01 via the intranasal route (50 µL in each nostril). On the day of challenge, 5 birds were randomly selected from the control group and culled to establish starting microbiota profile. On days 2, 4 and 10 post-challenge (p-ch), trachea samples were taken from 5 birds from both the control and infected groups. In Experiment 2, 20 SPF RIR chickens were kept in a cage together, and 5 birds were randomly selected and culled at days 7, 14, 21 and 28 of age to assess URT microbiota changes over time. These data were then compared with URT microbiota results obtained from Experiment 1 to further understand the changes that occurred during infection. RIR chickens were provided as day-old chicks from the National Avian Resource Facility (NARF) located at the Roslin Institute, Edinburgh, UK. The feed was provided ad libitum according to manufacturer’s instructions for the chicken age. All chicks moved from starter feed to grower at 3 weeks old. Control groups were housed in raised-floor pens whilst AIV-challenged chickens were housed in self-contained BioFlex
® B50 Rigid Body Poultry isolators (Bell Isolation Systems, Livingston, UK) maintained at negative pressure. All birds were swabbed daily from day of challenge until 8 days post infection to determine viral shedding. Swabbing was carried out with sterile polyester tipped swabs (Fisher Scientific, Loughborough, UK) which were transferred into viral transport media [
24], vortexed briefly, clarified by centrifugation and stored at −80 °C prior to virus detection. The URT samples, included anatomically: cranial larynx from proximal part, distally to syrinx, up to part where trachea divides into bronchi. Each trachea was then cut out from larynx with sterile scalpel. Proximal part of trachea samples was used for DNA/RNA extraction and snap-frozen, whereas distal trachea was used for histology examination and thus fixed in 4% buffered formaldehyde.
2.2. Virus Propagation
Recombinant A/chicken/Pakistan/UDL01/08 H9N2 virus was generated using reverse genetics as previously described [
25]. Virus stocks were produced via passage in 10-day-old embryonated chicken eggs; the allantoic fluid was harvested after 48 h and titrated by plaque assay on MDCK cells (ATCC). Madin-Darby Canine Kidney (MDCK) cells (ATCC) were maintained in DMEM (Gibco-Invitrogen, Inc., Waltham, MA, USA) supplemented with 10% fetal bovine serum (Biosera, Inc., Cholet, France), 1% penicillin/streptomycin (Sigma-Aldrich, Inc., St. Louis, MO, USA) and 1% non-essential aa (Sigma-Aldrich, Inc., St. Louis, MO, USA).
2.3. DNA Extraction and 16S rRNA Gene Amplification
Proximal part of trachea samples was cut and disrupted in the TissueLyser II (QIAGEN, Hilden, Germany) to homogenise the tissue. Each sample was then transferred to the PowerBead tubes provided in the DNeasy PowerSoil Kit (QIAGEN). These tubes were placed in the TissueLyser LT (QIAGEN) for next 10 min at 40 1/s. From this point onwards, the manufacturer’s instruction for the extraction of microbial genomic DNA using the DNeasy PowerSoil Kit was followed. Controls for DNA extraction reagents (negative control) and
E. coli DH5α (Thermo Fisher, Waltham, MA, USA) (positive control) were included. DNA from both controls followed the same DNA extraction protocol as described above. Controls were included to assess the level of possible contaminations and robustness of the method. The V2-V3 region of the 16S rRNA gene was amplified via a two-step nested PCR, using the protocol described by Glendinning et al. [
19]. This involved a V1-V4 region amplification using the primers 28F [5′-GAGTTTGATCNTGGCTCAG-3′] and 805R [5′-GACTACCAGGGTATCTAATC-3′]. This was followed by a short-cycle V2-V3 amplification using the primers 104F [5′-GGCGVACGGGTGAGTAA-3′] and 519R [5′-GTNTTACNGCGGCKGCTG-3′] with unique barcodes and Illumina adaptor sequences to prepare the gene segments for sequencing.
2.4. RNA Extraction and qRT-PCR
The supernatant from homogenised trachea tissue obtained at days 2 (n = 5) and 4 (n = 3) post-challenge was used for RNA extraction, using the QIAmp viral RNA minikit (Qiagen, Hilden, Germany) according to manufacturer instructions. For H9N2 influenza virus detection in trachea, quantitative analyses of matrix (M) were performed with primers IAV_F (5′-AGA TGA GTC TTC TAA CCG AGG TCG-3′) and IAV_R (5′-TGC AAA AAC ATC TTC AAG TCT CTG-3′) with IAV_probe (FAM-5′ TCA GGC CCC CTC AAA GCC GA-TAMRA-3′). qRT-PCR analysis was completed using the Superscript III Platinum one-step qRT-PCR kit (Life Technologies, Carlsbad, CA, USA). T7 RNA polymerase-derived transcripts from UDL-01 segment 7 were used for preparation of the standard curve. Cycling conditions were (1) 5 min at 50 °C, (2) a 2 min step at 95 °C and (3) 40 cycles of 3 s at 95 °C, 30 s of annealing and extension at 60 °C. Cycle threshold (CT) values were obtained using 7500 software v2.3.
