

Mouse Models for Human Herpesviruses


Abstract
1. General Considerations for the Development of In Vivo Models for Human Herpesviruses
2. Mice Infected with Promiscuous Human Alphaherpesviruses
3. Mouse Herpesvirus Orthologues as Models for Beta- and Gammaherpesviruses
3.1. MCMV as a Model for HCMV
3.2. Murine Roseolovirus (MRV) as a Model for HHV-6 and HHV-7
3.3. MHV-68 as a Model for EBV and KSHV
3.4. Limitations of Orthologue Viruses as Models for Human Herpes Viruses
4. Investigating Human Herpesviruses in Human Cell-Engrafted Mice
4.1. Engraftment of Functional Human Cells in Mice
4.1.1. Cell or Tissue Sources for Xenografts
4.1.2. Historical Overview of the Development of Humanized Mice
4.2. Herpesvirus Infection of the Hematopoietic Compartment in Xenografted Mice
4.2.1. HSV-1 and HSV-2 in Mice Engrafted Human Hematopoietic Cells
4.2.2. HCMV in Mice Engrafted Human Hematopoietic Cells
4.2.3. HHV-6A and HHV-6B in Mice Engrafted Human Hematopoietic Cells
4.2.4. EBV and KSHV in Mice-Engrafted Human Hematopoietic Cells
4.3. Herpesvirus Infection in Humanized Mouse Models Prepared from Tissue Xenografts
4.4. Herpesvirus Infection in Graft Models with Infection-Permissive Tissues in the Presence of a Reconstituted Human Immune System
5. Conclusions and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Connolly, S.A.; Jardetzky, T.S.; Longnecker, R. The Structural Basis of Herpesvirus Entry. Nat. Rev. Microbiol. 2021, 19, 110–121. [Google Scholar] [CrossRef] [PubMed]
- Spear, P.G.; Longnecker, R. Herpesvirus Entry: An Update. J. Virol. 2003, 77, 10179–10185. [Google Scholar] [CrossRef] [PubMed]
- Rajčáni, J.; Kúdelová, M. Gamma Herpesviruses: Pathogenesis of Infection and Cell Signaling. Folia Microbiol. 2003, 48, 291–318. [Google Scholar] [CrossRef]
- Boehmer, P.; Nimonkar, A. Herpes Virus Replication. IUBMB Life (Int. Union Biochem. Mol. Biol. Life) 2003, 55, 13–22. [Google Scholar] [CrossRef]
- Davison, A.J. Overview of Classification. In Human Herpesviruses; Arvin, A., Campadelli-Fiume, G., Mocarski, E., Moore, P.S., Roizman, B., Whitley, R., Yamanishi, K., Eds.; Cambridge University Press: Cambridge, UK, 2007. [Google Scholar]
- Gatherer, D.; Depledge, D.P.; Hartley, C.A.; Szpara, M.L.; Vaz, P.K.; Benkő, M.; Brandt, C.R.; Bryant, N.A.; Dastjerdi, A.; Doszpoly, A.; et al. ICTV Virus Taxonomy Profile: Herpesviridae 2021. J. Gen. Virol. 2021, 102, 001673. [Google Scholar] [CrossRef] [PubMed]
- Weber, F. Antiviral Innate Immunity: Introduction. In Encyclopedia of Virology; Elsevier: Amsterdam, The Netherlands, 2021; pp. 577–583. [Google Scholar]
- Cruz-Muñoz, M.E.; Fuentes-Pananá, E.M. Beta and Gamma Human Herpesviruses: Agonistic and Antagonistic Interactions with the Host Immune System. Front. Microbiol. 2018, 8, 2521. [Google Scholar] [CrossRef]
- Broussard, G.; Damania, B. Regulation of KSHV Latency and Lytic Reactivation. Viruses 2020, 12, 1034. [Google Scholar] [CrossRef]
- Stowe, R.; Kozlova, E.; Yetman, D.; Walling, D.; Goodwin, J.; Glaser, R. Chronic Herpesvirus Reactivation Occurs in Aging. Exp. Gerontol. 2007, 42, 563–570. [Google Scholar] [CrossRef]
- Worrall, G. Herpes Labialis. BMJ Clin. Evid. 2009, 2009, 1704. [Google Scholar]
- Sawtell, N.M.; Thompson, R.L. Rapid In Vivo Reactivation of Herpes Simplex Virus in Latently Infected Murine Ganglionic Neurons after Transient Hyperthermia. J. Virol. 1992, 66, 2150–2156. [Google Scholar] [CrossRef]
- Rosemarie, Q.; Sugden, B. Epstein–Barr Virus: How Its Lytic Phase Contributes to Oncogenesis. Microorganisms 2020, 8, 1824. [Google Scholar] [CrossRef] [PubMed]
- Aneja, K.K.; Yuan, Y. Reactivation and Lytic Replication of Kaposi’s Sarcoma-Associated Herpesvirus: An Update. Front. Microbiol. 2017, 8, 613. [Google Scholar] [CrossRef]
- Dubich, T.; Lieske, A.; Santag, S.; Beauclair, G.; Rückert, J.; Herrmann, J.; Gorges, J.; Büsche, G.; Kazmaier, U.; Hauser, H.; et al. An Endothelial Cell Line Infected by Kaposi’s Sarcoma–Associated Herpes Virus (KSHV) Allows the Investigation of Kaposi’s Sarcoma and the Validation of Novel Viral Inhibitors In Vitro and In Vivo. J. Mol. Med. 2019, 97, 311–324. [Google Scholar] [CrossRef] [PubMed]
- Lipps, C.; Badar, M.; Butueva, M.; Dubich, T.; Singh, V.V.; Rau, S.; Weber, A.; Kracht, M.; Köster, M.; May, T.; et al. Proliferation Status Defines Functional Properties of Endothelial Cells. Cell. Mol. Life Sci. 2017, 74, 1319–1333. [Google Scholar] [CrossRef]
- An, F.-Q.; Folarin, H.M.; Compitello, N.; Roth, J.; Gerson, S.L.; McCrae, K.R.; Fakhari, F.D.; Dittmer, D.P.; Renne, R. Long-Term-Infected Telomerase-Immortalized Endothelial Cells: A Model for Kaposi’s Sarcoma-Associated Herpesvirus Latency In Vitro and In Vivo. J. Virol. 2006, 80, 4833–4846. [Google Scholar] [CrossRef] [PubMed]
- Majorova, D.; Atkins, E.; Martineau, H.; Vokral, I.; Oosterhuis, D.; Olinga, P.; Wren, B.; Cuccui, J.; Werling, D. Use of Precision-Cut Tissue Slices as a Translational Model to Study Host-Pathogen Interaction. Front. Vet. Sci. 2021, 8, 686088. [Google Scholar] [CrossRef]
- Friedman, A.K.; Han, M. The Use of Herpes Simplex Virus in Ex Vivo Slice Culture. Curr. Protoc. Neurosci. 2015, 72, 4–36. [Google Scholar] [CrossRef]
- Baddal, B.; Marrazzo, P. Refining Host-Pathogen Interactions: Organ-on-Chip Side of the Coin. Pathogens 2021, 10, 203. [Google Scholar] [CrossRef]
- Sun, S.; Jin, L.; Zheng, Y.; Zhu, J. Modeling Human HSV Infection via a Vascularized Immune-Competent Skin-on-Chip Platform. Nat. Commun. 2022, 13, 5481. [Google Scholar] [CrossRef]
- Wilson, A.C. Impact of Cultured Neuron Models on α-Herpesvirus Latency Research. Viruses 2022, 14, 1209. [Google Scholar] [CrossRef]
- Thellman, N.M.; Botting, C.; Madaj, Z.; Triezenberg, S.J. An Immortalized Human Dorsal Root Ganglion Cell Line Provides a Novel Context To Study Herpes Simplex Virus 1 Latency and Reactivation. J. Virol. 2017, 91, e00080-17. [Google Scholar] [CrossRef] [PubMed]
- Lieber, D.; Hochdorfer, D.; Stoehr, D.; Schubert, A.; Lotfi, R.; May, T.; Wirth, D.; Sinzger, C. A Permanently Growing Human Endothelial Cell Line Supports Productive Infection with Human Cytomegalovirus under Conditional Cell Growth Arrest. Biotechniques 2015, 59, 127–136. [Google Scholar] [CrossRef] [PubMed]
- Crawford, L.B.; Diggins, N.L.; Caposio, P.; Hancock, M.H. Advances in Model Systems for Human Cytomegalovirus Latency and Reactivation. MBio 2022, 13, e01724-21. [Google Scholar] [CrossRef]
