

Individual Epitope-Specific CD8+ T Cell Immune Responses Are Shaped Differently during Chronic Viral Infection

 , ,
, ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Ethics Statement
2.2. Mice and Treatment
2.3. Synthetic Peptides and Dextramers
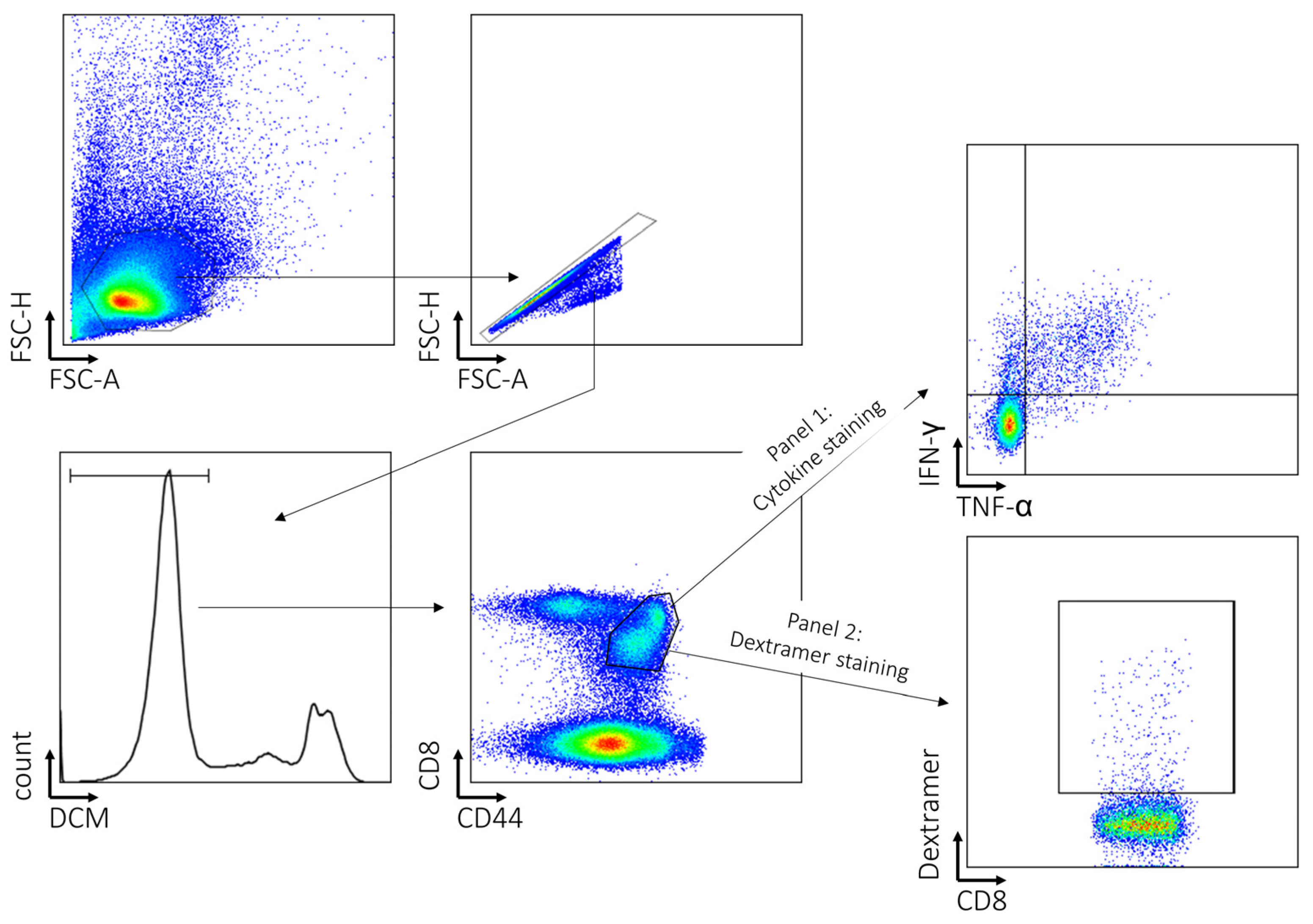
2.4. Cell Surface and Dextramer Staining for Flow Cytometry and Intracellular Cytokine Measurements
2.5. Next-Generation Sequencing of the Epitope-Specific T Cell Receptor Repertoire
