
A Multilayered Imaging and Microfluidics Approach for Evaluating the Effect of Fibrinolysis in Staphylococcus aureus Biofilm Formation

 ,
,  , , , , ,
, , , , ,  and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Ethics Statement and Clinical Samples
2.2. Staphylococcus aureus
2.3. IgG Depletion
2.4. IgG Titration
2.5. Microfluidic Device
2.6. Microfluidic Experimental Workflow and Live Cell Imaging
2.7. Analysis
3. Results
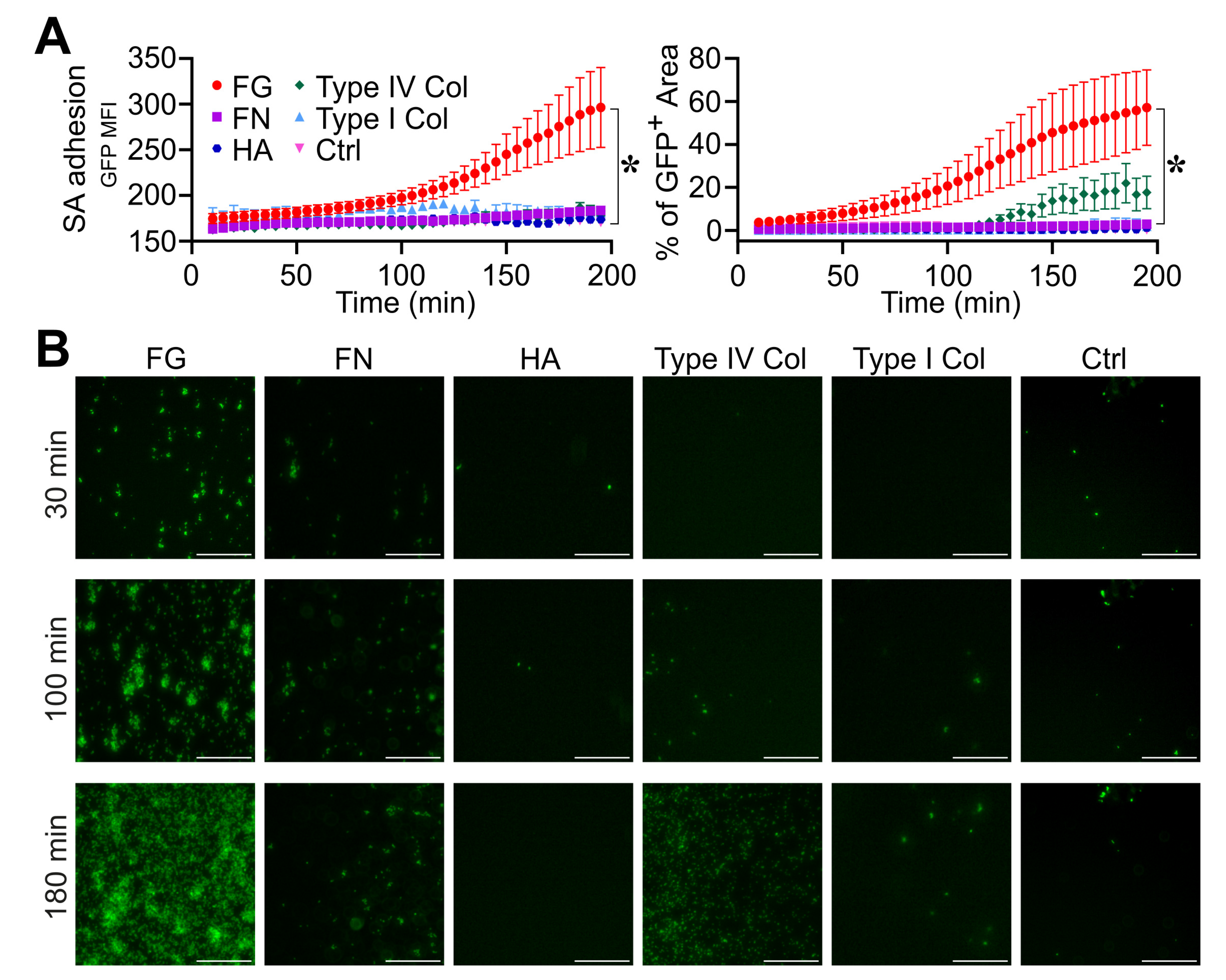
3.1. Fibrinogen Is Essential in the Different Phases of S. aureus Biofilm Formation
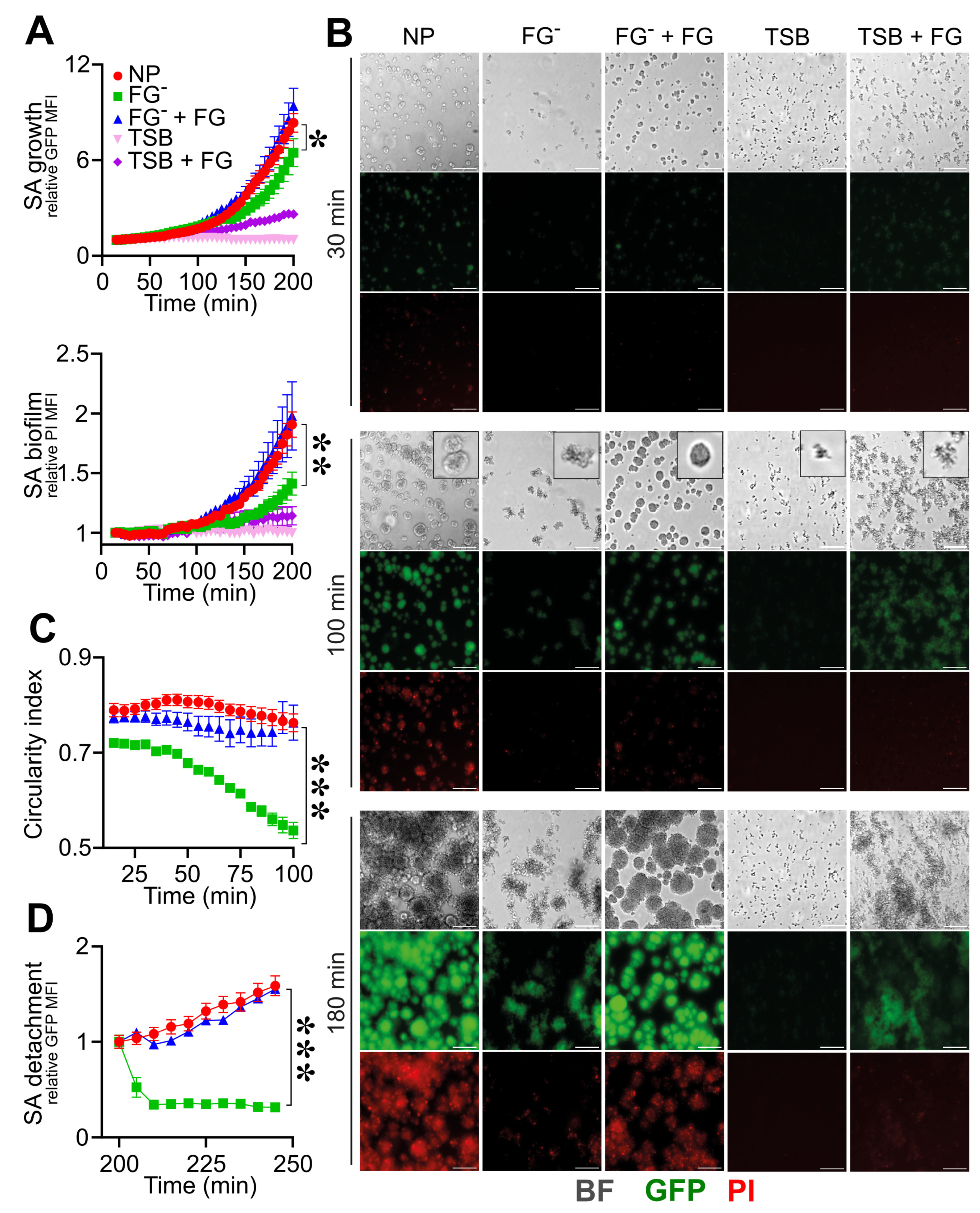
3.2. Triggering Fibrinolysis Interferes in the Formation of S. aureus Biofilm
3.3. The Reactivation of Fibrinolysis in S. aureus-Induced Sepsis Favors an IgG-Mediated Immune Response
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bergmann, S.; Hammerschmidt, S. Fibrinolysis and host response in bacterial infections. Thromb. Haemost. 2007, 98, 512–520. [Google Scholar] [CrossRef] [PubMed]
- Levi, M.; Van Der Poll, T.; Büller, H.R. Bidirectional relation between inflammation and coagulation. Circulation 2004, 109, 2698–2704. [Google Scholar] [CrossRef] [PubMed]
- Koyama, K.; Madoiwa, S.; Nunomiya, S.; Koinuma, T.; Wada, M.; Sakata, A.; Ohmori, T.; Mimuro, J.; Sakata, Y. Combination of thrombin-antithrombin complex, plasminogen activator inhibitor-1, and protein C activity for early identification of severe coagulopathy in initial phase of sepsis: A prospective observational study. Crit. Care 2014, 18, R13. [Google Scholar] [CrossRef]
- Milbrandt, E.B.; GenIMS Investigators; Reade, M.C.; Lee, M.; Shook, S.L.; Angus, D.C.; Kong, L.; Carter, M.; Yealy, D.M.; Kellum, J.A. Prevalence and significance of coagulation abnormalities in community-acquired pneumonia. Mol. Med. 2009, 15, 438–445. [Google Scholar] [CrossRef] [PubMed]
- Bokarewa, M.I.; Tarkowski, A. Thrombin generation and mortality during Staphylococcus aureus sepsis. Microb. Pathog. 2001, 30, 247–252. [Google Scholar] [CrossRef] [PubMed]
- Semple, J.W.; Italiano, J.E.; Freedman, J. Platelets and the immune continuum. Nat. Rev. Immunol. 2011, 11, 264–274. [Google Scholar] [CrossRef] [PubMed]
- Flick, M.J.; Du, X.; Witte, D.P.; Jiroušková, M.; Soloviev, D.A.; Busuttil, S.J.; Plow, E.F.; Degen, J.L. Leukocyte engagement of fibrin(ogen) via the integrin receptor αMβ2/Mac-1 is critical for host inflammatory response in vivo. J. Clin. Investig. 2004, 113, 1596–1606. [Google Scholar] [CrossRef] [PubMed]