- Fujiwara, S.; Nakamura, H. Animal Models for Gammaherpesvirus Infections: Recent Development in the Analysis of Virus-Induced Pathogenesis. Pathogens 2020, 9, 116. [Google Scholar] [CrossRef] [PubMed]
- Reynaud, J.M.; Horvat, B. Animal Models for Human Herpesvirus 6 Infection. Front. Microbiol. 2013, 4, 174. [Google Scholar] [CrossRef]
- Ye, L.; Qian, Y.; Yu, W.; Guo, G.; Wang, H.; Xue, X. Functional Profile of Human Cytomegalovirus Genes and Their Associated Diseases: A Review. Front. Microbiol. 2020, 11, 2104. [Google Scholar] [CrossRef]
- Mosier, D.E.; Gulizia, R.J.; Baird, S.M.; Wilson, D.B. Transfer of a Functional Human Immune System to Mice with Severe Combined Immunodeficiency. Nature 1988, 335, 256–259. [Google Scholar] [CrossRef]
- McCune, J.M.; Namikawa, R.; Kaneshima, H.; Shultz, L.D.; Lieberman, M.; Weissman, I.L. The SCID-Hu Mouse: Murine Model for the Analysis of Human Hematolymphoid Differentiation and Function. Science 1988, 241, 1632–1639. [Google Scholar] [CrossRef]
- Namikawa, R.; Weilbaecher, K.N.; Kaneshima, H.; Yee, E.J.; McCune, J.M. Long-Term Human Hematopoiesis in the SCID-Hu Mouse. J. Exp. Med. 1990, 172, 1055–1063. [Google Scholar] [CrossRef]
- Lan, P.; Tonomura, N.; Shimizu, A.; Wang, S.; Yang, Y.-G. Reconstitution of a Functional Human Immune System in Immunodeficient Mice through Combined Human Fetal Thymus/Liver and CD34+ Cell Transplantation. Blood 2006, 108, 487–492. [Google Scholar] [CrossRef]
- Melkus, M.W.; Estes, J.D.; Padgett-Thomas, A.; Gatlin, J.; Denton, P.W.; Othieno, F.A.; Wege, A.K.; Haase, A.T.; Garcia, J.V. Humanized Mice Mount Specific Adaptive and Innate Immune Responses to EBV and TSST-1. Nat. Med. 2006, 12, 1316–1322. [Google Scholar] [CrossRef] [PubMed]
- Cristóbal, L.; Asúnsolo, Á.; Sánchez, J.; Ortega, M.A.; Álvarez-Mon, M.; García-Honduvilla, N.; Buján, J.; Maldonado, A.A. Mouse Models for Human Skin Transplantation: A Systematic Review. Cells Tissues Organs 2021, 210, 250–259. [Google Scholar] [CrossRef] [PubMed]
- Wahl, A.; De, C.; Abad Fernandez, M.; Lenarcic, E.M.; Xu, Y.; Cockrell, A.S.; Cleary, R.A.; Johnson, C.E.; Schramm, N.J.; Rank, L.M.; et al. Precision Mouse Models with Expanded Tropism for Human Pathogens. Nat. Biotechnol. 2019, 37, 1163–1173. [Google Scholar] [CrossRef] [PubMed]
- Looker, K.J.; Magaret, A.S.; May, M.T.; Turner, K.M.E.; Vickerman, P.; Newman, L.M.; Gottlieb, S.L. First Estimates of the Global and Regional Incidence of Neonatal Herpes Infection. Lancet Glob. Health 2017, 5, e300–e309. [Google Scholar] [CrossRef]
- Solomon, T.; Michael, B.D.; Smith, P.E.; Sanderson, F.; Davies, N.W.S.; Hart, I.J.; Holland, M.; Easton, A.; Buckley, C.; Kneen, R.; et al. Management of Suspected Viral Encephalitis in Adults—Association of British Neurologists and British Infection Association National Guidelines. J. Infect. 2012, 64, 347–373. [Google Scholar] [CrossRef]
- Zhu, S.; Viejo-Borbolla, A. Pathogenesis and Virulence of Herpes Simplex Virus. Virulence 2021, 12, 2670–2702. [Google Scholar] [CrossRef]
- Mancini, M.; Vidal, S.M. Insights into the Pathogenesis of Herpes Simplex Encephalitis from Mouse Models. Mamm. Genome 2018, 29, 425–445. [Google Scholar] [CrossRef] [PubMed]
- Madavaraju, K.; Koganti, R.; Volety, I.; Yadavalli, T.; Shukla, D. Herpes Simplex Virus Cell Entry Mechanisms: An Update. Front. Cell. Infect. Microbiol. 2021, 10, 617578. [Google Scholar] [CrossRef]
- Spear, P.G. Herpes Simplex Virus: Receptors and Ligands for Cell Entry. Cell. Microbiol. 2004, 6, 401–410. [Google Scholar] [CrossRef]
- Laquerre, S.; Argnani, R.; Anderson, D.B.; Zucchini, S.; Manservigi, R.; Glorioso, J.C. Heparan Sulfate Proteoglycan Binding by Herpes Simplex Virus Type 1 Glycoproteins B and C, Which Differ in Their Contributions to Virus Attachment, Penetration, and Cell-to-Cell Spread. J. Virol. 1998, 72, 6119–6130. [Google Scholar] [CrossRef]
- Petermann, P.; Thier, K.; Rahn, E.; Rixon, F.J.; Bloch, W.; Özcelik, S.; Krummenacher, C.; Barron, M.J.; Dixon, M.J.; Scheu, S.; et al. Entry Mechanisms of Herpes Simplex Virus 1 into Murine Epidermis: Involvement of Nectin-1 and Herpesvirus Entry Mediator as Cellular Receptors. J. Virol. 2015, 89, 262–274. [Google Scholar] [CrossRef] [PubMed]
- Petermann, P.; Rahn, E.; Thier, K.; Hsu, M.-J.; Rixon, F.J.; Kopp, S.J.; Knebel-Mörsdorf, D. Role of Nectin-1 and Herpesvirus Entry Mediator as Cellular Receptors for Herpes Simplex Virus 1 on Primary Murine Dermal Fibroblasts. J. Virol. 2015, 89, 9407–9416. [Google Scholar] [CrossRef] [PubMed]
- Kollias, C.M.; Huneke, R.B.; Wigdahl, B.; Jennings, S.R. Animal Models of Herpes Simplex Virus Immunity and Pathogenesis. J. Neurovirol. 2015, 21, 8–23. [Google Scholar] [CrossRef] [PubMed]
- Kopp, S.J.; Banisadr, G.; Glajch, K.; Maurer, U.E.; Grünewald, K.; Miller, R.J.; Osten, P.; Spear, P.G. Infection of Neurons and Encephalitis after Intracranial Inoculation of Herpes Simplex Virus Requires the Entry Receptor Nectin-1. Proc. Natl. Acad. Sci. USA 2009, 106, 17916–17920. [Google Scholar] [CrossRef]
- Gebhardt, B.M.; Halford, W.P. Evidence That Spontaneous Reactivation of Herpes Virus Does Not Occur in Mice. Virol. J. 2005, 2, 67. [Google Scholar] [CrossRef]
- Webre, J.M.; Hill, J.M.; Nolan, N.M.; Clement, C.; McFerrin, H.E.; Bhattacharjee, P.S.; Hsia, V.; Neumann, D.M.; Foster, T.P.; Lukiw, W.J.; et al. Rabbit and Mouse Models of HSV-1 Latency, Reactivation, and Recurrent Eye Diseases. J. Biomed. Biotechnol. 2012, 2012, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Dasgupta, G.; BenMohamed, L. Of Mice and Not Humans: How Reliable Are Animal Models for Evaluation of Herpes CD8+-T Cell-Epitopes-Based Immunotherapeutic Vaccine Candidates? Vaccine 2011, 29, 5824–5836. [Google Scholar] [CrossRef]
- Pieknik, J.R.; Bertke, A.S.; Krause, P.R. Herpes Simplex Virus 2 in Autonomic Ganglia: Evidence for Spontaneous Reactivation. J. Virol. 2019, 93, e00227-19. [Google Scholar] [CrossRef]
- Sehl, J.; Hölper, J.E.; Klupp, B.G.; Baumbach, C.; Teifke, J.P.; Mettenleiter, T.C. An Improved Animal Model for Herpesvirus Encephalitis in Humans. PLoS Pathog. 2020, 16, e1008445. [Google Scholar] [CrossRef]
- Finnen, R.L.; Mizokami, K.R.; Banfield, B.W.; Cai, G.-Y.; Simpson, S.A.; Pizer, L.I.; Levin, M.J. Postentry Events Are Responsible for Restriction of Productive Varicella-Zoster Virus Infection in Chinese Hamster Ovary Cells. J. Virol. 2006, 80, 10325–10334. [Google Scholar] [CrossRef]
