Whole Genome Sequencing and Comparative Genomic Analysis of Chlamydia gallinacea Field Strains Isolated from Poultry in Poland

 , ,
, ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Samples
2.2. DNA Extraction and Chlamydia Identification
2.3. Isolation and Propagation in Cell Culture
2.4. Preparation of Genomic DNA for Illumina and Nanopore (MinION) Sequencing
2.5. Illumina and Nanopore Sequencing, Assembly, and Draft Annotation
2.6. Phylogenetic and Genome Analysis
3. Results
3.1. Identification and Isolation of Chlamydia gallinacea Strains
3.2. Chlamydia gallinacea Genomes
3.3. Phylogenetic and Genomic Analysis
3.3.1. ANIb and Tetra-Nucleotide Signatures
3.3.2. Analysis of 16S rRNA, 23S rRNA and ompA
3.3.3. Multi-Locus Sequence Typing
3.3.4. SNP-Based Analysis
3.3.5. Plasticity Zone
3.3.6. Plasmid Comparisons
3.3.7. Putative Virulence Factors and Effector Delivery System
3.3.8. Inclusion Membrane Proteins and Membrane-Associated Proteins
3.3.9. The Family of Polymorphic Membrane Proteins
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Vorimore, F.; Hölzer, M.; Liebler-Tenorio, E.M.; Barf, L.M.; Delannoy, S.; Vittecoq, M.; Wedlarski, R.; Lécu, A.; Scharf, S.; Blanchard, Y.; et al. Evidence for the existence of a new genus Chlamydiifrater gen. nov. inside the family Chlamydiaceae with two new species isolated from flamingo (Phoenicopterus roseus): Chlamydiifrater phoenicopteri sp. nov. and Chlamydiifrater volucris sp. nov. Syst. Appl. Microbiol. 2021, 44, 126200. [Google Scholar] [CrossRef] [PubMed]
- Sachse, K.; Bavoil, P.M.; Kaltenboeck, B.; Stephens, R.S.; Kuo, C.C.; Rosselló-Móra, R.; Horn, M. Emendation of the family Chlamydiaceae: Proposal of a single genus, Chlamydia, to include all currently recognized species. Syst. Appl. Microbiol. 2015, 38, 99–103. [Google Scholar] [CrossRef] [PubMed]
- Staub, E.; Marti, H.; Biondi, R.; Levi, A.; Donati, M.; Leonard, C.A.; Ley, S.D.; Pillonel, T.; Greub, G.; Seth-Smith, H.M.B.; et al. Novel Chlamydia species isolated from snakes are temperature-sensitive and exhibit decreased susceptibility to azithromycin. Sci. Rep. 2018, 8, 5660. [Google Scholar] [CrossRef]
- Borel, N.; Greub, G. International committee on systematics of prokaryotes (ICSP) subcommittee on the taxonomy of chlamydiae. Minutes of the closed meeting, 5 July 2018 Woudschoten, Zeist, The Netherlands. Int. J. Syst. Evol. Microbiol. 2019, 69, 2606–2608. [Google Scholar] [CrossRef]
- Laroucau, K.; Vorimore, F.; Aaziz, R.; Solmonson, L.; Hsia, R.C.; Bavoil, P.M.; Fach, P.; Hölzer, M.; Wuenschmann, A.; Sachse, K. Chlamydia buteonis, a new Chlamydia species isolated from a red-shouldered hawk. Syst. Appl. Microbiol. 2019, 42, 125997. [Google Scholar] [CrossRef] [PubMed]
- Chaiwattanarungruengpaisan, S.; Thongdee, M.; Anuntakarun, S.; Payungporn, S.; Arya, N.; Punchukrang, A.; Ramasoota, P.; Singhakaew, S.; Atithep, T.; Sariya, L. A new species of Chlamydia isolated from Siamese crocodiles (Crocodylus siamensis). PLoS ONE 2021, 16, e0252081. [Google Scholar] [CrossRef] [PubMed]
- Vorimore, F.; Hsia, R.-C.; Huot-Creasy, H.; Bastian, S.; Deruyter, L.; Passet, A.; Sachse, K.; Bavoil, P.M.; Myers, G.; Laroucau, K. Isolation of a New Chlamydia species from the Feral Sacred Ibis (Threskiornis aethiopicus): Chlamydia ibidis. PLoS ONE 2013, 8, e74823. [Google Scholar] [CrossRef]