2.6. Sequencing Analysis
2.7. Hydrogen Bond Calculations
2.8. Virus Titer Determination
2.9. Statistics
3. Results
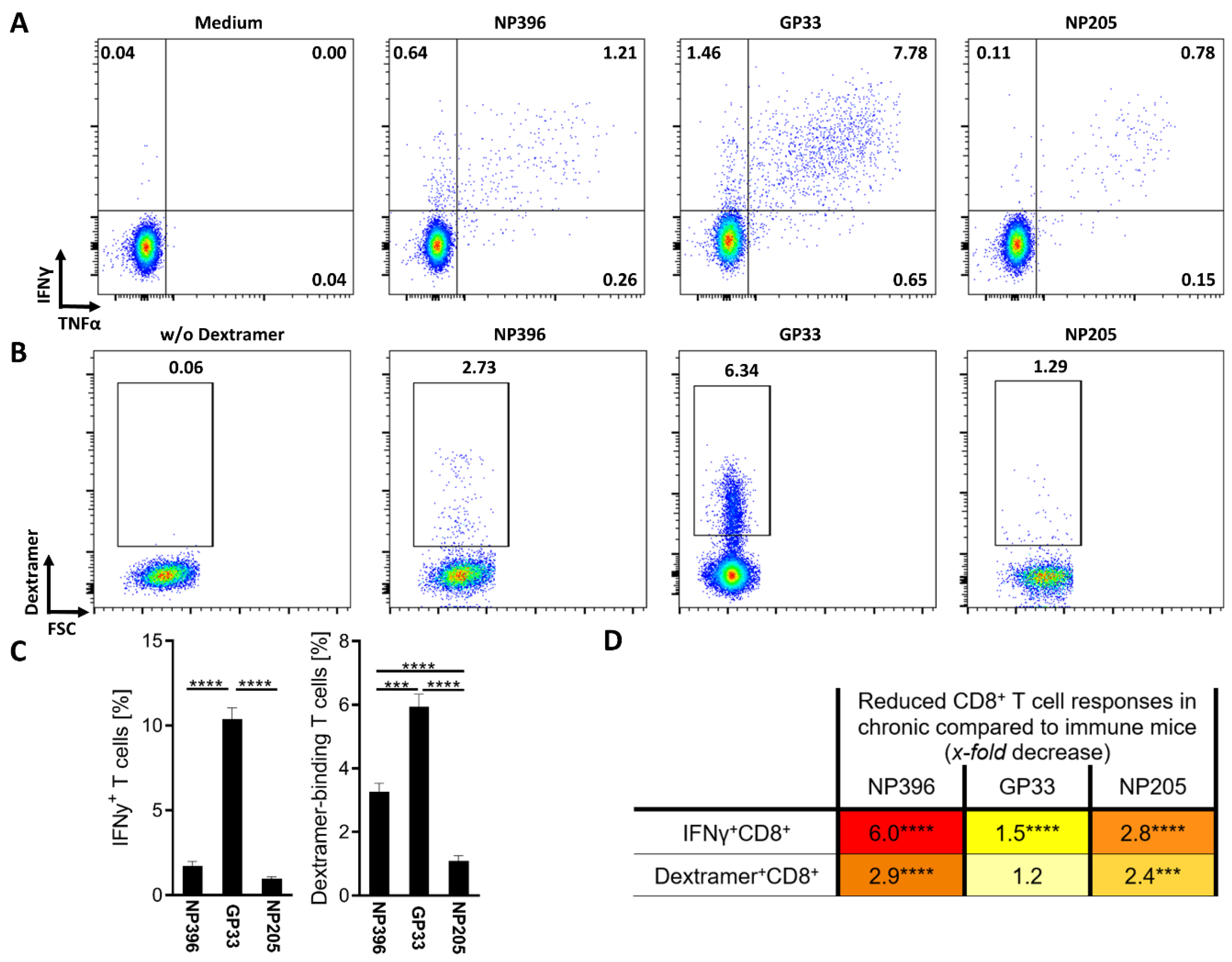
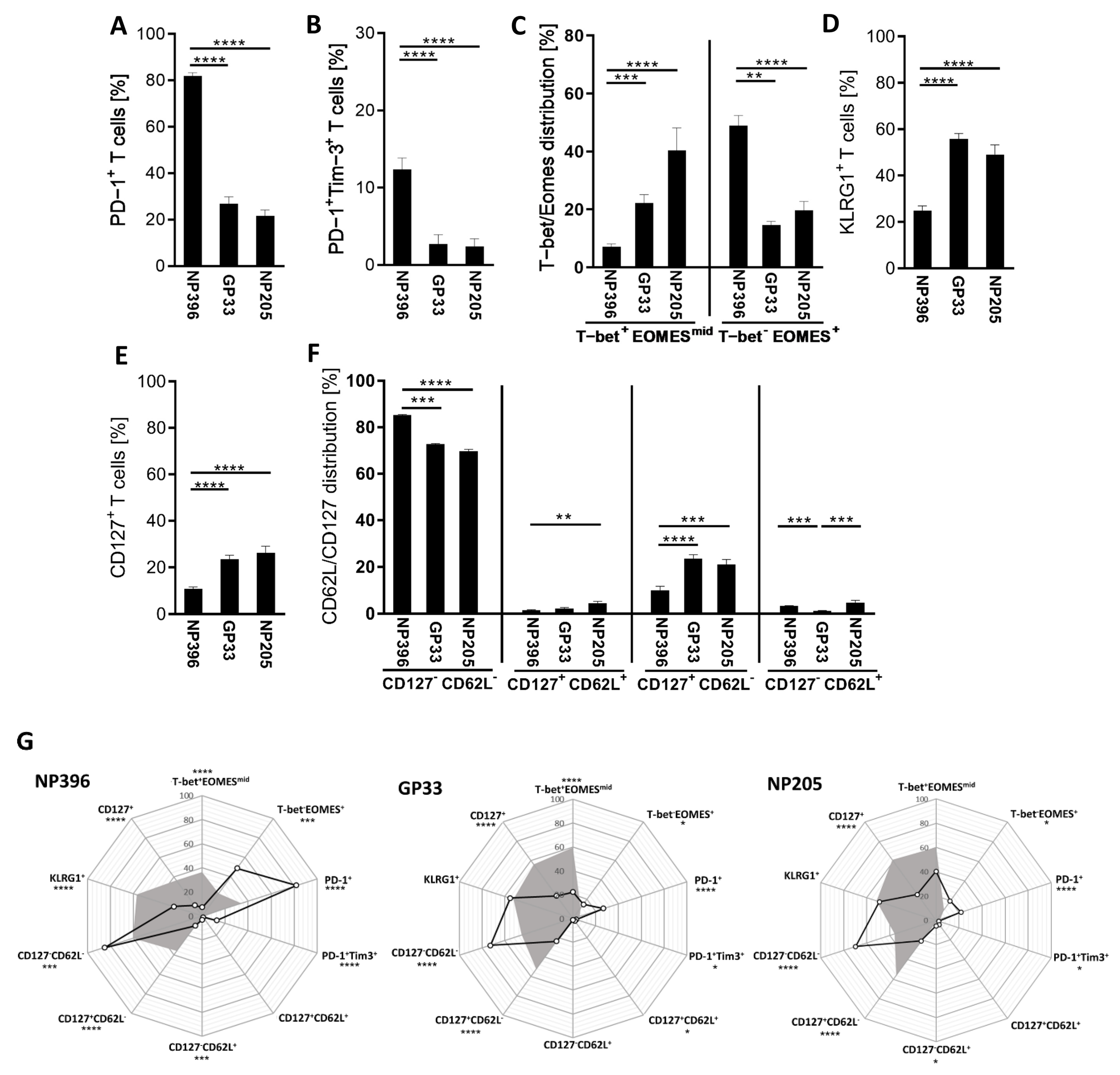