- Reynaud, J.M.; Jégou, J.-F.; Welsch, J.C.; Horvat, B. Human Herpesvirus 6A Infection in CD46 Transgenic Mice: Viral Persistence in the Brain and Increased Production of Proinflammatory Chemokines via Toll-Like Receptor 9. J. Virol. 2014, 88, 5421–5436. [Google Scholar] [CrossRef] [PubMed]
- Jurak, I.; Brune, W. Induction of Apoptosis Limits Cytomegalovirus Cross-Species Infection. EMBO J. 2006, 25, 2634–2642. [Google Scholar] [CrossRef] [PubMed]
- Lafemina, R.L.; Hayward, G.S. Differences in Cell Type-Specific Blocks to Immediate Early Gene Expression and DNA Replication of Human, Simian and Murine Cytomegalovirus. J. Gen. Virol. 1988, 69, 355–374. [Google Scholar] [CrossRef]
- Mocarski, E.S.; Shenk Griffiths, P.D.; Pass, R.F. Cytomegaloviruses. In Fields Virology; Knipe, D.M., Howley, P.M., Eds.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2013; pp. 1960–2014. [Google Scholar]
- Cosme, R.C.; Martínez, F.P.; Tang, Q. Functional Interaction of Nuclear Domain 10 and Its Components with Cytomegalovirus after Infections: Cross-Species Host Cells versus Native Cells. PLoS ONE 2011, 6, e19187. [Google Scholar] [CrossRef] [PubMed]
- Griffiths, P.D.; Walter, S. Cytomegalovirus. Curr. Opin. Infect. Dis. 2005, 18, 241–245. [Google Scholar] [CrossRef]
- Farrell, H.E.; Bruce, K.; Lawler, C.; Stevenson, P.G. Murine Cytomegalovirus Spread Depends on the Infected Myeloid Cell Type. J. Virol. 2019, 93, e00540-19. [Google Scholar] [CrossRef] [PubMed]
- Fisher, M.A.; Lloyd, M.L. A Review of Murine Cytomegalovirus as a Model for Human Cytomegalovirus Disease—Do Mice Lie? Int. J. Mol. Sci. 2020, 22, 214. [Google Scholar] [CrossRef]
- Berry, R.; Watson, G.M.; Jonjic, S.; Degli-Esposti, M.A.; Rossjohn, J. Modulation of Innate and Adaptive Immunity by Cytomegaloviruses. Nat. Rev. Immunol. 2020, 20, 113–127. [Google Scholar] [CrossRef]
- Loewendorf, A.; Benedict, C.A. Modulation of Host Innate and Adaptive Immune Defenses by Cytomegalovirus: Timing Is Everything. J. Intern. Med. 2010, 267, 483–501. [Google Scholar] [CrossRef]
- Seckert, C.K.; Grießl, M.; Büttner, J.K.; Freitag, K.; Lemmermann, N.A.; Hummel, M.A.; Liu, X.F.; Abecassis, M.I.; Angulo, A.; Messerle, M.; et al. Immune Surveillance of Cytomegalovirus Latency and Reactivation in Murine Models: Link to “Memory Inflation”. In Cytomegaloviruses: From Molecular Pathogenesis to Intervention, 1st ed.; Reddehase, M.J., Ed.; Caister Academic Press: Norfolk, UK, 2013. [Google Scholar]
- Seckert, C.K.; Renzaho, A.; Tervo, H.-M.; Krause, C.; Deegen, P.; Kühnapfel, B.; Reddehase, M.J.; Grzimek, N.K.A. Liver Sinusoidal Endothelial Cells Are a Site of Murine Cytomegalovirus Latency and Reactivation. J. Virol. 2009, 83, 8869–8884. [Google Scholar] [CrossRef]
- Seckert, C.K.; Grießl, M.; Büttner, J.K.; Scheller, S.; Simon, C.O.; Kropp, K.A.; Renzaho, A.; Kühnapfel, B.; Grzimek, N.K.A.; Reddehase, M.J. Viral Latency Drives ‘Memory Inflation’: A Unifying Hypothesis Linking Two Hallmarks of Cytomegalovirus Infection. Med. Microbiol. Immunol. 2012, 201, 551–566. [Google Scholar] [CrossRef] [PubMed]
- Goodrum, F. Human Cytomegalovirus Latency: Approaching the Gordian Knot. Annu. Rev. Virol. 2016, 3, 333–357. [Google Scholar] [CrossRef] [PubMed]
- Reddehase, M.J.; Lemmermann, N.A.W. Cellular Reservoirs of Latent Cytomegaloviruses. Med. Microbiol. Immunol. 2019, 208, 391–403. [Google Scholar] [CrossRef] [PubMed]
- Reddehase, M.J.; Podlech, J.; Grzimek, N.K. Mouse Models of Cytomegalovirus Latency: Overview. J. Clin. Virol. 2002, 25, 23–36. [Google Scholar] [CrossRef]
- Ssentongo, P.; Hehnly, C.; Birungi, P.; Roach, M.A.; Spady, J.; Fronterre, C.; Wang, M.; Murray-Kolb, L.E.; Al-Shaar, L.; Chinchilli, V.M.; et al. Congenital Cytomegalovirus Infection Burden and Epidemiologic Risk Factors in Countries with Universal Screening. JAMA Netw. Open 2021, 4, e2120736. [Google Scholar] [CrossRef]
- Woolf, N.K.; Jaquish, D.V.; Koehrn, F.J. Transplacental Murine Cytomegalovirus Infection in the Brain of SCID Mice. Virol. J. 2007, 4, 26. [Google Scholar] [CrossRef]
- Krstanović, F.; Britt, W.J.; Jonjić, S.; Brizić, I. Cytomegalovirus Infection and Inflammation in Developing Brain. Viruses 2021, 13, 1078. [Google Scholar] [CrossRef]
- Zhou, Y.-P.; Mei, M.-J.; Wang, X.-Z.; Huang, S.-N.; Chen, L.; Zhang, M.; Li, X.-Y.; Qin, H.-B.; Dong, X.; Cheng, S.; et al. A Congenital CMV Infection Model for Follow-up Studies of Neurodevelopmental Disorders, Neuroimaging Abnormalities, and Treatment. JCI Insight 2022, 7, e152551. [Google Scholar] [CrossRef]
- Koontz, T.; Bralic, M.; Tomac, J.; Pernjak-Pugel, E.; Bantug, G.; Jonjic, S.; Britt, W.J. Altered Development of the Brain after Focal Herpesvirus Infection of the Central Nervous System. J. Exp. Med. 2008, 205, 423–435. [Google Scholar] [CrossRef]
- Bradford, R.D.; Yoo, Y.-G.; Golemac, M.; Pugel, E.P.; Jonjic, S.; Britt, W.J. Murine CMV-Induced Hearing Loss Is Associated with Inner Ear Inflammation and Loss of Spiral Ganglia Neurons. PLoS Pathog. 2015, 11, e1004774. [Google Scholar] [CrossRef]
- Sung, C.Y.W.; Seleme, M.C.; Payne, S.; Jonjic, S.; Hirose, K.; Britt, W. Virus-Induced Cochlear Inflammation in Newborn Mice Alters Auditory Function. JCI Insight 2019, 4, e128878. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.J.; Zhao, G.; Penna, V.R.; Park, E.; Lauron, E.J.; Harvey, I.B.; Beatty, W.L.; Plougastel-Douglas, B.; Poursine-Laurent, J.; Fremont, D.H.; et al. A Murine Herpesvirus Closely Related to Ubiquitous Human Herpesviruses Causes T-Cell Depletion. J. Virol. 2017, 91, e02463-16. [Google Scholar] [CrossRef] [PubMed]
- Denner, J.; Bigley, T.M.; Phan, T.L.; Zimmermann, C.; Zhou, X.; Kaufer, B.B. Comparative Analysis of Roseoloviruses in Humans, Pigs, Mice, and Other Species. Viruses 2019, 11, 1108. [Google Scholar] [CrossRef] [PubMed]
- Rowe, W.P.; Capps, W.I. A New Mouse Virus Causing Necrosis of the Thymus in Newborn Mice. J. Exp. Med. 1961, 113, 831–844. [Google Scholar] [CrossRef]
- Bigley, T.M.; Yang, L.; Kang, L.-I.; Saenz, J.B.; Victorino, F.; Yokoyama, W.M. Disruption of Thymic Central Tolerance by Infection with Murine Roseolovirus Induces Autoimmune Gastritis. J. Exp. Med. 2022, 219, e20211403. [Google Scholar] [CrossRef]
- Bigley, T.M.; Xiong, M.; Ali, M.; Chen, Y.; Wang, C.; Serrano, J.R.; Eteleeb, A.; Harari, O.; Yang, L.; Patel, S.J.; et al. Murine Roseolovirus Does Not Accelerate Amyloid-β Pathology and Human Roseoloviruses Are Not over-Represented in Alzheimer Disease Brains. Mol. Neurodegener. 2022, 17, 10. [Google Scholar] [CrossRef]