- Taylor-Brown, A.; Bachmann, N.L.; Borel, N.; Polkinghorne, A. Culture-independent genomic characterisation of Candidatus Chlamydia sanzinia, a novel uncultivated bacterium infecting snakes. BMC Genom. 2016, 17, 710. [Google Scholar] [CrossRef]
- Taylor-Brown, A.; Spang, L.; Borel, N.; Polkinghorne, A. Culture-independent metagenomics supports discovery of uncultivable bacteria within the genus Chlamydia. Sci. Rep. 2017, 7, 10661. [Google Scholar] [CrossRef]
- Laroucau, K.; Ortega, N.; Vorimore, F.; Aaziz, R.; Mitura, A.; Szymańska-Czerwińska, M.; Cicerol, M.; Salinas, J.; Sachse, K.; Caro, M.R. Detection of a novel Chlamydia species in captive spur-thighed tortoises (Testudo graeca) in southeastern Spain and proposal of Candidatus Chlamydia testudinis. Syst. Appl. Microbiol. 2020, 43, 126071. [Google Scholar] [CrossRef]
- Sachse, K.; Laroucau, K.; Riege, K.; Wehner, S.; Dilcher, M.; Creasy, H.H.; Weidmann, M.; Myers, G.; Vorimore, F.; Vicari, N.; et al. Evidence for the existence of two new members of the family Chlamydiaceae and proposal of Chlamydia avium sp. nov. and Chlamydia gallinacea sp. nov. Syst. Appl. Microbiol. 2014, 37, 79–88. [Google Scholar] [CrossRef] [PubMed]
- Zocevic, A.; Vorimore, F.; Marhold, C.; Horvatek, D.; Wang, D.; Slavec, B.; Prentza, Z.; Stavianis, G.; Prukner-Radovcic, E.; Dovc, A.; et al. Molecular characterization of atypical Chlamydia and evidence of their dissemination in different European and Asian chicken flocks by specific real-time PCR. Environ. Microbiol. 2012, 14, 2212–2222. [Google Scholar] [CrossRef] [PubMed]
- Lagae, S.; Kalmar, I.; Laroucau, K.; Vorimore, F.; Vanrompay, D. Emerging Chlamydia psittaci infections in chickens and examination of transmission to humans. J. Med. Microbiol. 2014, 63, 399–407. [Google Scholar] [CrossRef] [PubMed]
- Szymańska-Czerwińska, M.; Mitura, A.; Zaręba, K.; Schnee, C.; Koncicki, A.; Niemczuk, K. Poultry in Poland as Chlamydiaceae carrier. J. Vet. Res. 2017, 61, 411–419. [Google Scholar] [CrossRef]
- Guo, W.; Li, J.; Kaltenboeck, B.; Gong, J.; Fan, W.; Wang, C. Chlamydia gallinacea not C. psittaci is the endemic chlamydial species in chicken Gallus gallus. Sci. Rep. 2016, 6, 19638. [Google Scholar] [CrossRef]
- Li, J.; Guo, W.; Kaltenboeck, B.; Sachse, K.; Yang, Y.; Lu, G.; Zhang, J.; Luan, L.; You, J.; Huang, K.; et al. Chlamydia pecorum is the endemic intestinal species in cattle while C. gallinacea, C. psittaci and C. pneumoniae associate with sporadic systemic infection. Vet. Microbiol. 2016, 193, 93–99. [Google Scholar] [CrossRef]
- Stokes, H.S.; Martens, J.M.; Chamings, A.; Walder, K.; Berg, M.L.; Segal, Y.; Bennett, A. Identification of Chlamydia gallinacea in a parrot and in free-range chickens in Australia. Aust. Vet. J. 2019, 97, 398–400. [Google Scholar] [CrossRef]
- Szymańska-Czerwińska, M.; Jodełko, A.; Zaręba-Marchewka, K.; Niemczuk, K. Experimental inoculation of chicken broilers with C. gallinacea strain 15-56/1. Sci. Rep. 2021, 11, 23856. [Google Scholar] [CrossRef]
- You, J.; Wu, Y.; Zhang, X.; Wang, X.; Gong, J.; Zhao, Z.; Zhang, J.; Zhang, J.; Sun, Z.; Li, J.; et al. Efficient fecal-oral and possible vertical, but not respiratory, transmission of emerging Chlamydia gallinacea in broilers. Vet. Microbiol. 2019, 230, 90–94. [Google Scholar] [CrossRef]
