
Looking at the Molecular Target of NS5A Inhibitors throughout a Population Highly Affected with Hepatitis C Virus

 ,
,  and
and 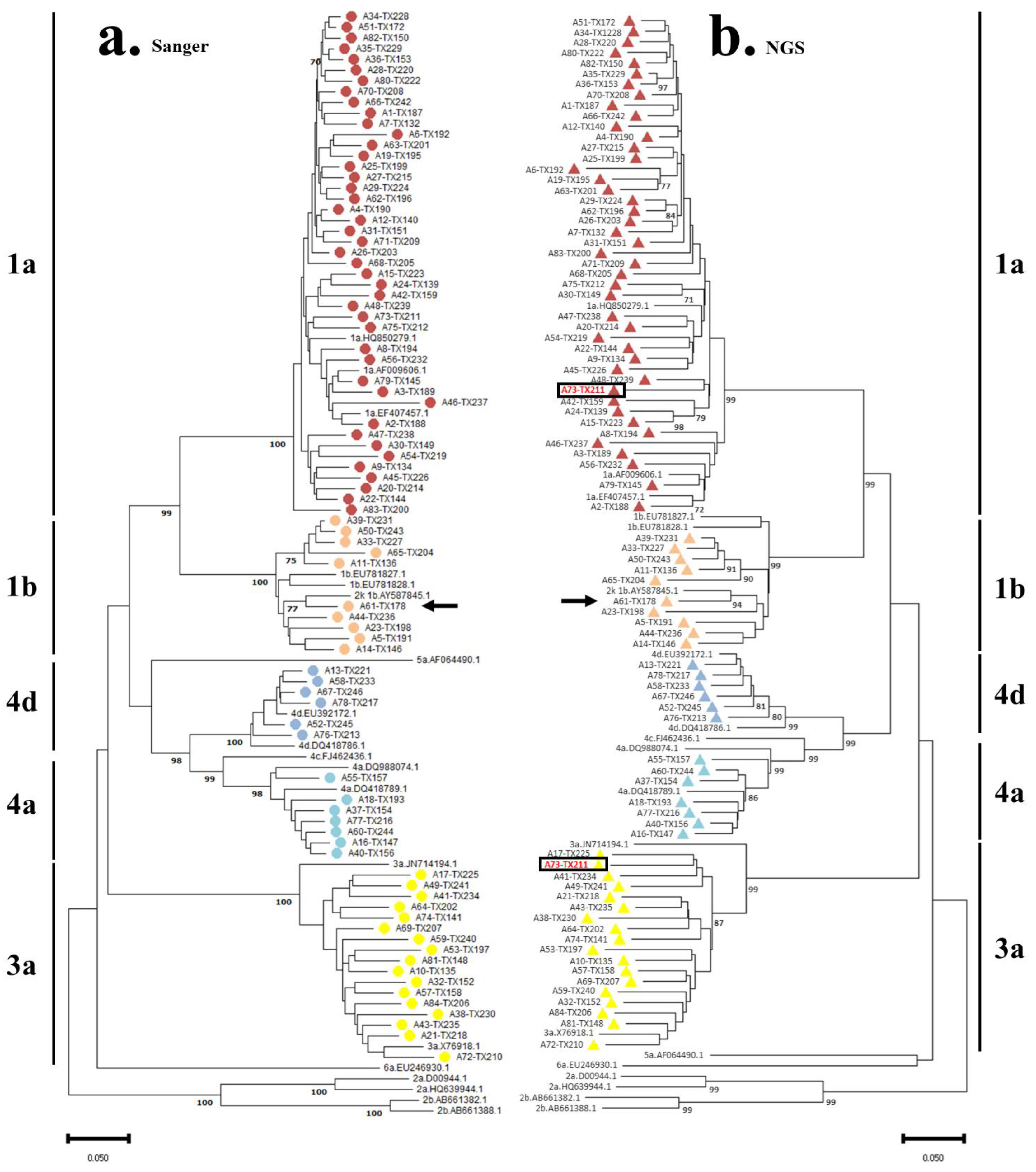{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Specimens and Study Population
2.2. HCV RNA Extraction, cDNA Synthesis and PCR Amplification
2.3. Products Detection, Purification and Sequencing Methods
2.4. Analysis of Sequencing Results
2.5. Phylogenetic Analysis
2.6. Mutation Analysis
2.7. Ethical Approval
3. Results
3.1. Population Characteristics
3.2. HCV Genotyping and Phylogenetic Analysis
3.3. Analysis of Resistance-Associated Mutations
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Martinez, M.A.; Franco, S. Therapy Implications of Hepatitis C Virus Genetic Diversity. Viruses 2021, 13, 41. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Global Hepatitis Report 2017. Available online: https://www.who.int/publications/i/item/9789241565455 (accessed on 5 September 2021).