- Barton, E.; Mandal, P.; Speck, S.H. Pathogenesis and Host Control of Gammaherpesviruses: Lessons from the Mouse. Annu. Rev. Immunol. 2011, 29, 351–397. [Google Scholar] [CrossRef]
- Rex, V.; Zargari, R.; Stempel, M.; Halle, S.; Brinkmann, M.M. The Innate and T-Cell Mediated Immune Response during Acute and Chronic Gammaherpesvirus Infection. Front. Cell. Infect. Microbiol. 2023, 13, 367. [Google Scholar] [CrossRef]
- Speck, S.H.; Ganem, D. Viral Latency and Its Regulation: Lessons from the γ-Herpesviruses. Cell Host Microbe 2010, 8, 100–115. [Google Scholar] [CrossRef]
- Salinas, E.; Gupta, A.; Sifford, J.M.; Oldenburg, D.G.; White, D.W.; Forrest, J.C. Conditional Mutagenesis In Vivo Reveals Cell Type- and Infection Stage-Specific Requirements for LANA in Chronic MHV68 Infection. PLoS Pathog. 2018, 14, e1006865. [Google Scholar] [CrossRef]
- Bennion, B.G.; Ingle, H.; Ai, T.L.; Miner, C.A.; Platt, D.J.; Smith, A.M.; Baldridge, M.T.; Miner, J.J. A Human Gain-of-Function STING Mutation Causes Immunodeficiency and Gammaherpesvirus-Induced Pulmonary Fibrosis in Mice. J. Virol. 2019, 93, e01806-18. [Google Scholar] [CrossRef] [PubMed]
- Dutia, B.M.; Allen, D.J.; Dyson, H.; Nash, A.A. Type I Interferons and IRF-1 Play a Critical Role in the Control of a Gammaherpesvirus Infection. Virology 1999, 261, 173–179. [Google Scholar] [CrossRef] [PubMed]
- Barton, E.S.; Lutzke, M.L.; Rochford, R.; Virgin, H.W. Alpha/Beta Interferons Regulate Murine Gammaherpesvirus Latent Gene Expression and Reactivation from Latency. J. Virol. 2005, 79, 14149–14160. [Google Scholar] [CrossRef] [PubMed]
- Schwerk, J.; Kemper, L.; Bussey, K.A.; Lienenklaus, S.; Weiss, S.; Čičin-Šain, L.; Kröger, A.; Kalinke, U.; Collins, C.M.; Speck, S.H.; et al. Type I Interferon Signaling Controls Gammaherpesvirus Latency In Vivo. Pathogens 2022, 11, 1554. [Google Scholar] [CrossRef]
- Rak, M.A.; Buehler, J.; Zeltzer, S.; Reitsma, J.; Molina, B.; Terhune, S.; Goodrum, F. Human Cytomegalovirus UL135 Interacts with Host Adaptor Proteins To Regulate Epidermal Growth Factor Receptor and Reactivation from Latency. J. Virol. 2018, 92, e00919-18. [Google Scholar] [CrossRef]
- Crawford, L.B.; Kim, J.H.; Collins-McMillen, D.; Lee, B.-J.; Landais, I.; Held, C.; Nelson, J.A.; Yurochko, A.D.; Caposio, P. Human Cytomegalovirus Encodes a Novel FLT3 Receptor Ligand Necessary for Hematopoietic Cell Differentiation and Viral Reactivation. MBio 2018, 9, e00682-18. [Google Scholar] [CrossRef]
- Caviness, K.; Bughio, F.; Crawford, L.B.; Streblow, D.N.; Nelson, J.A.; Caposio, P.; Goodrum, F. Complex Interplay of the UL136 Isoforms Balances Cytomegalovirus Replication and Latency. MBio 2016, 7, e01986. [Google Scholar] [CrossRef]
- Dong, S.; Forrest, J.C.; Liang, X. Murine Gammaherpesvirus 68: A Small Animal Model for Gammaherpesvirus-Associated Diseases; Springer Nature: Singapore, 2017; pp. 225–236. [Google Scholar]
- Husain, S.M.; Usherwood, E.J.; Dyson, H.; Coleclough, C.; Coppola, M.A.; Woodland, D.L.; Blackman, M.A.; Stewart, J.P.; Sample, J.T. Murine Gammaherpesvirus M2 Gene Is Latency-Associated and Its Protein a Target for CD8+ T Lymphocytes. Proc. Natl. Acad. Sci. USA 1999, 96, 7508–7513. [Google Scholar] [CrossRef]
- Maul, G.G.; Negorev, D. Differences between Mouse and Human Cytomegalovirus Interactions with Their Respective Hosts at Immediate Early Times of the Replication Cycle. Med. Microbiol. Immunol. 2008, 197, 241–249. [Google Scholar] [CrossRef]
- Ku, C.-C.; Zerboni, L.; Ito, H.; Graham, B.S.; Wallace, M.; Arvin, A.M. Varicella-Zoster Virus Transfer to Skin by T Cells and Modulation of Viral Replication by Epidermal Cell Interferon-α. J. Exp. Med. 2004, 200, 917–925. [Google Scholar] [CrossRef]
- Foreman, K.E.; Friborg, J.; Chandran, B.; Katano, H.; Sata, T.; Mercader, M.; Nabel, G.J.; Nickoloff, B.J. Injection of Human Herpesvirus-8 in Human Skin Engrafted on SCID Mice Induces Kaposi’s Sarcoma-like Lesions. J. Dermatol. Sci. 2001, 26, 182–193. [Google Scholar] [CrossRef]
- Kawahara, T.; Lisboa, L.F.; Cader, S.; Douglas, D.N.; Nourbakhsh, M.; Pu, C.H.; Lewis, J.T.; Churchill, T.A.; Humar, A.; Kneteman, N.M. Human Cytomegalovirus Infection in Humanized Liver Chimeric Mice. Hepatol. Res. 2013, 43, 679–684. [Google Scholar] [CrossRef]
- Lipps, C.; Klein, F.; Wahlicht, T.; Seiffert, V.; Butueva, M.; Zauers, J.; Truschel, T.; Luckner, M.; Köster, M.; MacLeod, R.; et al. Expansion of Functional Personalized Cells with Specific Transgene Combinations. Nat. Commun. 2018, 9, 994. [Google Scholar] [CrossRef] [PubMed]
- Yin, L.; Wang, X.-J.; Chen, D.-X.; Liu, X.-N.; Wang, X.-J. Humanized Mouse Model: A Review on Preclinical Applications for Cancer Immunotherapy. Am. J. Cancer Res. 2020, 10, 4568–4584. [Google Scholar] [PubMed]
- Chen, J.; Liao, S.; Xiao, Z.; Pan, Q.; Wang, X.; Shen, K.; Wang, S.; Yang, L.; Guo, F.; Liu, H.; et al. The Development and Improvement of Immunodeficient Mice and Humanized Immune System Mouse Models. Front. Immunol. 2022, 13, 1007579. [Google Scholar] [CrossRef] [PubMed]
- Flanagan, S.P. ‘Nude’, a New Hairless Gene with Pleiotropic Effects in the Mouse. Genet. Res. 1966, 8, 295–309. [Google Scholar] [CrossRef]
- Ganick, D.J.; Sarnwick, R.D.; Shahidi, N.T.; Manning, D.D. Inability of Intravenously Injected Monocellular Suspensions of Human Bone Marrow to Establish in the Nude Mouse. Int. Arch. Allergy Immunol. 1980, 62, 330–333. [Google Scholar] [CrossRef]
- Bosma, G.C.; Custer, R.P.; Bosma, M.J. A Severe Combined Immunodeficiency Mutation in the Mouse. Nature 1983, 301, 527–530. [Google Scholar] [CrossRef]
- Shultz, L.D.; Ishikawa, F.; Greiner, D.L. Humanized Mice in Translational Biomedical Research. Nat. Rev. Immunol. 2007, 7, 118–130. [Google Scholar] [CrossRef]
- Macchiarini, F.; Manz, M.G.; Palucka, A.K.; Shultz, L.D. Humanized Mice. J. Exp. Med. 2005, 202, 1307–1311. [Google Scholar] [CrossRef]
- Shultz, L.D.; Schweitzer, P.A.; Christianson, S.W.; Gott, B.; Schweitzer, I.B.; Tennent, B.; McKenna, S.; Mobraaten, L.; Rajan, T.V.; Greiner, D.L. Multiple Defects in Innate and Adaptive Immunologic Function in NOD/LtSz-Scid Mice. J. Immunol. 1995, 154, 180–191. [Google Scholar] [CrossRef] [PubMed]
- Fulop, G.M.; Phillips, R.A. The Scid Mutation in Mice Causes a General Defect in DNA Repair. Nature 1990, 347, 479–482. [Google Scholar] [CrossRef] [PubMed]