- Øhlenschlæger, T.; Garred, P.; Madsen, H.O.; Jacobsen, S. Mannose-Binding Lectin Variant Alleles and the Risk of Arterial Thrombosis in Systemic Lupus Erythematosus. N. Engl. J. Med. 2004, 351, 260–267. [Google Scholar] [CrossRef]
- Doni, A.; Garlanda, C.; Mantovani, A. Innate immunity, hemostasis and matrix remodeling: PTX3 as a link. Semin. Immunol. 2016, 28, 570–577. [Google Scholar] [CrossRef][Green Version]
- Engelmann, B.; Massberg, S. Thrombosis as an intravascular effector of innate immunity. Nat. Rev. Immunol. 2013, 13, 34–45. [Google Scholar] [CrossRef]
- Levi, M.; van der Poll, T. Coagulation and sepsis. Thromb. Res. 2017, 149, 38–44. [Google Scholar] [CrossRef] [PubMed]
- Doni, A.; D’Amico, G.; Morone, D.; Mantovani, A.; Garlanda, C. Humoral innate immunity at the crossroad between microbe and matrix recognition: The role of PTX3 in tissue damage. Semin. Cell Dev. Biol. 2017, 61, 31–40. [Google Scholar] [CrossRef] [PubMed]
- Foster, T.J. Immune evasion by staphylococci. Nat. Rev. Microbiol. 2005, 3, 948–958. [Google Scholar] [CrossRef] [PubMed]
- He, Y.-W.; Li, H.; Zhang, J.; Hsu, C.-L.; Lin, E.; Zhang, N.; Guo, J.; Forbush, K.A.; Bevan, M.J. The extracellular matrix protein mindin is a pattern-recognition molecule for microbial pathogens. Nat. Immunol. 2004, 5, 88–97. [Google Scholar] [CrossRef] [PubMed]
- Schack, L.; Stapulionis, R.; Christensen, B.; Kofod-Olsen, E.; Sørensen, U.B.S.; Vorup-Jensen, T.; Sørensen, E.S.; Höllsberg, P. Osteopontin Enhances Phagocytosis through a Novel Osteopontin Receptor, the αXβ2 Integrin. J. Immunol. 2009, 182, 6943–6950. [Google Scholar] [CrossRef]
- Sutherland, T.E.; Dyer, D.P.; Allen, J.E. The extracellular matrix and the immune system: A mutually dependent relationship. Science 2023, 379, eabp8964. [Google Scholar] [CrossRef]
- Jokinen, E.; Laine, J.; Huttunen, R.; Lyytikäinen, O.; Vuento, R.; Vuopio, J.; Syrjänen, J. Trends in incidence and resistance patterns of Staphylococcus aureus bacteremia. Infect. Dis. 2018, 50, 52–58. [Google Scholar] [CrossRef]
- Parsons, J.B.; Westgeest, A.C.; Conlon, B.P.; Fowler, V.G. Persistent Methicillin-Resistant Staphylococcus aureus Bacteremia: Host, Pathogen, and Treatment. Antibiotics 2023, 12, 455. [Google Scholar] [CrossRef]
- Nair, R.; Ammann, E.; Rysavy, M.; Schweizer, M.L. Mortality among Patients with Methicillin-Resistant Staphylococcus aureus USA300 versus Non-USA300 Invasive Infections: A Meta-Analysis. Infect. Control Hosp. Epidemiol. 2014, 35, 31–41. [Google Scholar] [CrossRef]