- Heijne, M.; Jelocnik, M.; Umanets, A.; Brouwer, M.S.M.; Dinkla, A.; Harders, F.; van Keulen, L.J.M.; Roest, H.J.; Schaafsma, F.; Velkers, F.C.; et al. Genetic and phenotypic analysis of the pathogenic potential of two novel Chlamydia gallinacea strains compared to Chlamydia psittaci. Sci. Rep. 2021, 11, 16516. [Google Scholar] [CrossRef]
- Laroucau, K.; Vorimore, F.; Aaziz, R.; Berndt, A.; Schubert, E.; Sachse, K. Isolation of a new chlamydial agent from infected domestic poultry coincided with cases of atypical pneumonia among slaughterhouse workers in France. Infect. Genet. Evol. 2009, 9, 1240–1247. [Google Scholar] [CrossRef] [PubMed]
- Marchino, M.; Rizzo, F.; Barzanti, P.; Sparasci, O.A.; Bottino, P.; Vicari, N.; Rigamonti, S.; Braghin, S.; Aaziz, R.; Vorimore, F.; et al. Chlamydia Species and Related Risk Factors in Poultry in North-Western Italy: Possible Bird-to-Human Transmission for C. gallinacea. Int. J. Environ. Res. Public. Health 2022, 19, 2174. [Google Scholar] [CrossRef] [PubMed]
- Shahbandeh, M. Meat Consumption Worldwide from 1990 to 2021, by Type. Available online: https://www.statista.com/statistics/274522/global-per-capita-consumption-of-meat/ (accessed on 30 November 2022).
- Biesek, J.; Banaszak, M.; Kądziołka, K.; Wlaźlak, S.; Adamski, M. Growth of broiler chickens, and physical features of the digestive system, and leg bones after aluminosilicates used. Sci. Rep. 2022, 12, 20425. [Google Scholar] [CrossRef]
- Guo, W.; Jelocnik, M.; Li, J.; Sachse, K.; Polkinghorne, A.; Pannekoek, Y.; Kaltenboeck, B.; Gong, J.; You, J.; Wang, C. From genomes to genotypes: Molecular epidemiological analysis of Chlamydia gallinacea reveals a high level of genetic diversity for this newly emerging chlamydial pathogen. BMC Genom. 2017, 18, 949. [Google Scholar] [CrossRef]
- Ehricht, R.; Slickers, P.; Goellner, S.; Hotzel, H.; Sachse, K. Optimized DNA microarray assay allows detection and genotyping of single PCR-amplifiable target copies. Mol. Cell. Probes 2006, 20, 60–63. [Google Scholar] [CrossRef]
- Laroucau, K.; Aaziz, R.; Meurice, L.; Servas, V.; Chossat, I.; Royer, H.; De Barbeyrac, B.; Vaillant, V.; Moyen, J.; Meziani, F.; et al. Outbreak of psittacosis in a group of women exposed to Chlamydia psittaci-infected chickens. Eurosurveillance 2015, 20, 21155. [Google Scholar] [CrossRef] [PubMed]
- Ménard, A.; Clerc, M.; Subtil, A.; Mégraud, F.; Bébéar, C.; de Barbeyrac, B. Development of a real-time PCR for the detection of Chlamydia psittaci. J. Med. Microbiol. 2006, 55, 471–473. [Google Scholar] [CrossRef]
- Zocevic, A.; Vorimore, F.; Vicari, N.; Gasparini, J.; Jacquin, L.; Sachse, K.; Magnino, S.; Laroucau, K. A real-time PCR assay for the detection of atypical strains of Chlamydiaceae from pigeons. PLoS ONE 2013, 8, e58741. [Google Scholar] [CrossRef]
- Pantchev, A.; Sting, R.; Bauerfeind, R.; Tyczka, J.; Sachse, K. Detection of all Chlamydophila and Chlamydia spp. of veterinary interest using species-specific real-time PCR assays. Comp. Immunol. Microbiol. Infect. Dis. 2010, 33, 473–484. [Google Scholar] [CrossRef]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 6 April 2022).