- Mosier, D.E.; Stell, K.L.; Gulizia, R.J.; Torbett, B.E.; Gilmore, G.L. Homozygous Scid/Scid;Beige/Beige Mice Have Low Levels of Spontaneous or Neonatal T Cell-Induced B Cell Generation. J. Exp. Med. 1993, 177, 191–194. [Google Scholar] [CrossRef] [PubMed]
- Shinkai, Y. RAG-2-Deficient Mice Lack Mature Lymphocytes Owing to Inability to Initiate V(D)J Rearrangement. Cell 1992, 68, 855–867. [Google Scholar] [CrossRef] [PubMed]
- Mombaerts, P.; Iacomini, J.; Johnson, R.S.; Herrup, K.; Tonegawa, S.; Papaioannou, V.E. RAG-1-Deficient Mice Have No Mature B and T Lymphocytes. Cell 1992, 68, 869–877. [Google Scholar] [CrossRef]
- Makino, S.; Kunimoto, K.; Muraoka, Y.; Mizushima, Y.; Katagiri, K.; Tochino, Y. Breeding of a Non-Obese, Diabetic Strain of Mice. Exp. Anim. 1980, 29, 1–13. [Google Scholar] [CrossRef]
- Hesselton, R.M.; Greiner, D.L.; Mordes, J.P.; Rajan, T.V.; Sullivan, J.L.; Shultz, L.D. High Levels of Human Peripheral Blood Mononuclear Cell Engraftment and Enhanced Susceptibility to Human Immunodeficiency Virus Type 1 Infection in NOD/LtSz-Scid/Scid Mice. J. Infect. Dis. 1995, 172, 974–982. [Google Scholar] [CrossRef]
- Pflumio, F.; Izac, B.; Katz, A.; Shultz, L.D.; Vainchenker, W.; Coulombel, L. Phenotype and Function of Human Hematopoietic Cells Engrafting Immune-Deficient CB17-Severe Combined Immunodeficiency Mice and Nonobese Diabetic-Severe Combined Immunodeficiency Mice after Transplantation of Human Cord Blood Mononuclear Cells. Blood 1996, 88, 3731–3740. [Google Scholar] [CrossRef]
- Traggiai, E.; Chicha, L.; Mazzucchelli, L.; Bronz, L.; Piffaretti, J.-C.; Lanzavecchia, A.; Manz, M.G. Development of a Human Adaptive Immune System in Cord Blood Cell-Transplanted Mice. Science 2004, 304, 104–107. [Google Scholar] [CrossRef]
- Sugamura, K.; Asao, H.; Kondo, M.; Tanaka, N.; Ishii, N.; Ohbo, K.; Nakamura, M.; Takeshita, T. The Interleukin-2 Receptor γ Chain: Its Role in the Multiple Cytokine Receptor Complexes and T Cell Development in XSCID. Annu. Rev. Immunol. 1996, 14, 179–205. [Google Scholar] [CrossRef]
- DiSanto, J.P.; Müller, W.; Guy-Grand, D.; Fischer, A.; Rajewsky, K. Lymphoid Development in Mice with a Targeted Deletion of the Interleukin 2 Receptor Gamma Chain. Proc. Natl. Acad. Sci. USA 1995, 92, 377–381. [Google Scholar] [CrossRef]
- Cao, X.; Shores, E.W.; Hu-Li, J.; Anver, M.R.; Kelsail, B.L.; Russell, S.M.; Drago, J.; Noguchi, M.; Grinberg, A.; Bloom, E.T.; et al. Defective Lymphoid Development in Mice Lacking Expression of the Common Cytokine Receptor γ Chain. Immunity 1995, 2, 223–238. [Google Scholar] [CrossRef] [PubMed]
- Ito, M.; Hiramatsu, H.; Kobayashi, K.; Suzue, K.; Kawahata, M.; Hioki, K.; Ueyama, Y.; Koyanagi, Y.; Sugamura, K.; Tsuji, K.; et al. NOD/SCID/Γcnull Mouse: An Excellent Recipient Mouse Model for Engraftment of Human Cells. Blood 2002, 100, 3175–3182. [Google Scholar] [CrossRef] [PubMed]
- Shultz, L.D.; Lyons, B.L.; Burzenski, L.M.; Gott, B.; Chen, X.; Chaleff, S.; Kotb, M.; Gillies, S.D.; King, M.; Mangada, J.; et al. Human Lymphoid and Myeloid Cell Development in NOD/LtSz- Scid IL2R γ Null Mice Engrafted with Mobilized Human Hemopoietic Stem Cells. J. Immunol. 2005, 174, 6477–6489. [Google Scholar] [CrossRef]
- Shultz, L.D.; Banuelos, S.; Lyons, B.; Samuels, R.; Burzenski, L.; Gott, B.; Lang, P.; Leif, J.; Appel, M.; Rossini, A.; et al. NOD/LtSz-Rag1 Null Pfp Null Mice: A New Model System with Increased Levels of Human Peripheral Leukocyte and Hematopoietic Stem-Cell Engraftment. Transplantation 2003, 76, 1036–1042. [Google Scholar] [CrossRef] [PubMed]
- Rongvaux, A.; Willinger, T.; Martinek, J.; Strowig, T.; Gearty, S.V.; Teichmann, L.L.; Saito, Y.; Marches, F.; Halene, S.; Palucka, A.K.; et al. Development and Function of Human Innate Immune Cells in a Humanized Mouse Model. Nat. Biotechnol. 2014, 32, 364–372. [Google Scholar] [CrossRef]
- Kwant-Mitchell, A.; Ashkar, A.A.; Rosenthal, K.L. Mucosal Innate and Adaptive Immune Responses against Herpes Simplex Virus Type 2 in a Humanized Mouse Model. J. Virol. 2009, 83, 10664–10676. [Google Scholar] [CrossRef]
- Koenig, J.; Theobald, S.J.; Stripecke, R. Modeling Human Cytomegalovirus in Humanized Mice for Vaccine Testing. Vaccines 2020, 8, 89. [Google Scholar] [CrossRef]
- Brown, J.M.; Kaneshima, H.; Mocarski, E.S. Dramatic Interstrain Differences in the Replication of Human Cytomegalovirus in SCID-Hu Mice. J. Infect. Dis. 1995, 171, 1599–1603. [Google Scholar] [CrossRef]
- Mocarski, E.S.; Bonyhadi, M.; Salimi, S.; McCune, J.M.; Kaneshima, H. Human Cytomegalovirus in a SCID-Hu Mouse: Thymic Epithelial Cells Are Prominent Targets of Viral Replication. Proc. Natl. Acad. Sci. USA 1993, 90, 104–108. [Google Scholar] [CrossRef]
- Crawford, L.B.; Tempel, R.; Streblow, D.N.; Kreklywich, C.; Smith, P.; Picker, L.J.; Nelson, J.A.; Caposio, P. Human Cytomegalovirus Induces Cellular and Humoral Virus-Specific Immune Responses in Humanized BLT Mice. Sci. Rep. 2017, 7, 937. [Google Scholar] [CrossRef] [PubMed]
- Volk, V.; Reppas, A.I.; Robert, P.A.; Spineli, L.M.; Sundarasetty, B.S.; Theobald, S.J.; Schneider, A.; Gerasch, L.; Deves Roth, C.; Klöss, S.; et al. Multidimensional Analysis Integrating Human T-Cell Signatures in Lymphatic Tissues with Sex of Humanized Mice for Prediction of Responses after Dendritic Cell Immunization. Front. Immunol. 2017, 8, 1709. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.S.; Goldman, D.C.; Bailey, A.S.; Pfaffle, D.L.; Kreklywich, C.N.; Spencer, D.B.; Othieno, F.A.; Streblow, D.N.; Garcia, J.V.; Fleming, W.H.; et al. Granulocyte-Colony Stimulating Factor Reactivates Human Cytomegalovirus in a Latently Infected Humanized Mouse Model. Cell Host Microbe 2010, 8, 284–291. [Google Scholar] [CrossRef]
- Umashankar, M.; Petrucelli, A.; Cicchini, L.; Caposio, P.; Kreklywich, C.N.; Rak, M.; Bughio, F.; Goldman, D.C.; Hamlin, K.L.; Nelson, J.A.; et al. A Novel Human Cytomegalovirus Locus Modulates Cell Type-Specific Outcomes of Infection. PLoS Pathog. 2011, 7, e1002444. [Google Scholar] [CrossRef] [PubMed]
- Crawford, L.B.; Caposio, P.; Kreklywich, C.; Pham, A.H.; Hancock, M.H.; Jones, T.A.; Smith, P.P.; Yurochko, A.D.; Nelson, J.A.; Streblow, D.N. Human Cytomegalovirus US28 Ligand Binding Activity Is Required for Latency in CD34+ Hematopoietic Progenitor Cells and Humanized NSG Mice. MBio 2019, 10, e01889-19. [Google Scholar] [CrossRef]