2.5. Histology
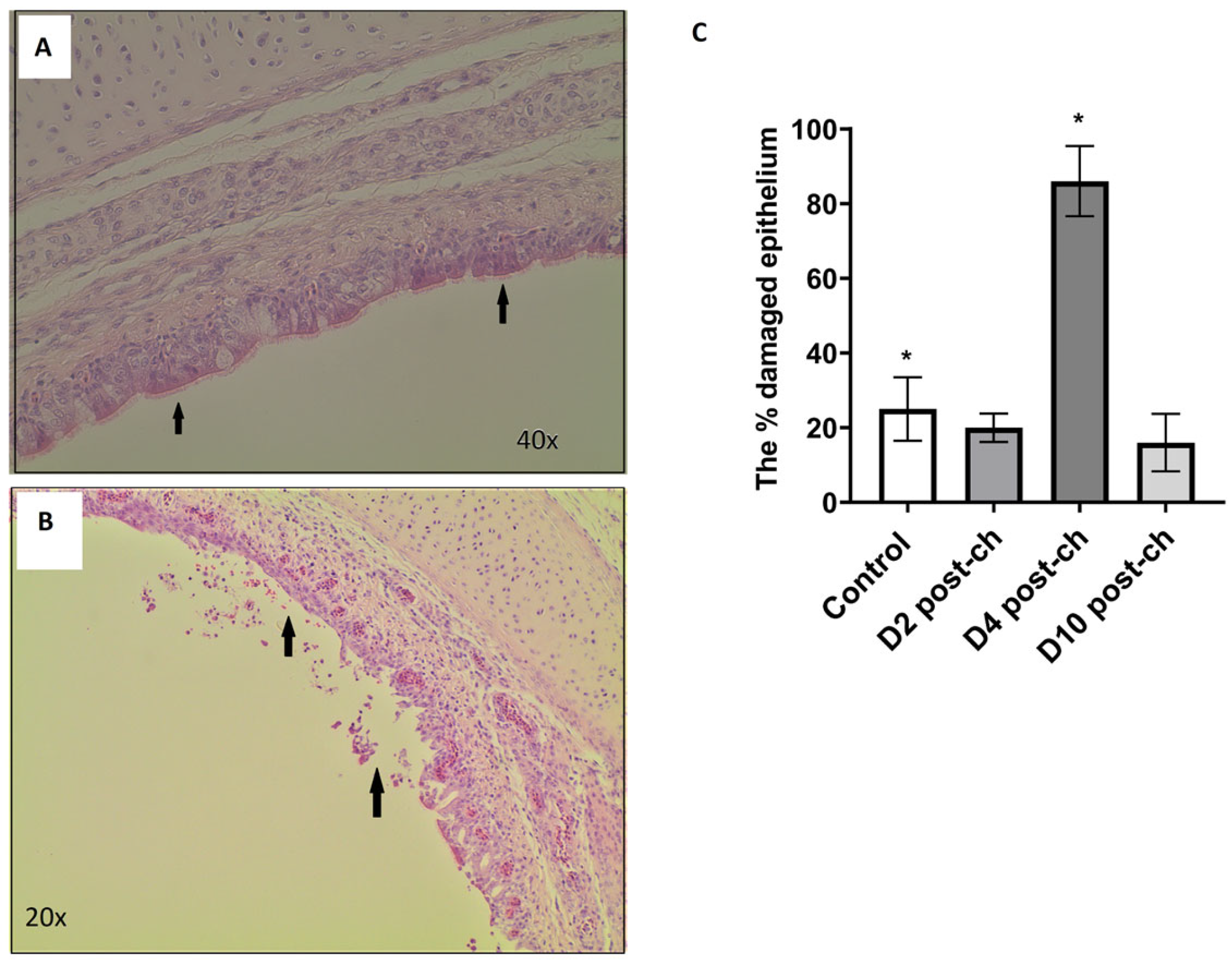
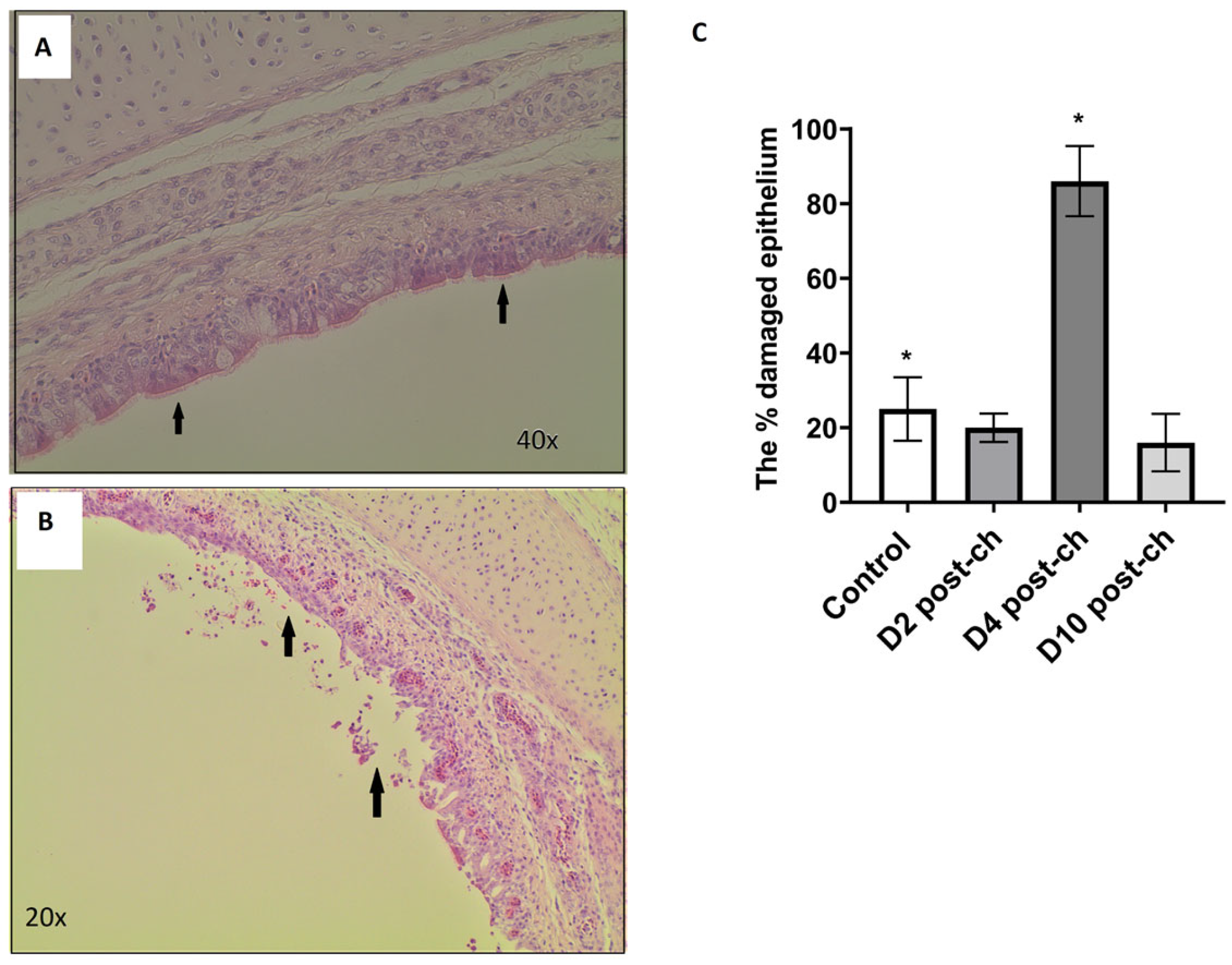
Trachea samples from controls were taken from a range of birds on days 2–10 (N = 6), whereas samples collected from H9N2-infected birds were taken at days 2, 4 and 10 post-challenge (5 samples at each time point tested). These samples were fixed in 4% buffered formaldehyde. In total, 21 samples were commercially prepared by ProPath® UK Limited (Hareford, UK). This included 15 samples from H9N2-infected group, five at each time point tested, accompanied by control samples, 2 at each time point tested. The tissues from 21 chickens were trimmed into cassettes as per Propath’s block list and Propath’s SOPs. The tissues were processed on a Tissue Tec VIP processor, dehydrated through a series of graded alcohols, cleared in toluene and infiltrated with poly wax. The processed tissues were then embedded in poly wax and sections cut at a nominal 4 µm, tissue sections were then placed on microscope glass slides. Slides were baked overnight in a 37 °C oven prior to staining. Slides from each of the 21 chicken trachea samples were dewaxed in xylene, hydrated through a series of graded alcohols and rinsed in running tap water. The slides were then stained in Mayer’s haematoxylin and washed in running tap water. The slides were then counter-stained with aqueous eosin and washed in two changes of running tap water. Finally, the slides were dehydrated through a series of graded alcohols, cleared in xylene and mounted with Pertex®. All stained slides from each of the 21 chickens were macroscopically and microscopically quality-checked by ProPath® UK Limited (Hareford, UK) for the following acceptance criteria: satisfactory staining and cover slipping; presence of correct tissue/tissue area on slide, absence of section artefact, e.g., scores, creases or chatter; the presence of lesions and abnormalities noted at necropsy; and consistency of embedding.