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. Fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- De Coster, W.; D’Hert, S.; Schultz, D.T.; Cruts, M.; Van Broeckhoven, C. NanoPack: Visualizing and processing long-read sequencing data. Bioinformatics 2018, 34, 2666–2669. [Google Scholar] [CrossRef] [PubMed]
- Wick, R.R.; Judd, L.M.; Gorrie, C.L.; Holt, K.E. Unicycler: Resolving bacterial genome assemblies from short and long sequencing reads. PLoS Comput. Biol. 2017, 13, e1005595. [Google Scholar] [CrossRef]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef]
- Seemann, T. Prokka: Rapid prokaryotic genome annotation. Bioinformatics 2014, 30, 2068–2069. [Google Scholar] [CrossRef] [PubMed]
- Richter, M.; Rosselló-Móra, R.; Glöckner, F.O.; Peplies, J. JSpeciesWS: A web server for prokaryotic species circumscription based on pairwise genome comparison. Bioinformatics 2015, 32, 929–931. [Google Scholar] [CrossRef]
- Kozlov, A.M.; Darriba, D.; Flouri, T.; Morel, B.; Stamatakis, A. RAxML-NG: A fast, scalable, and user-friendly tool for maximum likelihood phylogenetic inference. Bioinformatics 2019, 35, 4453–4455. [Google Scholar] [CrossRef]
- Letunic, I.; Bork, P. Interactive Tree of Life (iTOL) v5: An online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 2021, 49, W293–W296. [Google Scholar] [CrossRef]
- Hunt, M.; De Silva, N.; Otto, T.D.; Parkhill, J.; Keane, J.A.; Harris, S.R. Circlator: Automated circularization of genome assemblies using long sequencing reads. Genome Biol. 2015, 16, 294. [Google Scholar] [CrossRef]
- Katoh, K.; Misawa, K.; Kuma, K.; Miyata, T. MAFFT: A novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002, 30, 3059–3066. [Google Scholar] [CrossRef] [PubMed]
- Thompson, J.D.; Higgins, D.G.; Gibson, T.J. CLUSTAL W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic. Acids Res. 1994, 22, 4673–4680. [Google Scholar] [CrossRef]
- Nguyen, L.T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Trifinopoulos, J.; Nguyen, L.T.; von Haeseler, A.; Minh, B.Q. W-IQ-TREE: A fast online phylogenetic tool for maximum likelihood analysis. Nucleic Acids Res. 2016, 8, 232–235. [Google Scholar] [CrossRef]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef] [PubMed]
- Hoang, D.T.; Chernomor, O.; von Haeseler, A.; Minh, B.Q.; Vinh, L.S. UFBoot2: Improving the Ultrafast Bootstrap Approximation. Mol. Biol. Evol. 2018, 35, 518–522. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, M.J.; Petty, N.K.; Beatson, S.A. Easyfig: A genome comparison visualizer. Bioinformatics 2011, 27, 1009–1010. [Google Scholar] [CrossRef]