- Caposio, P.; van den Worm, S.; Crawford, L.; Perez, W.; Kreklywich, C.; Gilbride, R.M.; Hughes, C.M.; Ventura, A.B.; Ratts, R.; Marshall, E.E.; et al. Characterization of a Live-Attenuated HCMV-Based Vaccine Platform. Sci. Rep. 2019, 9, 19236. [Google Scholar] [CrossRef]
- Hakki, M.; Goldman, D.C.; Streblow, D.N.; Hamlin, K.L.; Krekylwich, C.N.; Fleming, W.H.; Nelson, J.A. HCMV Infection of Humanized Mice after Transplantation of G-CSF–Mobilized Peripheral Blood Stem Cells from HCMV-Seropositive Donors. Biol. Blood Marrow Transplant. 2014, 20, 132–135. [Google Scholar] [CrossRef] [PubMed]
- Tomić, A.; Varanasi, P.R.; Golemac, M.; Malić, S.; Riese, P.; Borst, E.M.; Mischak-Weissinger, E.; Guzmán, C.A.; Krmpotić, A.; Jonjić, S.; et al. Activation of Innate and Adaptive Immunity by a Recombinant Human Cytomegalovirus Strain Expressing an NKG2D Ligand. PLoS Pathog. 2016, 12, e1006015. [Google Scholar] [CrossRef]
- Theobald, S.J.; Khailaie, S.; Meyer-Hermann, M.; Volk, V.; Olbrich, H.; Danisch, S.; Gerasch, L.; Schneider, A.; Sinzger, C.; Schaudien, D.; et al. Signatures of T and B Cell Development, Functional Responses and PD-1 Upregulation after HCMV Latent Infections and Reactivations in Nod. Rag.Gamma Mice Humanized with Cord Blood CD34+ Cells. Front. Immunol. 2018, 9, 2734. [Google Scholar] [CrossRef]
- Dagna, L.; Pritchett, J.C.; Lusso, P. Immunomodulation and Immunosuppression by Human Herpesvirus 6A and 6B. Future Virol. 2013, 8, 273–287. [Google Scholar] [CrossRef]
- Virtanen, J.O.; Färkkilä, M.; Multanen, J.; Uotila, L.; Jääskeläinen, A.J.; Vaheri, A.; Koskiniemi, M. Evidence for Human Herpesvirus 6 Variant A Antibodies in Multiple Sclerosis: Diagnostic and Therapeutic Implications. J. Neurovirol. 2007, 13, 347–352. [Google Scholar] [CrossRef] [PubMed]
- Lusso, P.; Gallo, R.C. Human Herpesvirus 6 in AIDS. Immunol. Today 1995, 16, 67–71. [Google Scholar] [CrossRef] [PubMed]
- Gobbi, A.; Stoddart, C.A.; Malnati, M.S.; Locatelli, G.; Santoro, F.; Abbey, N.W.; Bare, C.; Linquist-Stepps, V.; Moreno, M.B.; Herndier, B.G.; et al. Human Herpesvirus 6 (HHV-6) Causes Severe Thymocyte Depletion in SCID-Hu Thy/Liv Mice. J. Exp. Med. 1999, 189, 1953–1960. [Google Scholar] [CrossRef] [PubMed]
- Tanner, A.; Carlson, S.A.; Nukui, M.; Murphy, E.A.; Berges, B.K. Human Herpesvirus 6A Infection and Immunopathogenesis in Humanized Rag2−/− Γc−/− Mice. J. Virol. 2013, 87, 12020–12028. [Google Scholar] [CrossRef]
- Cocco, M.; Bellan, C.; Tussiwand, R.; Corti, D.; Traggiai, E.; Lazzi, S.; Mannucci, S.; Bronz, L.; Palummo, N.; Ginanneschi, C.; et al. CD34+ Cord Blood Cell-Transplanted Rag2−/− Γc−/− Mice as a Model for Epstein-Barr Virus Infection. Am. J. Pathol. 2008, 173, 1369–1378. [Google Scholar] [CrossRef]
- Islas-Ohlmayer, M.; Padgett-Thomas, A.; Domiati-Saad, R.; Melkus, M.W.; Cravens, P.D.; Martin, M.D.P.; Netto, G.; Garcia, J.V. Experimental Infection of NOD/SCID Mice Reconstituted with Human CD34+ Cells with Epstein-Barr Virus. J. Virol. 2004, 78, 13891–13900. [Google Scholar] [CrossRef]
- Wahl, A.; Linnstaedt, S.D.; Esoda, C.; Krisko, J.F.; Martinez-Torres, F.; Delecluse, H.-J.; Cullen, B.R.; Garcia, J.V. A Cluster of Virus-Encoded MicroRNAs Accelerates Acute Systemic Epstein-Barr Virus Infection but Does Not Significantly Enhance Virus-Induced Oncogenesis In Vivo. J. Virol. 2013, 87, 5437–5446. [Google Scholar] [CrossRef]
- Heuts, F.; Rottenberg, M.E.; Salamon, D.; Rasul, E.; Adori, M.; Klein, G.; Klein, E.; Nagy, N. T Cells Modulate Epstein-Barr Virus Latency Phenotypes during Infection of Humanized Mice. J. Virol. 2014, 88, 3235–3245. [Google Scholar] [CrossRef]
- Strowig, T.; Gurer, C.; Ploss, A.; Liu, Y.-F.; Arrey, F.; Sashihara, J.; Koo, G.; Rice, C.M.; Young, J.W.; Chadburn, A.; et al. Priming of Protective T Cell Responses against Virus-Induced Tumors in Mice with Human Immune System Components. J. Exp. Med. 2009, 206, 1423–1434. [Google Scholar] [CrossRef]
- Yajima, M.; Imadome, K.; Nakagawa, A.; Watanabe, S.; Terashima, K.; Nakamura, H.; Ito, M.; Shimizu, N.; Honda, M.; Yamamoto, N.; et al. A New Humanized Mouse Model of Epstein-Barr Virus Infection That Reproduces Persistent Infection, Lymphoproliferative Disorder, and Cell-Mediated and Humoral Immune Responses. J. Infect. Dis. 2008, 198, 673–682. [Google Scholar] [CrossRef]
- Kuwana, Y.; Takei, M.; Yajima, M.; Imadome, K.-I.; Inomata, H.; Shiozaki, M.; Ikumi, N.; Nozaki, T.; Shiraiwa, H.; Kitamura, N.; et al. Epstein-Barr Virus Induces Erosive Arthritis in Humanized Mice. PLoS ONE 2011, 6, e26630. [Google Scholar] [CrossRef] [PubMed]
- Antsiferova, O.; Müller, A.; Rämer, P.C.; Chijioke, O.; Chatterjee, B.; Raykova, A.; Planas, R.; Sospedra, M.; Shumilov, A.; Tsai, M.-H.; et al. Adoptive Transfer of EBV Specific CD8+ T Cell Clones Can Transiently Control EBV Infection in Humanized Mice. PLoS Pathog. 2014, 10, e1004333. [Google Scholar] [CrossRef] [PubMed]
- Tsai, M.-H.; Raykova, A.; Klinke, O.; Bernhardt, K.; Gärtner, K.; Leung, C.S.; Geletneky, K.; Sertel, S.; Münz, C.; Feederle, R.; et al. Spontaneous Lytic Replication and Epitheliotropism Define an Epstein-Barr Virus Strain Found in Carcinomas. Cell Rep. 2013, 5, 458–470. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.-X.; Kang, G.; Kumar, P.; Lu, W.; Li, Y.; Zhou, Y.; Li, Q.; Wood, C. Humanized-BLT Mouse Model of Kaposi’s Sarcoma-Associated Herpesvirus Infection. Proc. Natl. Acad. Sci. USA 2014, 111, 3146–3151. [Google Scholar] [CrossRef]
- Ramos da Silva, S.; Elgui de Oliveira, D. HIV, EBV and KSHV: Viral Cooperation in the Pathogenesis of Human Malignancies. Cancer Lett. 2011, 305, 175–185. [Google Scholar] [CrossRef]
- Moffat, J.F.; Zerboni, L.; Sommer, M.H.; Heineman, T.C.; Cohen, J.I.; Kaneshima, H.; Arvin, A.M. The ORF47 and ORF66 Putative Protein Kinases of Varicella-Zoster Virus Determine Tropism for Human T Cells and Skin in the SCID-Hu Mouse. Proc. Natl. Acad. Sci. USA 1998, 95, 11969–11974. [Google Scholar] [CrossRef]
- Schaap-Nutt, A.; Sommer, M.; Che, X.; Zerboni, L.; Arvin, A.M. ORF66 Protein Kinase Function Is Required for T-Cell Tropism of Varicella-Zoster Virus In Vivo. J. Virol. 2006, 80, 11806–11816. [Google Scholar] [CrossRef]
- Schaap, A.; Fortin, J.-F.; Sommer, M.; Zerboni, L.; Stamatis, S.; Ku, C.-C.; Nolan, G.P.; Arvin, A.M. T-Cell Tropism and the Role of ORF66 Protein in Pathogenesis of Varicella-Zoster Virus Infection. J. Virol. 2005, 79, 12921–12933. [Google Scholar] [CrossRef]