Delafield’s haematoxylin- and eosin-stained slides were then analysed using a light microscope. The percentage of the inner epithelial layer that had either been degraded or had the cilia on its surface stripped away was estimated independently by two people, and the mean percentage damage of the two estimates was used in the statistical analysis. Two-tailed t-tests were used to assess significant differences between the control and infected groups at each time point * p < 0.05.
2.6. 16S rRNA Sequencing and Data Analysis
Libraries were analysed on a High Sensitivity DNA Chip on the Bioanalyzer (Agilent Technologies, Santa Clara, CA, USA) and Qubit dsDNA HS assay (Invitrogen, Waltham, MA, USA) and then loaded on the flow cell of the 500 cycle MiSeq Reagent Kit v2 (Illumina, Santa Clara, CA, USA) and pair-end sequencing (2 × 250 bp). Quantitative Insights into Microbial Ecology (QIIME) platform version qiime2-2019.10 was used to analyse sequencing dataset. Low-quality sequencing reads were quality-trimmed and denoised using DADA2 (trim position 15 for both forward and reverse sequencing reads generated). Potential chimeric sequences were removed using UCHIME, and the remaining reads were assigned to 16S rRNA operational taxonomic units (OTUs) based on 97% nucleotide similarity with the UCLUST algorithm and then classified taxonomically using the SILVA reference database (silva132-99-nb-classifer). Taxonomy was then collapsed to the genus level. The microbial community structure was estimated by microbial biodiversity (i.e., species richness and between-sample diversity). Shannon index, phylogenetic diversity and the observed number of species were used to evaluate alpha diversity, and the unweighted UniFrac distances were used to evaluate beta diversity. All these indices (alpha and beta diversity) were calculated using the QIIME pipeline.