- Simmonds, P. The origin of hepatitis C virus. Curr. Top. Microbiol. Immunol. 2013, 369, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Gale, M.; Foy, E.M. Evasion of intracellular host defence by hepatitis C virus. Nature 2005, 436, 939–945. [Google Scholar] [CrossRef] [PubMed]
- Smith, D.B.; Bukh, J.; Kuiken, C.; Muerhoff, A.S.; Rice, C.M.; Stapleton, J.T.; Simmonds, P. Classification and Genotype/Subtype Assignments of Hepatitis C Virus. ICTV. Available online: https://ictv.global/sg_wiki/flaviviridae/hepacivirus (accessed on 20 March 2022).
- Dietz, C.; Maasoumy, B. Direct-Acting Antiviral Agents for Hepatitis C Virus Infection-From Drug Discovery to Successful Implementation in Clinical Practice. Viruses 2022, 14, 1325. [Google Scholar] [CrossRef] [PubMed]
- Vega, S.; Neira, J.L.; Marcuello, C.; Lostao, A.; Abian, O.; Velazquez-Campoy, A. NS3 protease from hepatitis C virus: Biophysical studies on an intrinsically disordered protein domain. Int. J. Mol. Sci. 2013, 14, 13282–13306. [Google Scholar] [CrossRef]
- Calleja, J.L.; Uerena, S.; Perello, C.; Crespo, J. NS5A Resistance: Clinical implications and treatment possibilities. AIDS Rev. 2016, 18, 15–22. [Google Scholar]
- Sorbo, M.C.; Cento, V.; Di Maio, V.C.; Howe, A.Y.M.; Garcia, F.; Perno, C.F.; Ceccherini-Silberstein, F. Hepatitis C virus drug resistance associated substitutions and their clinical relevance: Update 2018. Drug Resist Updat. 2018, 37, 17–39. [Google Scholar] [CrossRef]
- Pádua, E.; Avó, A.P.; Almeida, C.; Água Doce, I.; Cortes Martins, H. Conhecer a Diversidade do Vírus da Hepatite C para Além da Frequência dos Genótipos em Amostras Analisadas entre 2009 e 2014 no Laboratório de Referência do Instituto Nacional de Saúde Dr. Ricardo Jorge. Acta Med. Port. 2015, 28, 695–701. [Google Scholar] [CrossRef]
- Pádua, E.; Avó, A.P.; Água Doce, I.; Almeida, C.; Martins, H.C. Subtipagem do vírus da Hepatite C por sequenciação: Um contributo para o conhecimento da diversidade genética. Obs. Bol. Epidemiológico INSA 2014, 3, 26–29. [Google Scholar]
- Silva, M.J.; Pereira, C.; Loureiro, R.; Balsa, C.; Lopes, P.; Água-Doce, I.; Belo, E.; Martins, H.C.; Coutinho, R.; Pádua, E. Hepatitis C in a Mobile Low-Threshold Methadone Program. Eur. J. Gastroenterol. Hepatol. 2017, 29, 657–662. [Google Scholar] [CrossRef]
- Andre-Garnier, E.; Besse, B.; Rodallec, A.; Ribeyrol, O.; Ferre, V.; Luco, C.; Le Guen, L.; Bourgeois, N.; Gournay, J.; Billaud, E.; et al. An NS5A single optimized method to determine genotype, subtype and resistance profiles of Hepatitis C strains. PLoS ONE 2017, 12, e0179562. [Google Scholar] [CrossRef] [PubMed]
- Hall, T.A. BIOEDIT: A user-friendly biological sequence alignment editor and analysis program for Windows 95/98/ NT. Nucleic Acids Symp Ser. 1999, 41, 95–98. [Google Scholar]
- Borges, V.; Pinheiro, M.; Pechirra, P.; Guiomar, R.; Gomes, J.P. INSaFLU: An automated open web-based bioinformatics suite “from-reads” for influenza whole-genome-sequencing-based surveillance. Genome Med. 2018, 10, 46. [Google Scholar] [CrossRef]
- Tamura, K.; Stecher, G.; Kumar, S. MEGA11: Molecular Evolutionary Genetics Analysis Version 11. Mol. Biol. Evol. 2021, 38, 3022–3027. [Google Scholar] [CrossRef]