- Pearson, W.R. An introduction to sequence similarity (“homology”) searching. Curr. Protoc. Bioinform. 2013, 42, 3.1.1–3.1.8. [Google Scholar] [CrossRef]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef] [PubMed]
- Hallgren, J.; Tsirigos, K.D.; Pedersen, M.D.; Armenteros, J.J.A.; Marcatili, P.; Nielsen, H.; Krogh, A.; Winther, O. DeepTMHMM predicts alpha and beta transmembrane proteins using deep neural networks. bioRxiv 2022, 04.08.2022, 487609. [Google Scholar] [CrossRef]
- Vasilevsky, S.; Stojanov, M.; Greub, G.; Baud, D. Chlamydial polymorphic membrane proteins: Regulation, function and potential vaccine candidates. Virulence 2016, 7, 11–22. [Google Scholar] [CrossRef]
- Pannekoek, Y.; Dickx, V.; Beeckman, D.S.; Jolley, K.A.; Keijzers, W.C.; Vretou, E.; Maiden, M.C.; Vanrompay, D.; van der Ende, A. Multi locus sequence typing of Chlamydia reveals an association between Chlamydia psittaci genotypes and host species. PLoS ONE 2010, 5, e14179. [Google Scholar] [CrossRef]
- Szabo, K.V.; O’Neill, C.E.; Clarke, I.N. Diversity in Chlamydial plasmids. PLoS ONE 2020, 15, e0233298. [Google Scholar] [CrossRef] [PubMed]
- Hölzer, M.; Barf, L.M.; Lamkiewicz, K.; Vorimore, F.; Lataretu, M.; Favaroni, A.; Schnee, C.; Laroucau, K.; Marz, M.; Sachse, K. Comparative Genome Analysis of 33 Chlamydia Strains Reveals Characteristic Features of Chlamydia psittaci and Closely Related Species. Pathogens 2020, 9, 899. [Google Scholar] [CrossRef] [PubMed]
- Borges, V.; Hyden, P.; Gomes, J.P.; Rattei, T. Chapter 12 Chlamydia Genomics. In Chlamydia Biology, 1st ed.; Tan, M., Hegemann, J.H., Sutterlin, C., Eds.; Caister Academic Press: Poole, UK, 2020; pp. 263–286. ISBN 978-1-912530-28-1. [Google Scholar] [CrossRef]
- Heather, J.M.; Chain, B. The sequence of sequencers: The history of sequencing DNA. Genomics 2016, 107, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Rang, F.J.; Kloosterman, W.P.; De Ridder, J. From squiggle to basepair: Computational approaches for improving nanopore sequencing read accuracy. Genome Biol. 2018, 19, 90. [Google Scholar] [CrossRef]
- Amarasinghe, S.L.; Su, S.; Dong, X.; Zappia, L.; Ritchie, M.E.; Gouil, Q. Opportunities and challenges in long-read sequencing data analysis. Genome Biol. 2020, 21, 30. [Google Scholar] [CrossRef]