2.7. Statistical Analysis of Sequencing Data
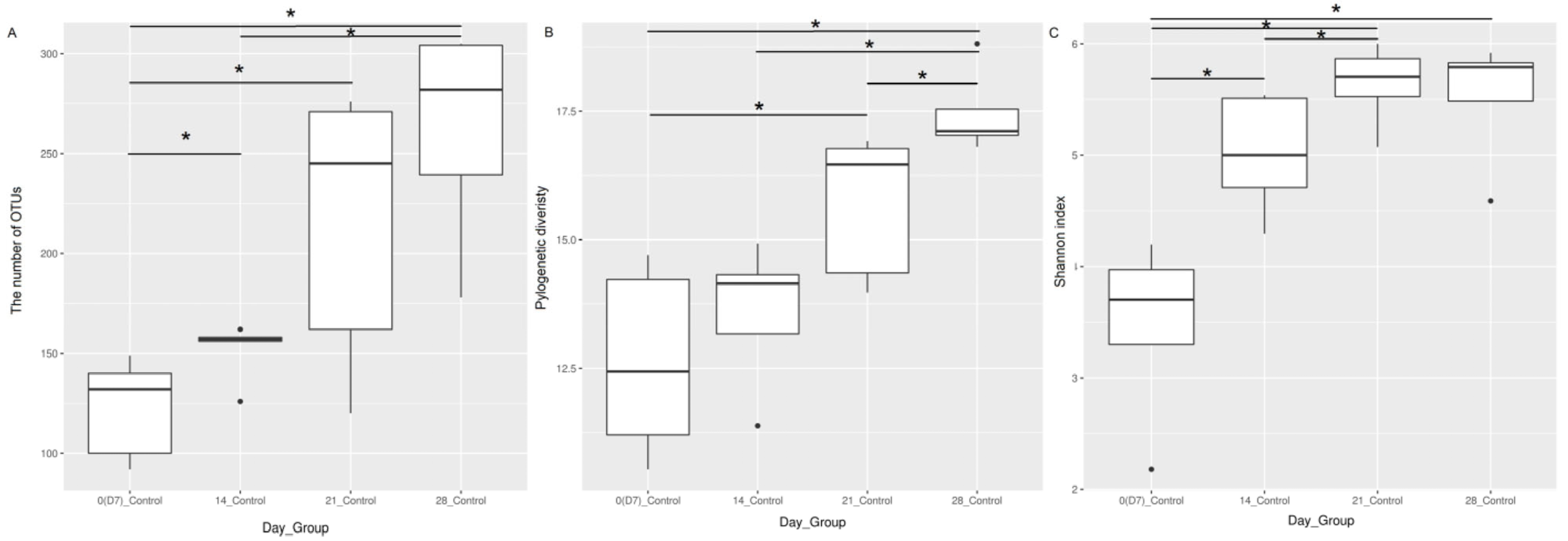
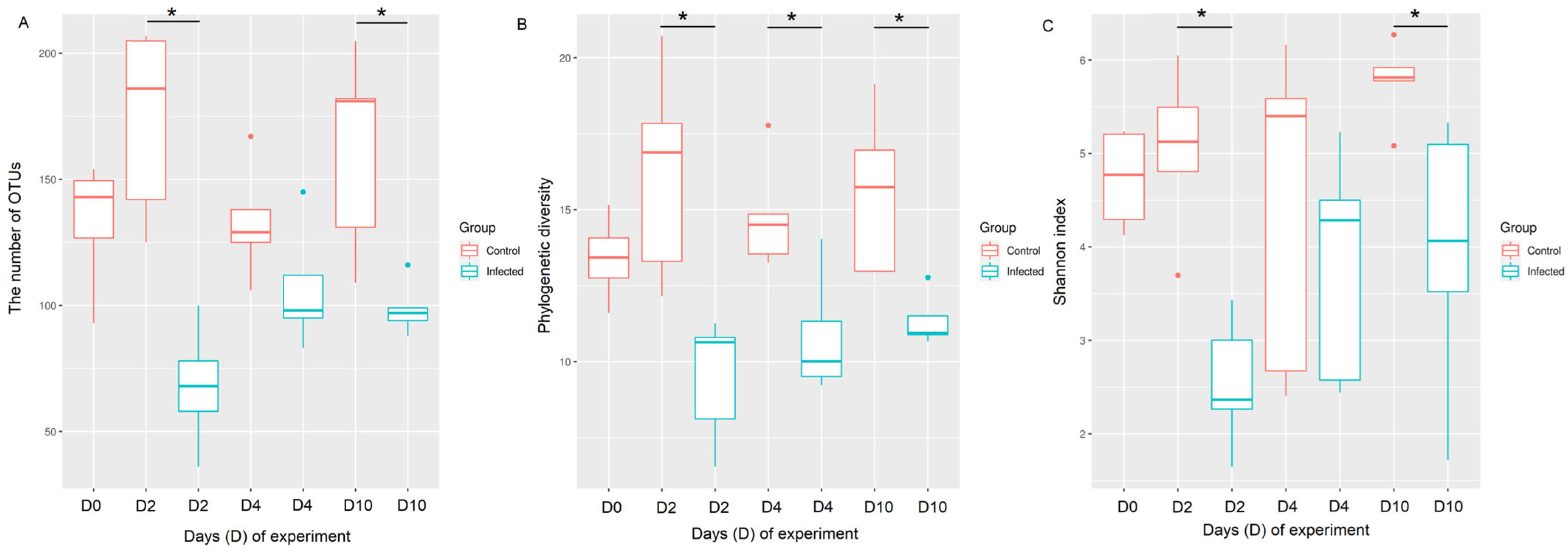
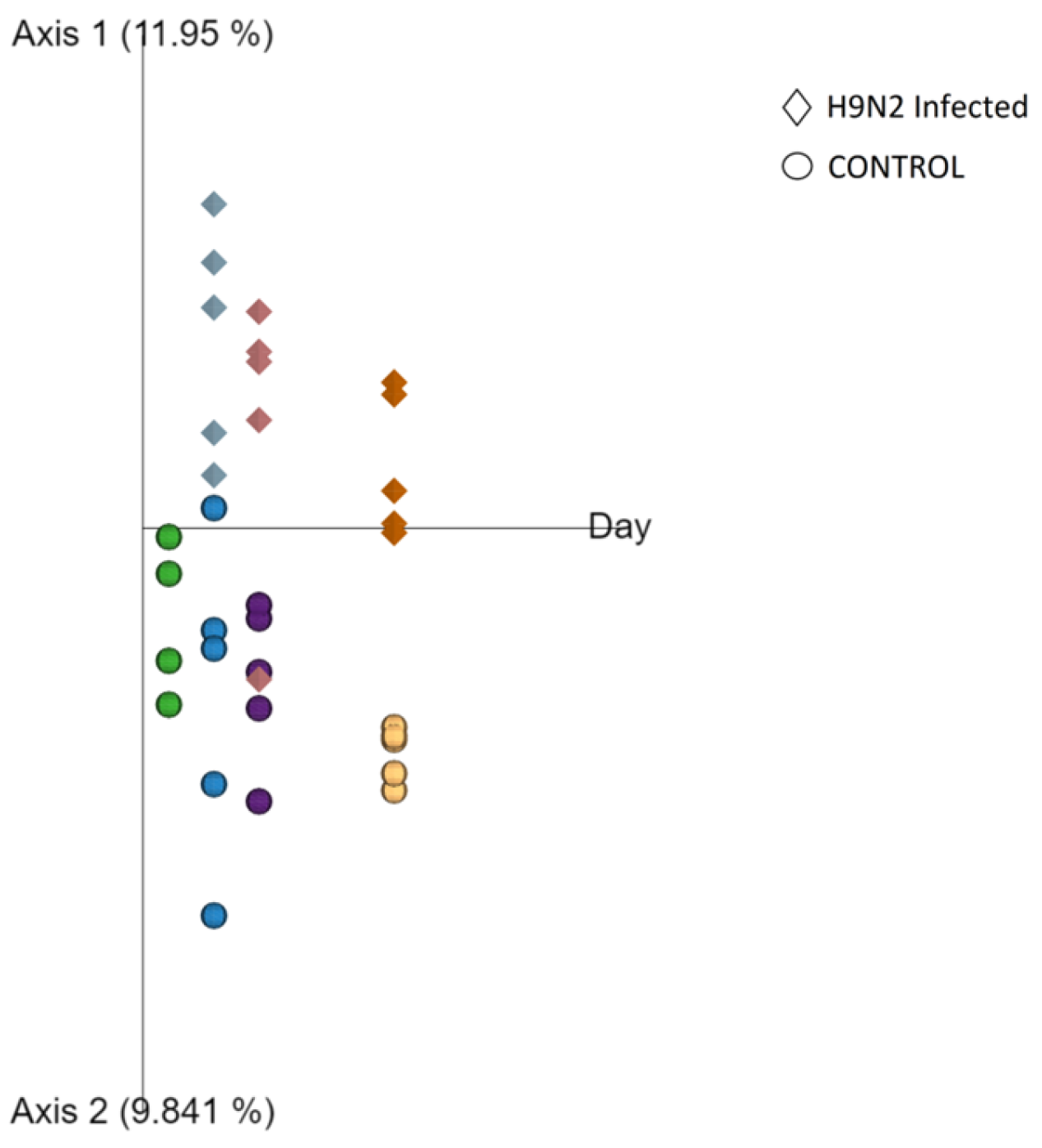
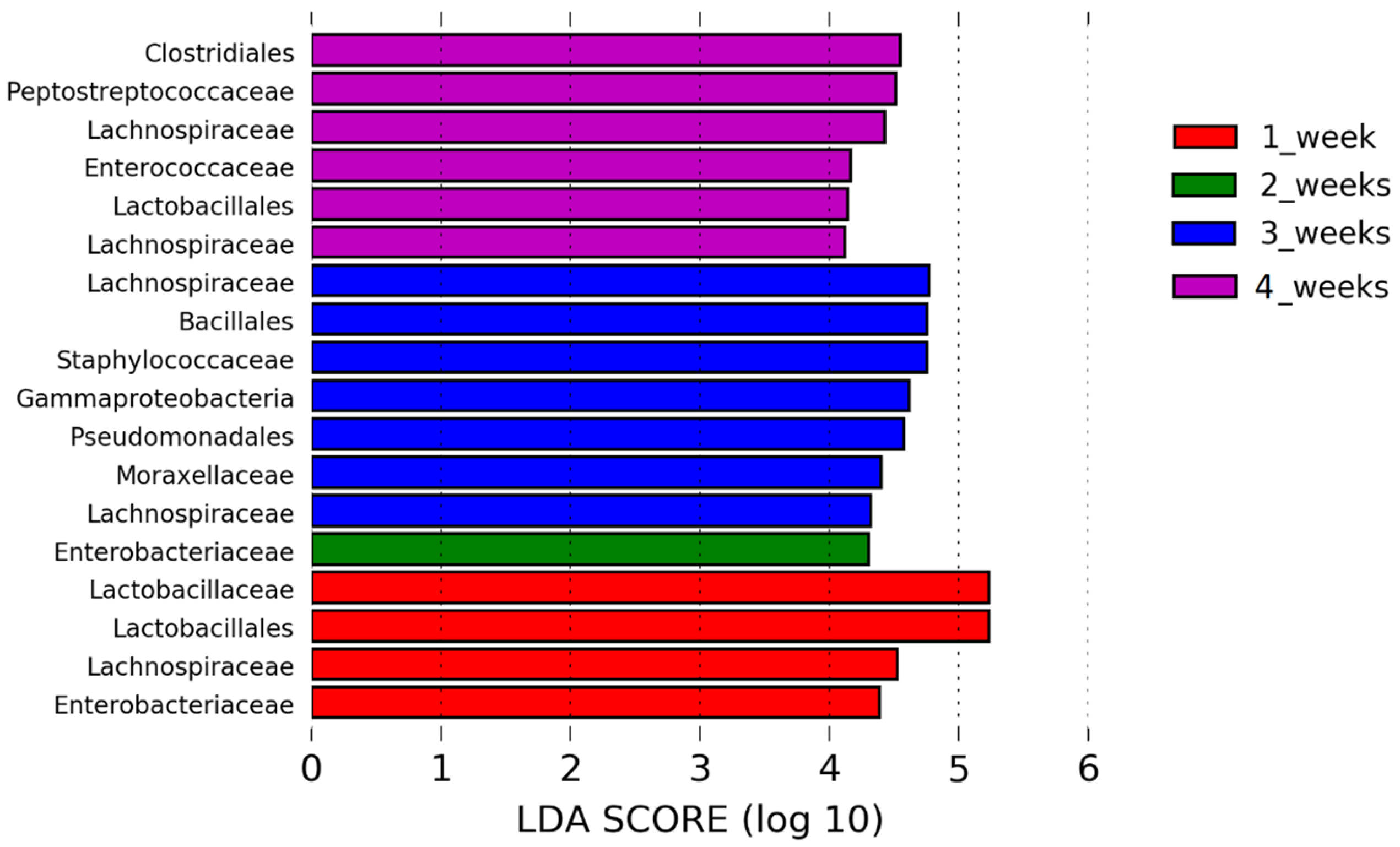
Kruskal–Wallis pairwise statistical testing was used to assess the differences in alpha diversity in terms of the observed number of operational taxonomic units (OTUs), Faith’s phylogenetic diversity and Shannon diversity. To assess beta diversity, analysis of variance (PERMANOVA) was performed using unweighted UniFrac distances. Principal-coordinate analysis (PCoA) graphs were constructed to visualize similarity between the samples. The linear discriminant analysis effect size (LEfSe) algorithm was used to identify differentially abundant taxa between the groups at genus level. For LEfSe analysis, depending on the experiments, different groups were assigned as comparison classes and were analysed by day. Briefly, in Experiment 1, RIR control and RIR AIV-infected groups were assigned as comparison classes and assessed at day 0, day 2, day 4 and day 10 post-challenge. In Experiment 2, control groups were assigned as comparison classes at days 7, 14, 21 and 28 and analysed all against all. LEfSe identified features that were statistically different between assigned groups and then compared the features using the non-parametric factorial Kruskal–Wallis sum-rank test (alpha value of 0.05) and linear discriminant analysis (LDA) cut-off value of 4.0.
4. Discussion
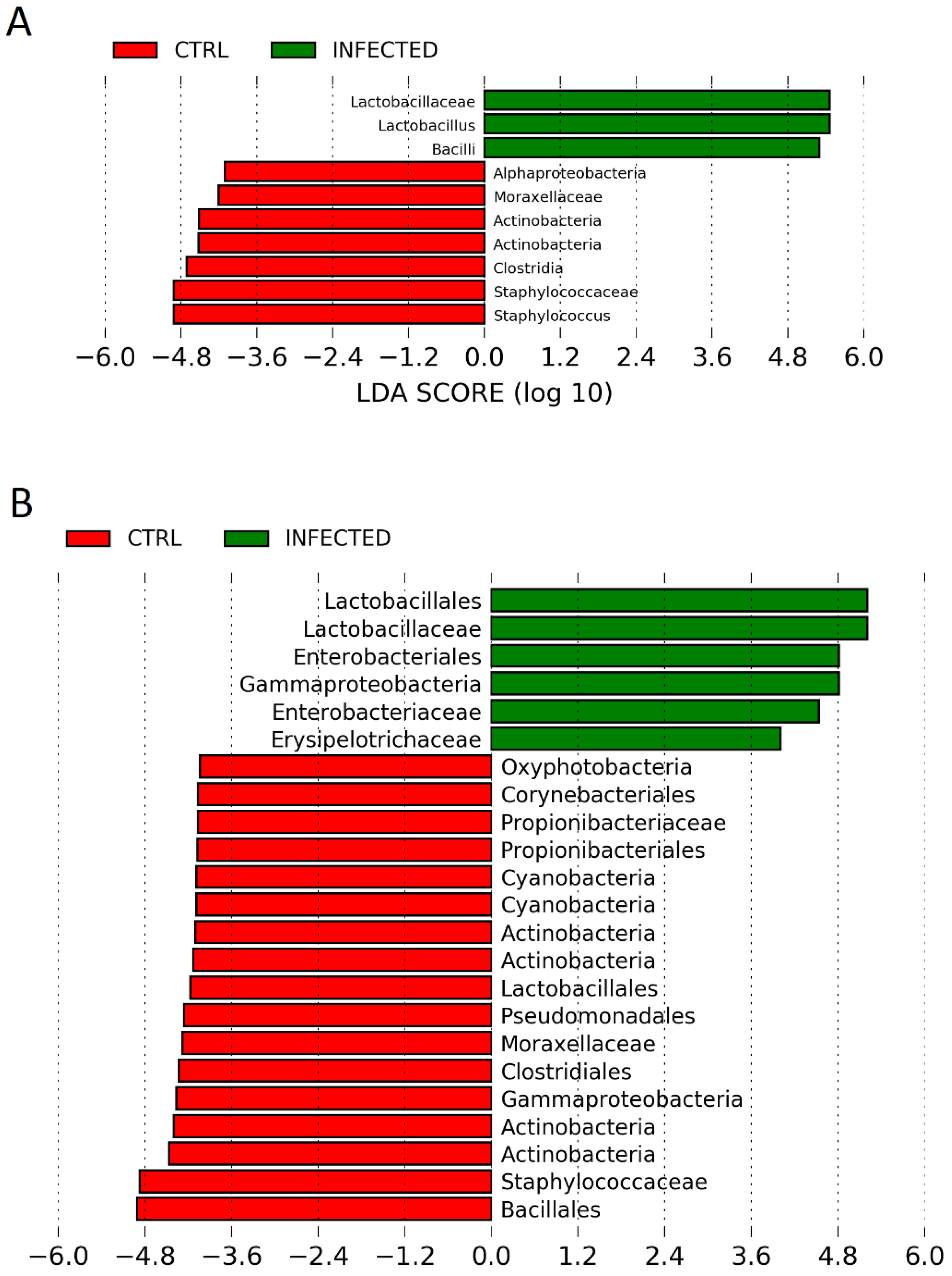