- Naruya, S.; Masatoshi, N. The neighbor-joining method: A new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 1987, 4, 406–425. [Google Scholar] [CrossRef]
- Kimura, M. A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J. Mol. Evol. 1980, 16, 111–120. [Google Scholar] [CrossRef] [PubMed]
- Felsenstein, J. Confidence Limits on Phylogenies: An Approach Using the Bootstrap. Evolution 1985, 39, 783. [Google Scholar] [CrossRef]
- Pawlotsky, J.-M. Hepatitis C Virus Resistance to Direct-Acting Antiviral Drugs in Interferon-Free Regimens. Gastroenterology 2016, 151, 70–86. [Google Scholar] [CrossRef] [PubMed]
- Perales, C.; Chen, Q.; Soria, M.E.; Gregori, J.; Garcia-Cehic, D.; Nieto-Aponte, L.; Castells, L.; Imaz, A.; Llorens-Revull, M.; Domingo, E.; et al. Baseline hepatitis C virus resistance-associated substitutions present at frequencies lower than 15% may be clinically significant. Infect Drug Resist. 2018, 11, 2207–2210. [Google Scholar] [CrossRef] [PubMed]
- Christensen, K.T.; Pierard, F.; Beuselinck, K.; Bonsall, D.; Bowden, R.; Lagrou, K.; Nevens, F.; Schrooten, Y.; Simmonds, P.; Vandamme, A.M.; et al. Full-genome next-generation sequencing of hepatitis C virus to assess the accuracy of genotyping by the commercial assay LiPA and the prevalence of resistance-associated substitutions in a Belgian cohort. J. Clin. Virol. 2022, 155, 105252. [Google Scholar] [CrossRef]
- Viazov, S.; Ross, S.S.; Kyuregyan, K.K.; Timm, J.; Neumann-Haefelin, C.; Isaeva, O.V.; Popova, O.E.; Dmitriev, P.N.; El Sharkawi, F.; Thimme, R.; et al. Hepatitis C virus recombinants are rare even among intravenous drug users. J. Med. Virol. 2010, 82, 232–238. [Google Scholar] [CrossRef]
- Del Campo, J.A.; Parra-Sánchez, M.; Figueruela, B.; García-Rey, S.; Quer, J.; Gregori, J.; Bernal, S.; Grande, L.; Palomares, J.C.; Romero-Gómez, M. Hepatitis C virus deep sequencing for sub-genotype identification in mixed infections: A real-life experience. Int. J. Infect Dis. 2018, 67, 114–117. [Google Scholar] [CrossRef] [PubMed]
- Pham, S.T.; Bull, R.A.; Bennett, J.M.; Rawlinson, W.D.; Dore, G.J.; Lloyd, A.R.; White, P.A. Frequent multiple hepatitis C virus infections among injection drug users in a prison setting. Hepatology 2010, 52, 1564–1572. [Google Scholar] [CrossRef] [PubMed]
- Kalinina, O.; Norder, H.; Mukomolov, S.; Magnius, L.O. A Natural Intergenotypic Recombinant of Hepatitis C Virus Identified in St. Petersburg. J. Virol. 2002, 76, 4034–4043. [Google Scholar] [CrossRef]
- Avó, A.P.; Água-Doce, I.; Andrade, A.; Pádua, E. Hepatitis C Virus Subtyping Based on Sequencing of the C/E1 and NS5B Genomic Regions in Comparison to a Commercially Available Line Probe Assay. J. Med. Virol. 2013, 85, 815–822. [Google Scholar] [CrossRef] [PubMed]
- Llamosas-Falcón, L.; Shield, K.D.; Gelovany, M.; Hasan, O.S.M.; Manthey, J.; Monteiro, M.; Walsh, N.; Rehm, J. Impact of alcohol on the progression of HCV-related liver disease: A systematic review and meta-analysis. J. Hepatol. 2021, 75, 536–546. [Google Scholar] [CrossRef]
- Eltahla, A.A.; Leung, P.; Pirozyan, M.R.; Rodrigo, C.; Grebely, J.; Applegate, T.; Maher, L.; Luciani, F.; Lloyd, A.R.; Bull, R.A. Dynamic evolution of hepatitis C virus resistance-associated substitutions in the absence of antiviral treatment. Sci. Rep. 2017, 7, 41719. [Google Scholar] [CrossRef]