- Stryjewski, M.E.; Corey, G.R. Methicillin-resistant Staphylococcus aureus: An evolving pathogen. Clin. Infect. Dis. 2014, 58 (Suppl. S1), S10–S19. [Google Scholar] [CrossRef]
- Liesenborghs, L.; Verhamme, P.; Vanassche, T. Staphylococcus aureus, master manipulator of the human hemostatic system. J. Thromb. Haemost. 2018, 16, 441–454. [Google Scholar] [CrossRef] [PubMed]
- Ko, Y.P.; Flick, M.J. Fibrinogen Is at the Interface of Host Defense and Pathogen Virulence in Staphylococcus aureus Infection. Semin. Thromb. Hemost. 2016, 42, 408–421. [Google Scholar] [CrossRef]
- Thomer, L.; Schneewind, O.; Missiakas, D. Pathogenesis of Staphylococcus aureus Bloodstream Infections. Annu. Rev. Pathol. Mech. Dis. 2016, 11, 343–364. [Google Scholar] [CrossRef] [PubMed]
- Cheng, A.G.; McAdow, M.; Kim, H.K.; Bae, T.; Missiakas, D.M.; Schneewind, O. Contribution of coagulases towards Staphylococcus aureus disease and protective immunity. PLoS Pathog. 2010, 6, e1001036. [Google Scholar] [CrossRef] [PubMed]
- Kwiecinski, J.; Peetermans, M.; Liesenborghs, L.; Na, M.; Björnsdottir, H.; Zhu, X.; Jacobsson, G.; Johansson, B.R.; Geoghegan, J.A.; Foster, T.J.; et al. Staphylokinase control of Staphylococcus aureus biofilm formation and detachment through host plasminogen activation. J. Infect. Dis. 2016, 213, 139–148. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Li, J.; Hagström, E.; Ny, T. Protective effects of plasmin(ogen) in a mouse model of Staphylococcus aureus-induced arthritis. Arthritis Rheum. 2008, 58, 764–772. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, S.D.; Deleo, F.R. A MRSA-terious enemy among us: Boosting MRSA vaccines. Nat. Med. 2011, 17, 168–169. [Google Scholar] [CrossRef]
- Agarwal, A.; Singh, K.P.; Jain, A. Medical significance and management of staphylococcal biofilm. FEMS Immunol. Med. Microbiol. 2010, 58, 147–160. [Google Scholar] [CrossRef]
- Rodrigues, L.R. Inhibition of bacterial adhesion on medical devices. Adv. Exp. Med. Biol. 2011, 715, 351–367. [Google Scholar] [CrossRef]
- Arciola, C.R.; Campoccia, D.; Speziale, P.; Montanaro, L.; Costerton, J.W. Biofilm formation in Staphylococcus implant infections. A review of molecular mechanisms and implications for biofilm-resistant materials. Biomaterials 2012, 33, 5967–5982. [Google Scholar] [CrossRef]