We demonstrated that the chicken upper respiratory (URT) microbiota is altered at the acute and recovery phases of AIV H9N2 infection. Both, alpha and beta diversity indices were continuously compromised following AIV infection. Furthermore, the composition of bacteria taxa also differs, suggesting that different bacteria taxa might play a role in the acute and recovery phases from AIV infection. The presence of the viral M-gene in the tracheas of chickens infected with H9N2 AIV was seen during the acute, infectious phase (days 2 and 4 post-challenge), verifying the replication of influenza A virus in tracheas at the time points tested. In addition, the highest levels of oral viral shedding were also detected at day 2 post-challenge which ceased by day 6 [
19], providing further evidence of active infection. During the acute phase of infection, the most significant losses in species richness, evenness and phylogenetic diversity were seen, which continued to be compromised also at the recovery phase of infection. In our previous study [
19], we saw a similar trend where all colon microbiota of H9N2-AIV-infected birds lost their overall richness at the acute phase of infection; however, species richness was restored in the recovery phase through the predominant bacteria becoming more dominant within the microbiota which we did not see in URT microbiota. Therefore, the typical levels of diversity in the upper respiratory microbiome are not quickly restored after the infectious phase.
The major bacterial families that characterise the healthy upper respiratory microbiota during maturation are
Lactobacillaceae,
Staphylococcaceae,
Lachnospiraceae,
Enterobacteriaceae and
Ruminococcaceae. This is in agreement with previous studies, which have shown that members of
Lactobacillales are the most abundant in the respiratory microbiota, along with others like
Enterobacteriales [
20,
26,
27]. It has also been shown previously that