- Bachmann, N.L.; Polkinghorne, A.; Timms, P. Chlamydia genomics: Providing novel insights into chlamydial biology. Trends Microbiol. 2014, 22, 464–472. [Google Scholar] [CrossRef]
- Nunes, A.; Gomes, J.P. Evolution, phylogeny, and molecular epidemiology of Chlamydia. Infect. Genet. Evol. 2014, 23, 49–64. [Google Scholar] [CrossRef]
- Zaręba-Marchewka, K.; Szymańska-Czerwińska, M.; Niemczuk, K. Chlamydiae—What’s New? J. Vet. Res. 2020, 64, 461–467. [Google Scholar] [CrossRef]
- Zaręba-Marchewka, K.; Szymańska-Czerwińska, M.; Livingstone, M.; Longbottom, D.; Niemczuk, K. Whole Genome Sequencing and Comparative Genome Analyses of Chlamydia abortus Strains of Avian Origin Suggests That Chlamydia abortus Species Should Be Expanded to Include Avian and Mammalian Subgroups. Pathogens 2021, 10, 1405. [Google Scholar] [CrossRef]
- Rajaram, K.; Giebel, A.M.; Toh, E.; Hu, S.; Newman, J.H.; Morrison, S.G.; Kari, L.; Morrison, R.P.; Nelson, D.E. Mutational analysis of the Chlamydia muridarum plasticity zone. Infect. Immun. 2015, 83, 2870–2881. [Google Scholar] [CrossRef]
- Rockey, D.D. Unraveling the basic biology and clinical significance of the chlamydial plasmid. J. Exp. Med. 2011, 208, 2159–2162. [Google Scholar] [CrossRef] [PubMed]
- Pawlikowska-Warych, M.; Śliwa-Dominiak, J.; Deptuła, W. Chlamydial plasmids and bacteriophages. Acta Biochim. Pol. 2015, 62, 1–6. [Google Scholar] [CrossRef]
- Tampakaki, A.P.; Fadouloglou, V.E.; Gazi, A.D.; Panopoulos, N.J.; Kokkinidis, M. Conserved features of type III secretion. Cell. Microbiol. 2004, 6, 805–816. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Lesser, C.F.; Lory, S. The essential role of the CopN protein in Chlamydia pneumoniae intracellular growth. Nature 2008, 456, 112–115. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, P.H.; Lutter, E.I.; Hackstadt, T. Chlamydia trachomatis inclusion membrane protein MrcA interacts with the inositol 1,4,5-trisphosphate receptor type 3 (ITPR3) to regulate extrusion formation. PLoS Pathog. 2018, 14, e1006911. [Google Scholar] [CrossRef]
- Pennini, M.E.; Perrinet, S.; Dautry-Varsat, A.; Subtil, A. Histone methylation by NUE, a novel nuclear effector of the intracellular pathogen Chlamydia trachomatis. PLoS Pathog. 2010, 6, e1000995. [Google Scholar] [CrossRef]