- Howden, B.P.; Giulieri, S.G.; Lung, T.W.F.; Baines, S.L.; Sharkey, L.K.; Lee, J.Y.H.; Hachani, A.; Monk, I.R.; Stinear, T.P. Staphylococcus aureus host interactions and adaptation. Nat. Rev. Microbiol. 2023, 21, 380–395. [Google Scholar] [CrossRef]
- Knott, S.; Curry, D.; Zhao, N.; Metgud, P.; Dastgheyb, S.S.; Purtill, C.; Harwood, M.; Chen, A.F.; Schaer, T.P.; Otto, M.; et al. Staphylococcus aureus Floating Biofilm Formation and Phenotype in Synovial Fluid Depends on Albumin, Fibrinogen, and Hyaluronic Acid. Front. Microbiol. 2021, 12, 655873. [Google Scholar] [CrossRef] [PubMed]
- Archer, N.K.; Mazaitis, M.J.; Costerton, J.W.; Leid, J.G.; Powers, M.E.; Shirtliff, M.E. Staphylococcus aureus biofilms: Properties, regulation and roles in human disease. Virulence 2011, 2, 445–459. [Google Scholar] [CrossRef] [PubMed]
- Zapotoczna, M.; McCarthy, H.; Rudkin, J.K.; O’Gara, J.P.; O’Neill, E. An essential role for coagulase in Staphylococcus aureus biofilm development reveals new therapeutic possibilities for device-related infections. J. Infect. Dis. 2015, 212, 1883–1893. [Google Scholar] [CrossRef] [PubMed]
- Bryers, J.D. Medical biofilms. Biotechnol. Bioeng. 2008, 100, 1–18. [Google Scholar] [CrossRef]
- Akiyama, H.; Ueda, M.; Kanzaki, H.; Tada, J.; Arata, J. Biofilm formation of Staphylococcus aureus strains isolated from impetigo and furuncle: Role of fibrinogen and fibrin. J. Dermatol. Sci. 1997, 16, 2–10. [Google Scholar] [CrossRef]
- Yawata, Y.; Nguyen, J.; Stocker, R.; Rusconi, R. Microfluidic studies of biofilm formation in dynamic environments. J. Bacteriol. 2016, 198, 2589–2595. [Google Scholar] [CrossRef]
- Yuan, L.; Straub, H.; Shishaeva, L.; Ren, Q. Microfluidics for Biofilm Studies. Annu. Rev. Anal. Chem. 2023, 16, 139–159. [Google Scholar] [CrossRef]
- Aleklett, K.; Kiers, E.T.; Ohlsson, P.; Shimizu, T.S.; Caldas, V.E.; Hammer, E.C. Build your own soil: Exploring microfluidics to create microbial habitat structures. ISME J. 2018, 12, 312–319. [Google Scholar] [CrossRef]
- Takahashi, K.; Li, X.; Kunoh, T.; Nagasawa, R.; Takeshita, N.; Utada, A.S. Novel Insights into Microbial Behavior Gleaned Using Microfluidics. Microbes Environ. 2023, 38, ME22089. [Google Scholar] [CrossRef]
- Pidwill, G.R.; Pyrah, J.F.; Sutton, J.A.F.; Best, A.; Renshaw, S.A.; Foster, S.J. Clonal population expansion of Staphylococcus aureus occurs due to escape from a finite number of intraphagocyte niches. Sci. Rep. 2023, 13, 1188. [Google Scholar] [CrossRef] [PubMed]