Lactobacillaceae members are early colonisers of the chicken respiratory tract but become less abundant as the microbiota becomes more diverse [
20]. This is supported in our findings, as
Lactobacillaceae is the most abundant at 7 days of age, but it becomes less prominent over time as species evenness advances over time. However, the composition of bacteria in the URT can change during the H9N2 AIV infection as we showed in this study. In the acute phase of H9N2 AIV infection,
Lactobacillaceae highly dominates the upper respiratory microbiota. Furthermore,
Lactobacillaceae were still predominant bacteria at the recovery phase of infection suggesting an important role of this family in surviving influenza infection.
Lactobacillaceae is a family of Gram-positive lactic acid bacteria that has been shown to act as a probiotic [
28]. Members of the
Lactobacillaceae family, particularly the genus
Lactobacillus, have been shown to regulate the immune system and can activate innate and adaptive immune responses [
28,
29]. Interestingly, in our previous study on the colon microbiota, we noticed that although the
Firmicutes phylum was the most differentially abundant between infected and non-infected individuals, the
Lactobacillales was missing in the colon at the recovery phase of infection, and this trend was seen in two genetically distinct chicken breeds infected with H9N2 AIV [
19]. The exact reason why
Lactobacillaceae is so enriched in the acute and recovery phases of H9N2 AIV infection in the chicken upper respiratory tract and missing at the recovery phase in colon microbiota needs to be further investigated. However, this is the first report showing that there might be some potential interactions and cross-talk between gut-respiratory microbiota during active viral infection in chickens.