- Moffat, J.F.; Zerboni, L.; Kinchington, P.R.; Grose, C.; Kaneshima, H.; Arvin, A.M. Attenuation of the Vaccine Oka Strain of Varicella-Zoster Virus and Role of Glycoprotein C in Alphaherpesvirus Virulence Demonstrated in the SCID-Hu Mouse. J. Virol. 1998, 72, 965–974. [Google Scholar] [CrossRef]
- Kern, E.R. Pivotal Role of Animal Models in the Development of New Therapies for Cytomegalovirus Infections. Antiviral Res. 2006, 71, 164–171. [Google Scholar] [CrossRef]
- Lloyd, M.G.; Smith, N.A.; Tighe, M.; Travis, K.L.; Liu, D.; Upadhyaya, P.K.; Kinchington, P.R.; Chan, G.C.; Moffat, J.F. A Novel Human Skin Tissue Model To Study Varicella-Zoster Virus and Human Cytomegalovirus. J. Virol. 2020, 94, e01082-20. [Google Scholar] [CrossRef] [PubMed]
- Epstein, L.G.; Cvetkovich, T.A.; Lazar, E.S.; DiLoreto, D.; Saito, Y.; James, H.; del Cerro, C.; Kaneshima, H.; McCune, J.M.; Britt, W.J.; et al. Human Neural Xenografts: Progress in Developing an In-Vivo Model to Study Human Immunodeficiency Virus (HIV) and Human Cytomegalovirus (HCMV) Infection. Adv. Neuroimmunol. 1994, 4, 257–260. [Google Scholar] [CrossRef] [PubMed]
- Da Cunha, T.; Wu, G.Y. Cytomegalovirus Hepatitis in Immunocompetent and Immunocompromised Hosts. J. Clin. Transl. Hepatol. 2021, 9, 106. [Google Scholar] [CrossRef] [PubMed]
- Toupance, O.; Bouedjoro-Camus, M.-C.; Carquin, J.; Novella, J.-L.; Lavaud, S.; Wynckel, A.; Jolly, D.; Chanard, J. Cytomegalovirus-Related Disease and Risk of Acute Rejection in Renal Transplant Recipients: A Cohort Study with Case-Control Analyses. Transpl. Int. 2000, 13, 413–419. [Google Scholar] [CrossRef] [PubMed]
- Heim, C.; Müller, P.P.; Tandler, R.; Cherikh, W.S.; Toll, A.E.; Stehlik, J.; Weyand, M.; Khush, K.K.; Ensminger, S.M. Cytomegalovirus Donor Seropositivity Negatively Affects Survival after Heart Transplantation. Transplantation 2022, 106, 1243–1252. [Google Scholar] [CrossRef]
- Abele-Ohl, S.; Leis, M.; Wollin, M.; Mahmoudian, S.; Hoffmann, J.; Müller, R.; Heim, C.; Spriewald, B.M.; Weyand, M.; Stamminger, T.; et al. Human Cytomegalovirus Infection Leads to Elevated Levels of Transplant Arteriosclerosis in a Humanized Mouse Aortic Xenograft Model. Am. J. Transplant. 2012, 12, 1720–1729. [Google Scholar] [CrossRef]
- Dubich, T.; Dittrich, A.; Bousset, K.; Geffers, R.; Büsche, G.; Köster, M.; Hauser, H.; Schulz, T.F.; Wirth, D. 3D Culture Conditions Support Kaposi’s Sarcoma Herpesvirus (KSHV) Maintenance and Viral Spread in Endothelial Cells. J. Mol. Med. 2021, 99, 425–438. [Google Scholar] [CrossRef]
- Beauclair, G.; Naimo, E.; Dubich, T.; Rückert, J.; Koch, S.; Dhingra, A.; Wirth, D.; Schulz, T.F. Targeting Kaposi’s Sarcoma-Associated Herpesvirus ORF21 Tyrosine Kinase and Viral Lytic Reactivation by Tyrosine Kinase Inhibitors Approved for Clinical Use. J. Virol. 2020, 94, e01791-19. [Google Scholar] [CrossRef] [PubMed]
- Sato, K.; Misawa, N.; Nie, C.; Satou, Y.; Iwakiri, D.; Matsuoka, M.; Takahashi, R.; Kuzushima, K.; Ito, M.; Takada, K.; et al. A Novel Animal Model of Epstein-Barr Virus–Associated Hemophagocytic Lymphohistiocytosis in Humanized Mice. Blood 2011, 117, 5663–5673. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Chen, X.; Dowbaj, A.M.; Sljukic, A.; Bratlie, K.; Lin, L.; Fong, E.L.S.; Balachander, G.M.; Chen, Z.; Soragni, A.; et al. Organoids. Nat. Rev. Methods Prim. 2022, 2, 94. [Google Scholar] [CrossRef] [PubMed]
- Dang, J.; Tiwari, S.K.; Lichinchi, G.; Qin, Y.; Patil, V.S.; Eroshkin, A.M.; Rana, T.M. Zika Virus Depletes Neural Progenitors in Human Cerebral Organoids through Activation of the Innate Immune Receptor TLR3. Cell Stem Cell 2016, 19, 258–265. [Google Scholar] [CrossRef] [PubMed]
- Garcez, P.P.; Loiola, E.C.; Madeiro da Costa, R.; Higa, L.M.; Trindade, P.; Delvecchio, R.; Nascimento, J.M.; Brindeiro, R.; Tanuri, A.; Rehen, S.K. Zika Virus Impairs Growth in Human Neurospheres and Brain Organoids. Science 2016, 352, 816–818. [Google Scholar] [CrossRef] [PubMed]
- Ettayebi, K.; Crawford, S.E.; Murakami, K.; Broughman, J.R.; Karandikar, U.; Tenge, V.R.; Neill, F.H.; Blutt, S.E.; Zeng, X.-L.; Qu, L.; et al. Replication of Human Noroviruses in Stem Cell–Derived Human Enteroids. Science 2016, 353, 1387–1393. [Google Scholar] [CrossRef]
- Yin, Y.; Bijvelds, M.; Dang, W.; Xu, L.; van der Eijk, A.A.; Knipping, K.; Tuysuz, N.; Dekkers, J.F.; Wang, Y.; de Jonge, J.; et al. Modeling Rotavirus Infection and Antiviral Therapy Using Primary Intestinal Organoids. Antiviral Res. 2015, 123, 120–131. [Google Scholar] [CrossRef] [PubMed]
- Sachs, N.; Papaspyropoulos, A.; Zomer-van Ommen, D.D.; Heo, I.; Böttinger, L.; Klay, D.; Weeber, F.; Huelsz-Prince, G.; Iakobachvili, N.; Amatngalim, G.D.; et al. Long-Term Expanding Human Airway Organoids for Disease Modeling. EMBO J. 2019, 38, e100300. [Google Scholar] [CrossRef]
- Lamers, M.M.; Beumer, J.; van der Vaart, J.; Knoops, K.; Puschhof, J.; Breugem, T.I.; Ravelli, R.B.G.; Paul van Schayck, J.; Mykytyn, A.Z.; Duimel, H.Q.; et al. SARS-CoV-2 Productively Infects Human Gut Enterocytes. Science 2020, 369, 50–54. [Google Scholar] [CrossRef]
- Zhao, B.; Ni, C.; Gao, R.; Wang, Y.; Yang, L.; Wei, J.; Lv, T.; Liang, J.; Zhang, Q.; Xu, W.; et al. Recapitulation of SARS-CoV-2 Infection and Cholangiocyte Damage with Human Liver Ductal Organoids. Protein Cell 2020, 11, 771–775. [Google Scholar] [CrossRef]
- O’Brien, B.S.; Mokry, R.L.; Schumacher, M.L.; Pulakanti, K.; Rao, S.; Terhune, S.S.; Ebert, A.D. Downregulation of Neurodevelopmental Gene Expression in IPSC-Derived Cerebral Organoids upon Infection by Human Cytomegalovirus. iScience 2022, 25, 104098. [Google Scholar] [CrossRef]
- Ingber, D.E. Human Organs-on-Chips for Disease Modelling, Drug Development and Personalized Medicine. Nat. Rev. Genet. 2022, 23, 467–491. [Google Scholar] [CrossRef]


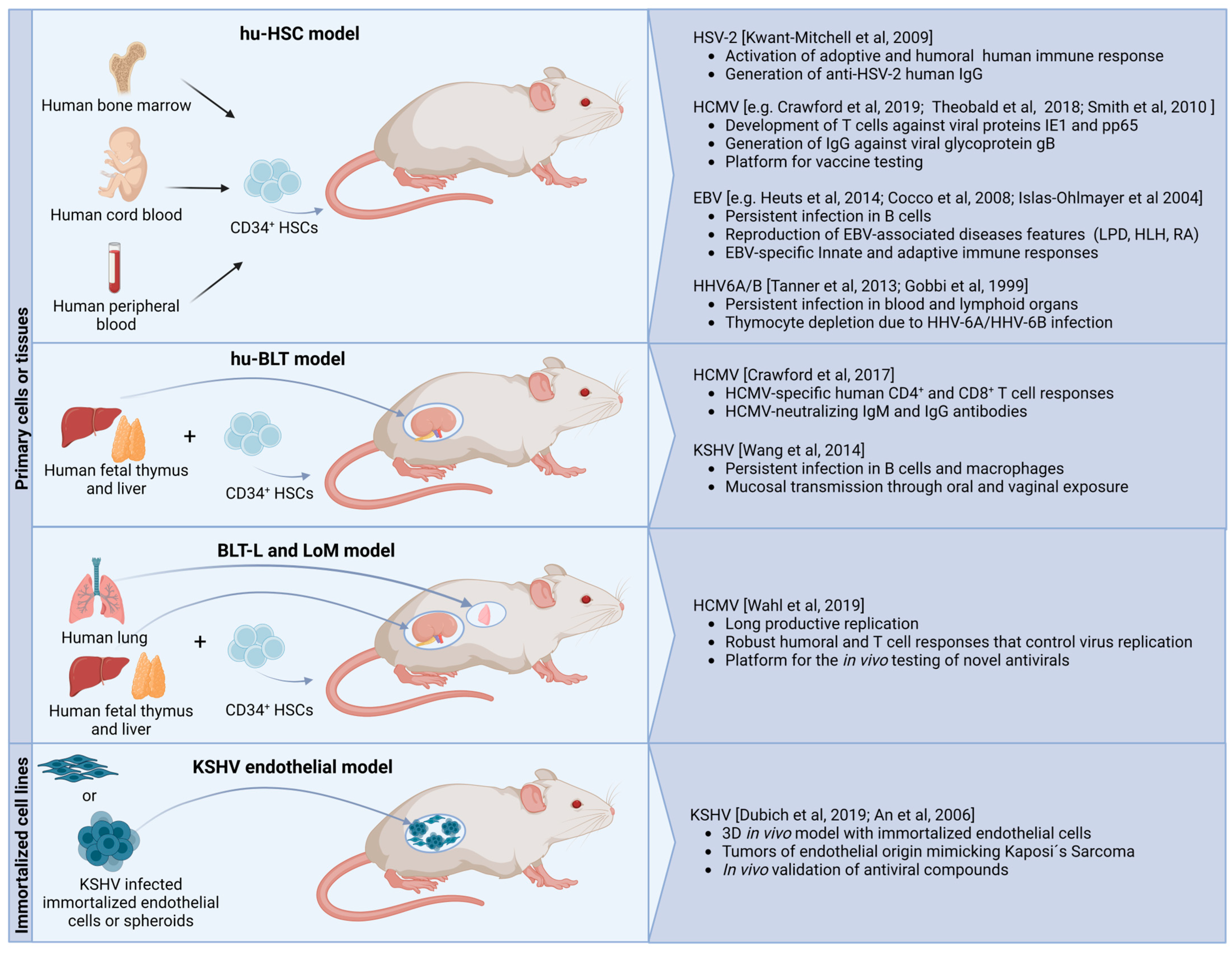{kind=link}
| Virus Names | Transmission Media | Tropism in Human Cells | Infection Associated Pathophysiology in Humans (Examples) | Animal Homologues (Examples) | |
|---|---|---|---|---|---|
| Alphaherpesvirinae | HHV-1/ Herpes simplex virus (HSV)-1 | Body fluids (e.g., saliva, mucus, wound fluid) | Epithelial cells (mucosa, skin), neurons | Cold sores, stromal keratitis, genital herpes, HSE, meningitis, eczema herpeticum, pneumonitis | Bovine herpesvirus 1 (BHV-1) Suid Herpesvirus 1 (SuHV-1) |
| HHV-2/ Herpes simplex virus (HSV)-2 | Body fluids (e.g., saliva, mucus, wound fluid) | ||||