- Belgiovine, C.; Pellegrino, L.; Bulgarelli, A.; Lauta, F.C.; Di Claudio, A.; Ciceri, R.; Cancellara, A.; Calcaterra, F.; Mavilio, D.; Grappiolo, G.; et al. Interaction of Bacteria, Immune Cells, and Surface Topography in Periprosthetic Joint Infections. Int. J. Mol. Sci. 2023, 24, 9028. [Google Scholar] [CrossRef] [PubMed]
- Savorana, G.; Słomka, J.; Stocker, R.; Rusconi, R.; Secchi, E. A microfluidic platform for characterizing the structure and rheology of biofilm streamers. Soft Matter 2022, 18, 3878–3890. [Google Scholar] [CrossRef] [PubMed]
- Secchi, E.; Savorana, G.; Vitale, A.; Eberl, L.; Stocker, R.; Rusconi, R. The structural role of bacterial eDNA in the formation of biofilm streamers. Proc. Natl. Acad. Sci. USA 2022, 119, e2113723119. [Google Scholar] [CrossRef] [PubMed]
- Qin, D.; Xia, Y.; Whitesides, G.M. Soft lithography for micro- and nanoscale patterning. Nat. Protoc. 2010, 5, 491–502. [Google Scholar] [CrossRef] [PubMed]
- Boulos, L.; Prévost, M.; Barbeau, B.; Coallier, J.; Desjardins, R. LIVE/DEAD® BacLight(TM): Application of a new rapid staining method for direct enumeration of viable and total bacteria in drinking water. J. Microbiol. Methods 1999, 37, 77–86. [Google Scholar] [CrossRef] [PubMed]
- Whitchurch, C.B.; Tolker-Nielsen, T.; Ragas, P.C.; Mattick, J.S. Extracellular DNA required for bacterial biofilm formation. Science 2002, 295, 1487. [Google Scholar] [CrossRef] [PubMed]
- Allesen-Holm, M.; Barken, K.B.; Yang, L.; Klausen, M.; Webb, J.S.; Kjelleberg, S.; Molin, S.; Givskov, M.; Tolker-Nielsen, T. A characterization of DNA release in Pseudomonas aeruginosa cultures and biofilms. Mol. Microbiol. 2006, 59, 1114–1128. [Google Scholar] [CrossRef]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef]
- Berg, S.; Kutra, D.; Kroeger, T.; Straehle, C.N.; Kausler, B.X.; Haubold, C.; Schiegg, M.; Ales, J.; Beier, T.; Rudy, M.; et al. ilastik: Interactive machine learning for (bio)image analysis. Nat. Methods 2019, 16, 1226–1232. [Google Scholar] [CrossRef]
- R. C. Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2021. [Google Scholar]
- Otto, M. Staphylococcal infections: Mechanisms of biofilm maturation and detachment as critical determinants of pathogenicity. Annu. Rev. Med. 2013, 64, 175–188. [Google Scholar] [CrossRef] [PubMed]
- Paharik, A.E.; Horswill, A.R. The staphylococcal biofilm: Adhesins, regulation, and host response. Microbiol. Spectr. 2016, 4, 529–566. [Google Scholar] [CrossRef] [PubMed]