The next question we asked in this study was whether the reason for the dysbiosis of the respiratory microbiota observed during the H9N2 AIV infection relates to the physical disruption of the epithelial cell lining of the respiratory tract, which in turn could potentially cause the dysbiosis of the microbial communities that reside on it. No difference in epithelial cell lining was observed between the control and infected groups at days 2 and 10 post-challenge, suggesting that there must be another factor/s that corresponded to microbiota dysbiosis at this time point tested. We hypothesised that the reason for dysbiosis particularly at the acute phase of infection might be connected to an innate immune response elicited towards the replicating influenza A virus which could potentially directly impair the presence of some commensal bacteria, or it might have created an inflammatory environment where only some types of bacteria can survive. This suggests that bacteria might be either killed directly by immune responses or bacteria that are better adapted to persist in harsher conditions are able to thrive, while other species die out. The association between damage to the inner epithelial layer and H9N2 AIV infection was seen at day 4 post-challenge; however, this is not substantially associated with differences in microbial composition between the control and infected chickens observed in this study. The damage to the tissues in infected samples could be connected to the antiviral mechanism regulated by an interplay between different cytokine and interferon types. Morris et al. (2023) showed the upregulation of genes responsible for defence and inflammatory responses at day 5 in lungs following the H9N2 challenge in chickens [
30].