| HHV-3/ Varicella zoster virus (VZV) | Direct contact, aerosols, vesicular fluids | T cells, skin epithelial cells, neurons | Varicella (chickenpox), Zoster (shingles), postherpetic neuralgia, meningoencephalitis, myelitis, vasculopathy, keratitis, retinopathy, visceral and gastrointestinal disorders | Cercopithecine alphaherpesvirus 9 (CeHV-9) Simian varicella virus (SVV) | |
| Betaherpesvirinae | HHV-5/ Cytomegalovirus (HCMV) | Body fluids (e.g., saliva, semen, breast milk, mucus, transfusions) | PBMCs (monocytes, CD34+ progenitor cells, dendritic cells), epithelial cells, endothelial cells, fibroblasts, smooth muscle cells | Congenital infections: mental retardation, hearing loss, miscarriage. Transplant patients: graft rejection, hepatitis, pneumonitis, retinitis, cardiovascular diseases | Murine cytomegalovirus (MCMV) |
| HHV-6A/ Roseolovirus | Saliva, (congenital transmission in case of chromosomal integration of viral DNA) | CD4+ T cells, PBMCs | Exanthema subitum (HHV-6B), febrile seizures, encephalitis, hepatitis, colitis, pneumonitis, GVHD, graft rejection, myelitis, neurological disorders, and oncogenesis | Murine roseolovirus (MRV) Porcine roseolovirus (PRV) | |
| HHV-6B/ Roseolovirus | |||||
| HHV-7/ Roseolovirus | Body fluids (e.g., saliva, breast milk) | CD4+ T cells | Exanthema subitem, encephalitis, meningitis, acutemyelitis, Guillain–Barré syndrome | ||
| Gammaherpesvirinae | HHV-4/ Epstein-Barr virus (EBV) | Body fluids (e.g., saliva, semen, blood) | Epithelial cells, B cells | Infectious mononucleosis, Burkitt’s lymphoma, nasopharyngeal carcinoma, gastric carcinoma, T cell lymphomas | Murine gammaherpesvirus 68 (MHV-68) Rhesus monkey rhadinovirus (RRV) |
| HHV-8/ Kaposi’s sarcoma-associated herpesvirus (KSHV) | Body fluids (e.g., saliva, mucus, genital secretion, semen, breast milk, blood) | B cells, endothelial cells, epithelial cells, keratinocytes, fibroblasts, dendritic cells, monocytes/macrophages | Kaposi’s sarcoma (KS), primary effusion lymphoma (PEL), multicentric Castleman’s disease (MCD) |
| Virus | Establishment of Model | Achievements/Features | Limitations/Challenges |
|---|---|---|---|
| Human viruses productively infecting mice | |||
| HSV-1 HSV-2 | Infection of wildtype or genetically modified mice via different infection routes | Latency Innate immunity Role of viral genes in vivo T cell activation | Lack of spontaneous reactivation Lack of human immune system |
| Orthologue viruses in mouse as native host | |||
| MCMV | Infection of wildtype or genetically modified mice via different infection routes | Cellular tropism Congenital infection | Role of private genes Human immune response Immune evasion Route of infection |
| MHV-68 | Infection of wildtype or genetically modified mice via different infection routes | Lymphocytes tropism Chronic infection and latency Innate immunity | Endothelial cell tropism Route of infection Differences in latency program Lack of KS lesions |
| MRV | Infection of wildtype neonates | T cell tropism | Latent infection Infection in adult animals |
| Model | Establishment of Model | Genetic Background | Features | Limitations | References (Examples) |
|---|---|---|---|---|---|
| hu-PBL | IP injection of human PBMCs | SCID, NOD-SCID, NSG, BRG | T cell engraftment | No multilineage hematopoiesis No primary immune response Development of early GvHD | [29] |
| hu-HSC | Injection of human CD34+ cells from cord blood or fetal liver | SCID, NOD-SCID, NSG, BRG, NRG | Multilineage hematopoiesis Primary immune response | No HLA restriction Inadequate innate immune system | [30] |
| SCID- hu Thy/Liv | Implantation of human fetal thymus and liver fragments | SCID | Multilineage hematopoiesis | Immature T cells Low myeloid cells repopulation No B cells | [31] |
| hu-BLT | Implantation of human fetal thymus, liver fragments, and human CD34+ cells from fetal liver | NOD-SCID, NSG, NRG | Multilineage hematopoiesis Primary immune response HLA T cells restriction Functional memory T cells | Human fetal tissue Development of late GvHD Lower myeloid cells repopulation Non-functional NK cells | [32,33] |
| Skin graft models | Subcutaneous implantation of human fetal or adult skin tissue | SCID, SCID-beige athymic nude, NOD-SCID, NSG, BRG | Resident human immune cells Modeling multiple skin-related diseases Drug screening Fast and simple readouts | Reduced vascularization Need for additional reconstruction of human immune response Human fetal tissue | [34] |
| LoM | Subcutaneous implantation of human fetal lung tissue | NSG | Vascularized human lung implants Human cytokines and chemokines | Few hematopoietic cells detected Human fetal tissue | [35] |
| BLT-L | Implantation of human fetal thymus, liver fragments, and human CD34+ cells from fetal liver Subcutaneous implantation of human fetal lung tissue | NSG | Multilineage hematopoiesis Primary immune response HLA T cells restriction Functional memory T cells Vascularized human lung implants Human cytokines and chemokines | Human fetal tissue | [35] |
| Strain | Characteristics | References |
|---|---|---|
| Athymic Nude NU(NCr)-Foxn1nu | T cell deficiency | [101] |
| SCID Prkdcscid | T and B cell deficiency | [30] |
| SCID/beige Lystbg Prkdcscid | T and B cell deficiency Reduced NK cell activity | [108] |
| NOD-SCID NOD Prkdcscid | T and B cell deficiency Phagocytic tolerance | [106,112] |
| NOG/NSG NOD-SCID IL2rg−/− | T, B, and NK cell deficiency Phagocytic tolerance | [118,119] |
| NRG NOD-Rag2−/−IL2rg−/− | T, B, and NK cell deficiency Phagocytic tolerance | [120] |
| BRG BALB/c-Rag2−/−Il2rg−/− | T, B, and NK cell deficiency Phagocytic tolerance | [114] |
| MISTRG M-CSFh/hIL-3/GM-CSFh/hSIRPah/hTPOh/hRAG2−/−IL2Rg−/− | T, B, and NK cell deficiency Phagocytic tolerance Expression of human cytokines | [121] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kutle, I.; Dittrich, A.; Wirth, D. Mouse Models for Human Herpesviruses. Pathogens 2023, 12, 953. https://doi.org/10.3390/pathogens12070953
Kutle I, Dittrich A, Wirth D. Mouse Models for Human Herpesviruses. Pathogens. 2023; 12(7):953. https://doi.org/10.3390/pathogens12070953
Chicago/Turabian StyleKutle, Ivana, Anne Dittrich, and Dagmar Wirth. 2023. "Mouse Models for Human Herpesviruses" Pathogens 12, no. 7: 953. https://doi.org/10.3390/pathogens12070953
APA StyleKutle, I., Dittrich, A., & Wirth, D. (2023). Mouse Models for Human Herpesviruses. Pathogens, 12(7), 953. https://doi.org/10.3390/pathogens12070953