- Wolff, B.J.; Morrison, S.S.; Pesti, D.; Ganakammal, S.R.; Srinivasamoorthy, G.; Changayil, S.; Weil, M.R.; MacCannell, D.; Rowe, L.; Frace, M.; et al. Chlamydia psittaci comparative genomics reveals intraspecies variations in the putative outer membrane and type III secretion system genes. Microbiol. Soc. 2015, 161, 1378–1391. [Google Scholar] [CrossRef]
- Tolchard, J.; Walpole, S.J.; Miles, A.J.; Maytum, R.; Eaglen, L.A.; Hackstadt, T.; Wallace, B.A.; Blumenschein, T.M.A. The intrinsically disordered Tarp protein from chlamydia binds actin with a partially preformed helix. Sci. Rep. 2018, 8, 1960. [Google Scholar] [CrossRef]
- Mojica, S.A.; Hovis, K.M.; Frieman, M.B.; Tran, B.; Hsia, R.C.; Ravel, J.; Jenkins-Houk, C.; Wilson, K.L.; Bavoil, P.M. SINC, a type III secreted protein of Chlamydia psittaci, targets the inner nuclear membrane of infected cells and uninfected neighbors. Mol. Biol. Cell 2015, 26, 1918–1934. [Google Scholar] [CrossRef] [PubMed]
- Knittler, M.R.; Sachse, K. Chlamydia psittaci: Update on an underestimated zoonotic agent. Pathog. Dis. 2015, 73, 1–15. [Google Scholar] [CrossRef]
- Sixt, B.S.; Bastidas, R.J.; Finethy, R.; Baxter, R.M.; Carpenter, V.K.; Kroemer, G.; Coers, J.; Valdivia, R.H. The Chlamydia trachomatis Inclusion Membrane Protein CpoS Counteracts STING-Mediated Cellular Surveillance and Suicide Programs. Cell Host Microbe 2017, 21, 113–121. [Google Scholar] [CrossRef] [PubMed]
- Stanhope, R.; Flora, E.; Bayne, C.; Derre, I. IncV, a FFAT motif-containing Chlamydia protein, tethers the endoplasmic reticulum to the pathogen-containing vacuole. Proc. Natl. Acad. Sci. USA 2017, 114, 12039–12044. [Google Scholar] [CrossRef] [PubMed]
- Dehoux, P.; Flores, R.; Dauga, C.; Zhong, G.; Subtil, A. Multi-genome identification and characterization of chlamydiae-specific type III secretion substrates: The Inc proteins. BMC Genom. 2011, 12, 109. [Google Scholar] [CrossRef] [PubMed]
- Bugalhão, J.N.; Luís, M.P.; Pereira, I.S.; da Cunha, M.; Pais, S.V.; Mota, L.J. The Chlamydia trachomatis inclusion membrane protein CT006 associates with lipid droplets in eukaryotic cells. PLoS ONE 2022, 17, e0264292. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
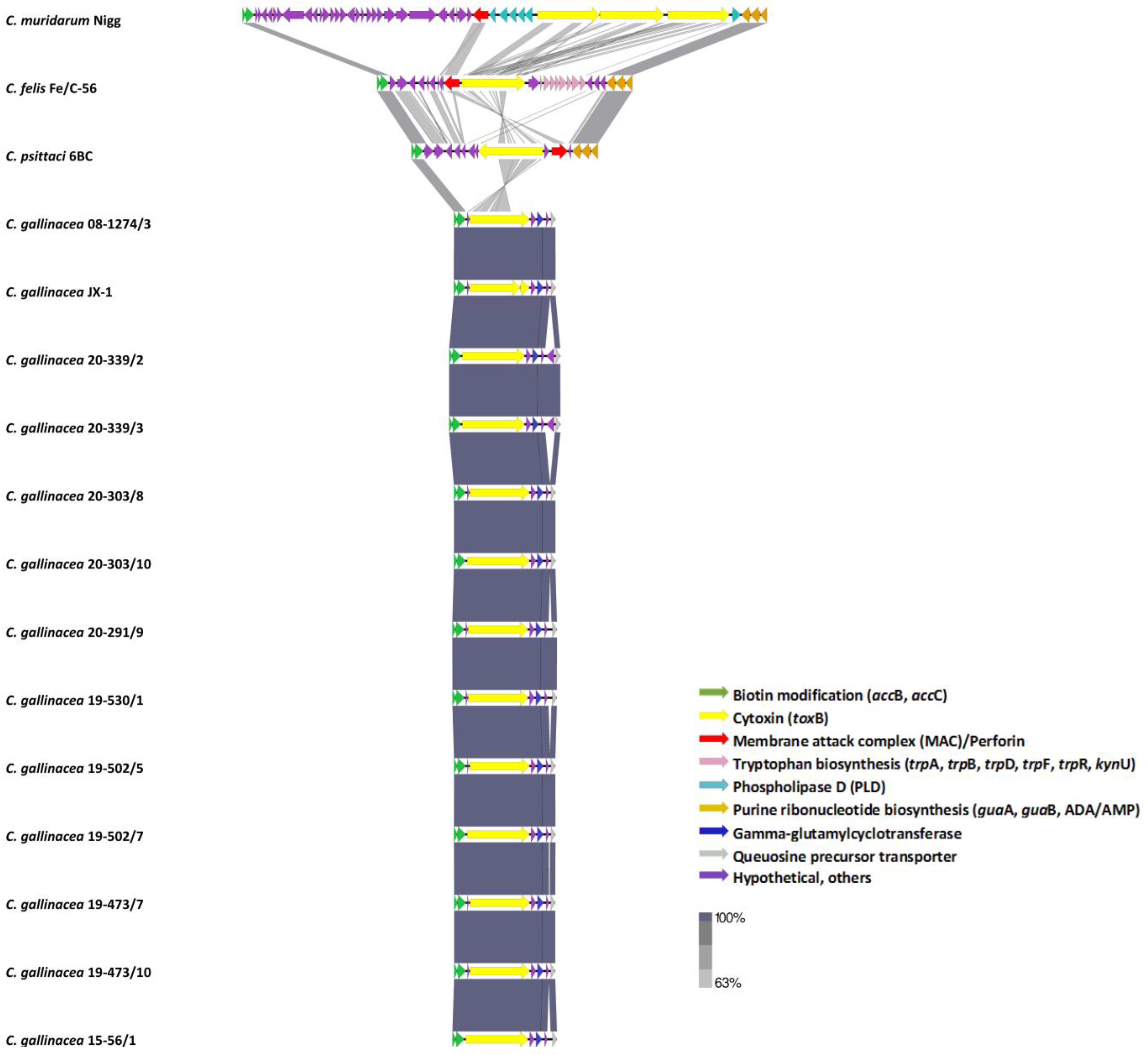{kind=link}
| No. of Flock | Sample | Real-Time PCR (Ct Value) | |||
|---|---|---|---|---|---|
| ID | Type | Origin | Chlamydiaceae 23S rRNA | C. gallinacea | |
| 20-339 | 20-3392 | cloacal swab | chicken | 27.95 | 27.23 |
| 20-339/3 | cloacal swab | chicken | 29.03 | 28.13 | |
| 20-303 | 20-303/8 | cloacal swab | chicken | 28.31 | 26.35 |