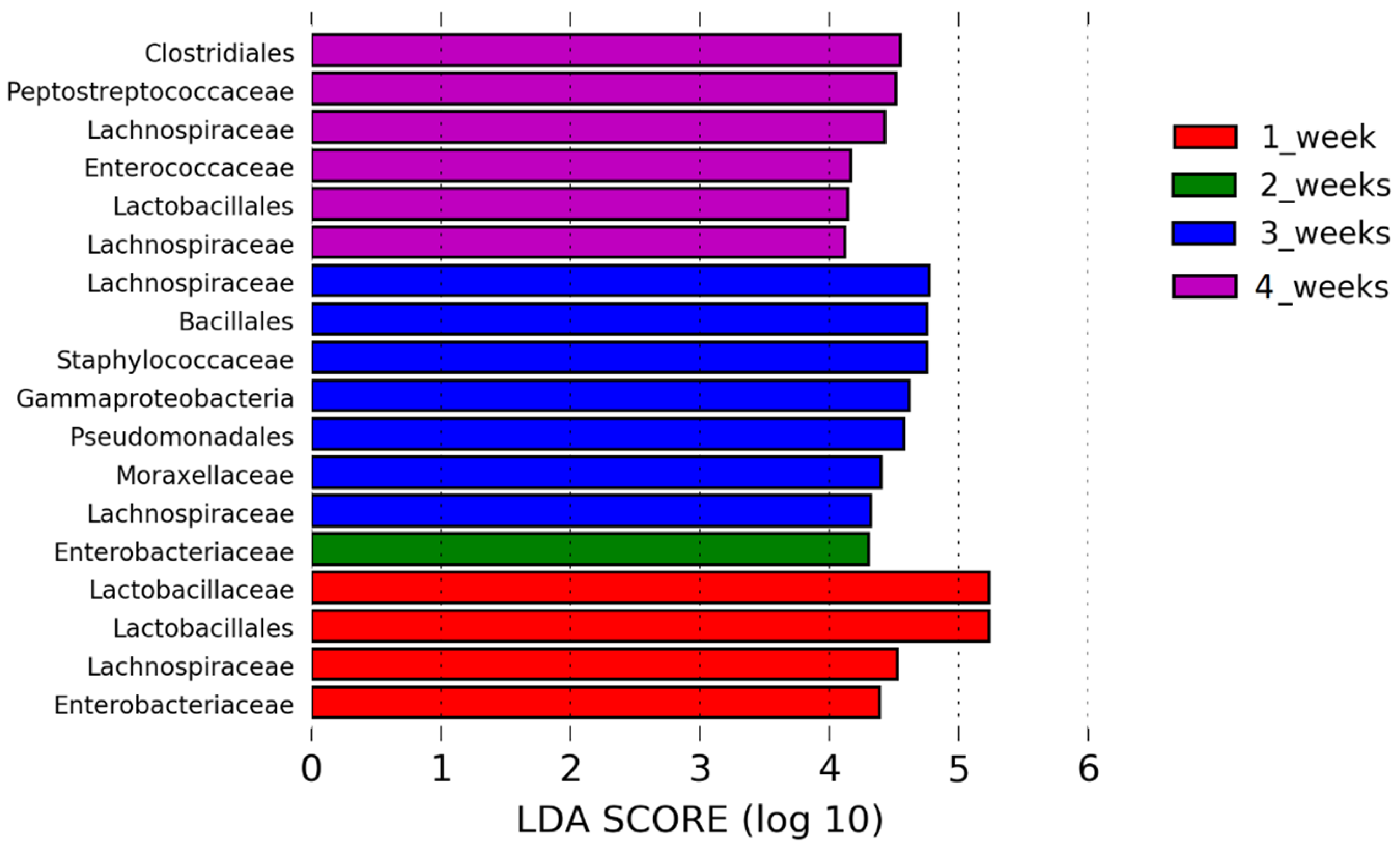
In addition to assessing changes in the URT microbiome, during the AIV H9N2 infection, we also showed that one of the factors that determines the composition of the healthy respiratory microbiota in the URT is age, as we observed alpha diversity indices increasing over time and differential enrichment of bacterial taxa as chickens matured up to 28 days of age. A significant positive correlation between alpha diversity indices and increasing age was observed, with the most significant period being between day 7 and days 14, 21 and 28. Beta diversity analysis supported our finding that microbial diversity is shaped by ageing. Interestingly, no significant changes in beta diversity were found between days 14 and 21 of age or in alpha diversity indices (OTUs and phylogenetic diversity) at this time point tested, indicating that the least substantial changes occur between weeks two and three of age. Although our control Experiment 2 finished at the bird age of 28 days, we observed significant separation between day 0 and 10 control groups in our challenge experiment (Experiment 1). This provides further evidence for age being a factor that influences the respiratory microbiota, as it suggests that the microbial composition in the respiratory tract significantly evolved during maturation from days 21 to 31 of age, which goes further beyond the timeframe of age we studied in chickens for Experiment 2. It was previously shown that the respiratory tract of chickens is populated by a diverse community of commensal bacteria, whose composition can be altered by a number of host- and environment-related factors [
20,
22,
26].
In conclusion, this is the first study showing alteration of upper respiratory tract microbiota flowing the H9N2 AIV infection in chickens. We observed that alpha and beta diversity in the respiratory microbiota changes in both the acute and recovery phases of influenza A virus infection and that there is dysbiosis in the abundance of different bacterial taxa at different time points. A lack of homeostasis in the upper respiratory tract seems to be connected to different factors and might change over the time of infection. Dysbiosis might be more associated with anti-viral response, which could then directly or indirectly (by changing the environment) affect microbiota on mucosal surfaces, and less with physical damage caused to the inner epithelial layer cells by the replicating virus. The family Lactobacillaceae is most strongly associated with infected chickens compared to the controls during the acute phase and continues to be a major player in the respiratory microbiota into the recovery phase, while the abundance of less dominant families is reduced to levels lower than in control chickens. Although the reason why the Lactobacillaceae family was the main player during the AIV infection in the upper respiratory tract of chickens is unknown and needs to be investigated further, this study might suggest that supplementation with Lactobacillaceae could potentially aid recovery from H9N2 influenza infection.


 ,
, 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}