- Aiyer, K.S.; Vijayakumar, B.S.; Vishwanathan, A.S. The enigma of biofilms. Curr. Sci. 2018, 115, 204–205. [Google Scholar] [CrossRef]
- Thomas, S.; Liu, W.; Arora, S.; Ganesh, V.; Ko, Y.P.; Höök, M. The complex fibrinogen interactions of the Staphylococcus aureus coagulases. Front. Cell. Infect. Microbiol. 2019, 9, 106. [Google Scholar] [CrossRef]
- Ayache, S.; Panelli, M.C.; Byrne, K.M.; Slezak, S.; Leitman, S.F.; Marincola, F.M.; Stroncek, D.F. Comparison of proteomic profiles of serum, plasma, and modified media supplements used for cell culture and expansion. J. Transl. Med. 2006, 4, 40. [Google Scholar] [CrossRef]
- Pietrocola, G.; Campoccia, D.; Motta, C.; Montanaro, L.; Arciola, C.R.; Speziale, P. Colonization and Infection of Indwelling Medical Devices by Staphylococcus aureus with an Emphasis on Orthopedic Implants. Int. J. Mol. Sci. 2022, 23, 5958. [Google Scholar] [CrossRef] [PubMed]
- Das, S.; Kumar, A. Formation and post-formation dynamics of bacterial biofilm streamers as highly viscous liquid jets. Sci. Rep. 2014, 4, 7126. [Google Scholar] [CrossRef] [PubMed]
- Jørgensen, N.P.; Zobek, N.; Dreier, C.; Haaber, J.; Ingmer, H.; Larsen, O.H.; Meyer, R.L. Streptokinase treatment reverses biofilm-associated antibiotic resistance in Staphylococcus aureus. Microorganisms 2016, 4, 36. [Google Scholar] [CrossRef]
- Hogan, S.; O’Gara, J.P.; O’Neill, E. Novel treatment of Staphylococcus aureus device-related infections using fibrinolytic agents. Antimicrob. Agents Chemother. 2018, 62, e02008–e02017. [Google Scholar] [CrossRef]
- Kwiecinski, J.; Na, M.; Jarneborn, A.; Jacobsson, G.; Peetermans, M.; Verhamme, P.; Jin, T. Tissue plasminogen activator coating on implant surfaces reduces Staphylococcus aureus biofilm formation. Appl. Environ. Microbiol. 2016, 82, 394–401. [Google Scholar] [CrossRef]
- Chapin, J.C.; Hajjar, K.A. Fibrinolysis and the control of blood coagulation. Blood Rev. 2015, 29, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Roilides, E.; Simitsopoulou, M.; Katragkou, A.; Walsh, T.J. How Biofilms Evade Host Defenses. Microbiol. Spectr. 2015, 3, 287–300. [Google Scholar] [CrossRef] [PubMed]
- Degen, J.L.; Bugge, T.H.; Goguen, J.D. Fibrin and fibrinolysis in infection and host defense. J. Thromb. Haemost. 2007, 5 (Suppl. S1), 24–31. [Google Scholar] [CrossRef] [PubMed]