- Sarrazin, C. The importance of resistance to direct antiviral drugs in HCV infection in clinical practice. J. Hepatol. 2016, 64, 486–504. [Google Scholar] [CrossRef]
- Papaluca, T.; O’Keefe, J.; Bowden, S.; Doyle, J.S.; Stoove, M.; Hellard, M.; Thompson, A.J. Prevalence of baseline HCV NS5A resistance associated substitutions in genotype 1a, 1b and 3 infection in Australia. J. Clin. Virol. 2019, 120, 84–87. [Google Scholar] [CrossRef]
- Lontok, E.; Harrington, P.; Howe, A.; Kieffer, T.; Lennerstrand, J.; Lenz, O.; McPhee, F.; Mo, H.; Parkin, N.; Pilot-Matias, T.; et al. Hepatitis C virus drug resistance-associated substitutions: State of the art summary. Hepatology 2015, 62, 1623–1632. [Google Scholar] [CrossRef]
- Aissa Larousse, J.; Trimoulet, P.; Recordon Pinson, P.; Tauzin, B.; Azzouz, M.M.; Ben Mami, N.; Cheikh, I.; Triki, H.; Fleury, H. Prevalence of hepatitis C virus (HCV) variants resistant to NS5A inhibitors in naïve patients infected with HCV genotype 1 in Tunisia. Virol. J. 2015, 12, 84. [Google Scholar] [CrossRef] [PubMed]
- McPhee, F.; Hernandez, D.; Zhou, N. Effect of Minor Populations of NS5A and NS5B Resistance-Associated Variants on HCV Genotype-3 Response to Daclatasvir plus Sofosbuvir, with or without Ribavirin. Antivir. Ther. 2017, 22, 237–246. [Google Scholar] [CrossRef] [PubMed]
- Manns, M.; Samuel, D.; Gane, E.J.; Mutimer, D.; McCaughan, G.; Buti, M.; Prieto, M.; Calleja, J.L.; Peck-Radosavljevic, M.; Müllhaupt, B.; et al. Ledipasvir and sofosbuvir plus ribavirin in patients with genotype 1 or 4 hepatitis C virus infection and advanced liver disease: A multicentre, open-label, randomised, phase 2 trial. Lancet Infect Dis. 2016, 16, 685–697. [Google Scholar] [CrossRef]
- Schnell, G.; Tripathi, R.; Beyer, J.; Reisch, T.; Krishnan, P.; Dekhtyar, T.; Irvin, M.; Hall, C.; Yu, Y.; Mobashery, N.; et al. Characterization of demographics and NS5A genetic diversity for hepatitis C virus genotype 4-infected patients with or without cirrhosis treated with ombitasvir/paritaprevir/ritonavir. J. Viral Hepat. 2018, 25, 1078–1088. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ramos, D.; Pinto, M.; Sousa Coutinho, R.; Silva, C.; Quina, M.; Gomes, J.P.; Pádua, E. Looking at the Molecular Target of NS5A Inhibitors throughout a Population Highly Affected with Hepatitis C Virus. Pathogens 2023, 12, 754. https://doi.org/10.3390/pathogens12060754
Ramos D, Pinto M, Sousa Coutinho R, Silva C, Quina M, Gomes JP, Pádua E. Looking at the Molecular Target of NS5A Inhibitors throughout a Population Highly Affected with Hepatitis C Virus. Pathogens. 2023; 12(6):754. https://doi.org/10.3390/pathogens12060754
Chicago/Turabian StyleRamos, Diogo, Miguel Pinto, Rodrigo Sousa Coutinho, Carolina Silva, Miriam Quina, João Paulo Gomes, and Elizabeth Pádua. 2023. "Looking at the Molecular Target of NS5A Inhibitors throughout a Population Highly Affected with Hepatitis C Virus" Pathogens 12, no. 6: 754. https://doi.org/10.3390/pathogens12060754
APA StyleRamos, D., Pinto, M., Sousa Coutinho, R., Silva, C., Quina, M., Gomes, J. P., & Pádua, E. (2023). Looking at the Molecular Target of NS5A Inhibitors throughout a Population Highly Affected with Hepatitis C Virus. Pathogens, 12(6), 754. https://doi.org/10.3390/pathogens12060754