| 20-303/10 | cloacal swab | chicken | 25.88 | 23.59 | |
| 20-291 | 20-291/9 | cloacal swab | chicken | 29.61 | 27.73 |
| 19-530 | 19-530/1 | cloacal swab | chicken | 31.16 | 30.35 |
| 19-502 | 19-502/5 | cloacal swab | chicken | 28.19 | 26.98 |
| 19-502/7 | cloacal swab | chicken | 26.61 | 25.21 | |
| 19-473 | 19-473/7 | cloacal swab | chicken | 33.04 | 31.68 |
| 19-473/10 | cloacal swab | chicken | 27.99 | 26.94 | |
| 15-56 | 15-56/1 | cloacal swab | chicken | 23.26 | 21.80 |
| Sample | gatA | oppA | hflX | gidA | enoA | hemN | fumC | ST |
|---|---|---|---|---|---|---|---|---|
| 20-339/2 | 44 | 81 | 38 | 45 | 36 | 29 | 28 | 321 |
| 20-339/3 | 44 | 81 | 38 | 45 | 36 | 29 | 28 | 321 |
| 20-303/8 | 44 | 36 | 38 | 45 | 110 | 83 | 28 | 322 |
| 20-303/10 | 44 | 36 | 38 | 45 | 110 | 83 | 28 | 322 |
| 20-291/9 | 44 | 36 | 40 | 45 | 36 | 29 | 28 | 323 |
| 19-530/1 | 44 | 36 | 103 | 45 | 36 | 29 | 28 | 324 |
| 19-502/5 | 44 | 36 | 102 | 45 | 36 | 29 | 28 | 325 |
| 19-502/7 | 44 | 88 | 38 | 45 | 36 | 84 | 28 | 326 |
| 19-473/7 | 44 | 36 | 38 | 45 | 36 | 29 | 89 | 327 |
| 19-473/10 | 44 | 36 | 38 | 45 | 36 | 29 | 89 | 327 |
| 15-56/1 | 44 | 36 | 102 | 45 | 111 | 29 | 28 | 328 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zaręba-Marchewka, K.; Bomba, A.; Scharf, S.; Niemczuk, K.; Schnee, C.; Szymańska-Czerwińska, M. Whole Genome Sequencing and Comparative Genomic Analysis of Chlamydia gallinacea Field Strains Isolated from Poultry in Poland. Pathogens 2023, 12, 891. https://doi.org/10.3390/pathogens12070891
Zaręba-Marchewka K, Bomba A, Scharf S, Niemczuk K, Schnee C, Szymańska-Czerwińska M. Whole Genome Sequencing and Comparative Genomic Analysis of Chlamydia gallinacea Field Strains Isolated from Poultry in Poland. Pathogens. 2023; 12(7):891. https://doi.org/10.3390/pathogens12070891
Chicago/Turabian StyleZaręba-Marchewka, Kinga, Arkadiusz Bomba, Sabine Scharf, Krzysztof Niemczuk, Christiane Schnee, and Monika Szymańska-Czerwińska. 2023. "Whole Genome Sequencing and Comparative Genomic Analysis of Chlamydia gallinacea Field Strains Isolated from Poultry in Poland" Pathogens 12, no. 7: 891. https://doi.org/10.3390/pathogens12070891
APA StyleZaręba-Marchewka, K., Bomba, A., Scharf, S., Niemczuk, K., Schnee, C., & Szymańska-Czerwińska, M. (2023). Whole Genome Sequencing and Comparative Genomic Analysis of Chlamydia gallinacea Field Strains Isolated from Poultry in Poland. Pathogens, 12(7), 891. https://doi.org/10.3390/pathogens12070891