- Panigada, M.; Zacchetti, L.; L’acqua, C.; Cressoni, M.; Anzoletti, M.B.; Bader, R.; Protti, A.; Consonni, D.; D’angelo, A.; Gattinoni, L. Assessment of fibrinolysis in sepsis patients with urokinase modified thromboelastography. PLoS ONE 2015, 10, e0136463. [Google Scholar] [CrossRef]
- Fitzgerald, J.R.; Foster, T.J.; Cox, D. The interaction of bacterial pathogens with platelets. Nat. Rev. Microbiol. 2006, 4, 445–457. [Google Scholar] [CrossRef] [PubMed]
- Tsuruta, Y.; Park, Y.-J.; Siegal, G.P.; Liu, G.; Abraham, E. Involvement of Vitronectin in Lipopolysaccaride-Induced Acute Lung Injury. J. Immunol. 2007, 179, 7079–7086. [Google Scholar] [CrossRef] [PubMed]
- Millien, V.O.; Lu, W.; Shaw, J.; Yuan, X.; Mak, G.; Roberts, L.; Song, L.-Z.; Knight, J.M.; Creighton, C.J.; Luong, A.; et al. Cleavage of fibrinogen by proteinases elicits allergic responses through toll-like receptor 4. Science 2013, 341, 792–796. [Google Scholar] [CrossRef]
- Gerold, G.; Ajaj, K.A.; Bienert, M.; Laws, H.J.; Zychlinsky, A.; de Diego, J.L. A Toll-like receptor 2-integrin β3 complex senses bacterial lipopeptides via vitronectin. Nat. Immunol. 2008, 9, 761–768. [Google Scholar] [CrossRef]
- Naik-Mathuria, B.; Pilling, D.; Crawford, J.R.; Gay, A.N.; Smith, C.W.; Gomer, R.H.; Olutoye, O.O. Serum amyloid P inhibits dermal wound healing. Wound Repair Regen. 2008, 16, 266–273. [Google Scholar] [CrossRef]
- Schilcher, K.; Horswill, A.R. Staphylococcal Biofilm Development: Structure, Regulation, and Treatment Strategies. Microbiol. Mol. Biol. Rev. 2020, 84, e00026-19. [Google Scholar] [CrossRef]
- Kwiecinski, J.; Jacobsson, G.; Karlsson, M.; Zhu, X.; Wang, W.; Bremell, T.; Josefsson, E.; Jin, T. Staphylokinase promotes the establishment of Staphylococcus aureus skin infections while decreasing disease severity. J. Infect. Dis. 2013, 208, 990–999. [Google Scholar] [CrossRef] [PubMed]
- Idrees, M.; Sawant, S.; Karodia, N.; Rahman, A. Staphylococcus aureus biofilm: Morphology, genetics, pathogenesis and treatment strategies. Int. J. Environ. Res. Public Health 2021, 18, 7602. [Google Scholar] [CrossRef] [PubMed]
- Tipoe, T.L.; Wu, W.K.K.; Chung, L.; Gong, M.; Dong, M.; Liu, T.; Roever, L.; Ho, J.; Wong, M.C.S.; Chan, M.T.V.; et al. Plasminogen activator inhibitor 1 for predicting sepsis severity and mortality outcomes: A systematic review and meta-analysis. Front. Immunol. 2018, 9, 1218. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, F.C.F.; Manolov, V.; Morgenstern, J.; Fleming, T.; Heitmeier, S.; Uhle, F.; Al-Saeedi, M.; Hackert, T.; Bruckner, T.; Schöchl, H.; et al. Acute fibrinolysis shutdown occurs early in septic shock and is associated with increased morbidity and mortality: Results of an observational pilot study. Ann. Intensive Care 2019, 9, 19. [Google Scholar] [CrossRef] [PubMed]
- Hermans, P.W.M.; Hazelzet, J.A. Plasminogen activator inhibitor type 1 gene polymorphism and sepsis. Clin. Infect. Dis. 2005, 41 (Suppl. S7), S453–S458. [Google Scholar] [CrossRef]
- Iba, T.; Thachil, J. Clinical significance of measuring plasminogen activator inhibitor-1 in sepsis. J. Intensive Care 2017, 5, 56. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Lorenz, U.; Ohlsen, K.; Karch, H.; Hecker, M.; Thiede, A.; Hacker, J. Human antibody response during sepsis against targets expressed by methicillin resistant Staphylococcus aureus. FEMS Immunol. Med. Microbiol. 2000, 29, 145–153. [Google Scholar] [CrossRef]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Parente, R.; Fumagalli, M.R.; Di Claudio, A.; Cárdenas Rincón, C.L.; Erreni, M.; Zanini, D.; Iapichino, G.; Protti, A.; Garlanda, C.; Rusconi, R.; et al. A Multilayered Imaging and Microfluidics Approach for Evaluating the Effect of Fibrinolysis in Staphylococcus aureus Biofilm Formation. Pathogens 2023, 12, 1141. https://doi.org/10.3390/pathogens12091141
Parente R, Fumagalli MR, Di Claudio A, Cárdenas Rincón CL, Erreni M, Zanini D, Iapichino G, Protti A, Garlanda C, Rusconi R, et al. A Multilayered Imaging and Microfluidics Approach for Evaluating the Effect of Fibrinolysis in Staphylococcus aureus Biofilm Formation. Pathogens. 2023; 12(9):1141. https://doi.org/10.3390/pathogens12091141
Chicago/Turabian StyleParente, Raffaella, Maria Rita Fumagalli, Alessia Di Claudio, Cindy Lorena Cárdenas Rincón, Marco Erreni, Damiano Zanini, Giacomo Iapichino, Alessandro Protti, Cecilia Garlanda, Roberto Rusconi, and et al. 2023. "A Multilayered Imaging and Microfluidics Approach for Evaluating the Effect of Fibrinolysis in Staphylococcus aureus Biofilm Formation" Pathogens 12, no. 9: 1141. https://doi.org/10.3390/pathogens12091141
APA StyleParente, R., Fumagalli, M. R., Di Claudio, A., Cárdenas Rincón, C. L., Erreni, M., Zanini, D., Iapichino, G., Protti, A., Garlanda, C., Rusconi, R., & Doni, A. (2023). A Multilayered Imaging and Microfluidics Approach for Evaluating the Effect of Fibrinolysis in Staphylococcus aureus Biofilm Formation. Pathogens, 12(9), 1141. https://doi.org/10.3390/pathogens12091141