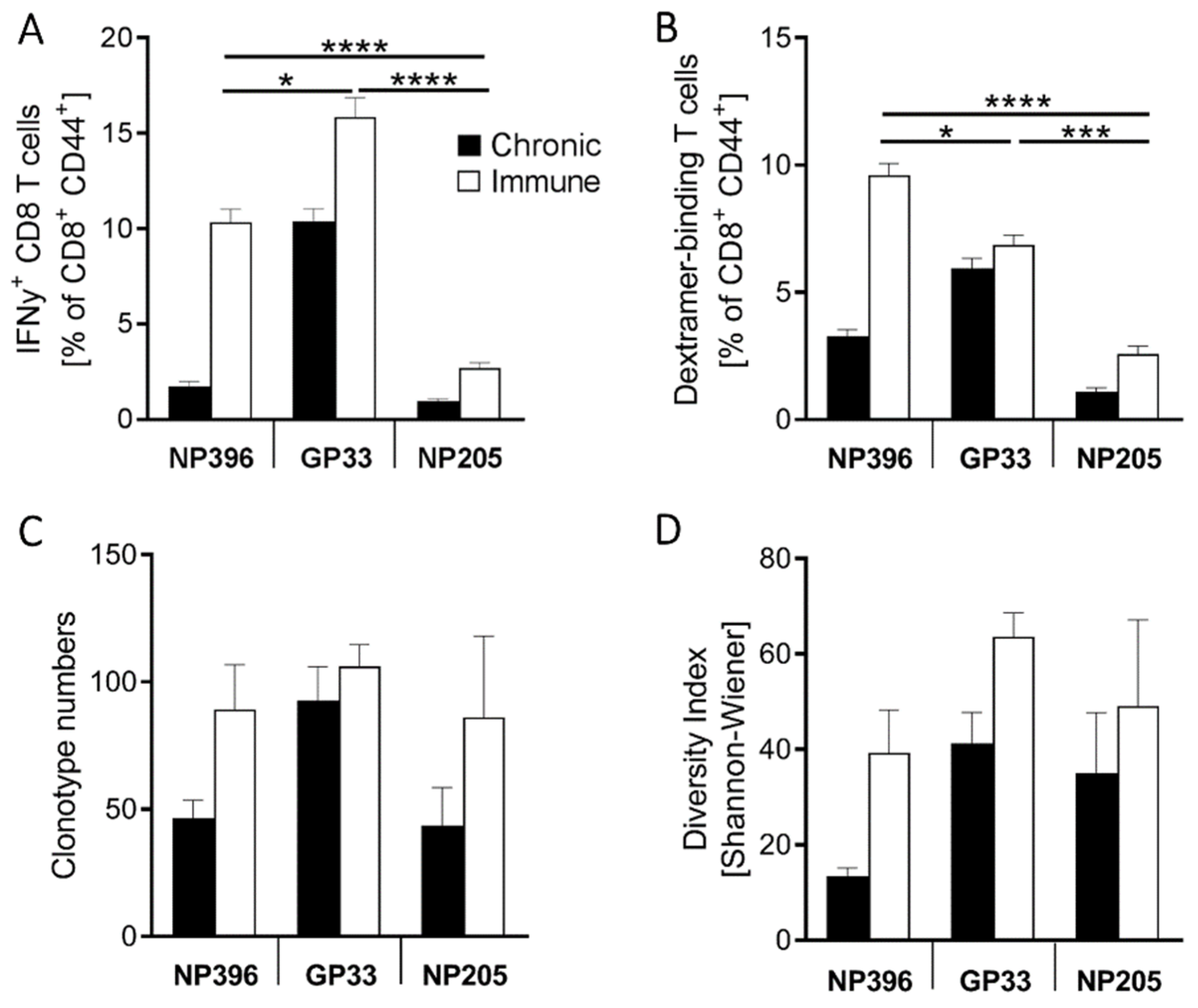
3.1. NP396-, GP33- and NP205-Specific T Cell Responses Can Be Used to Analyze TCR Repertoires of Differentially Exhausted Epitope-Specific Responses in Chronic LCMV Infection
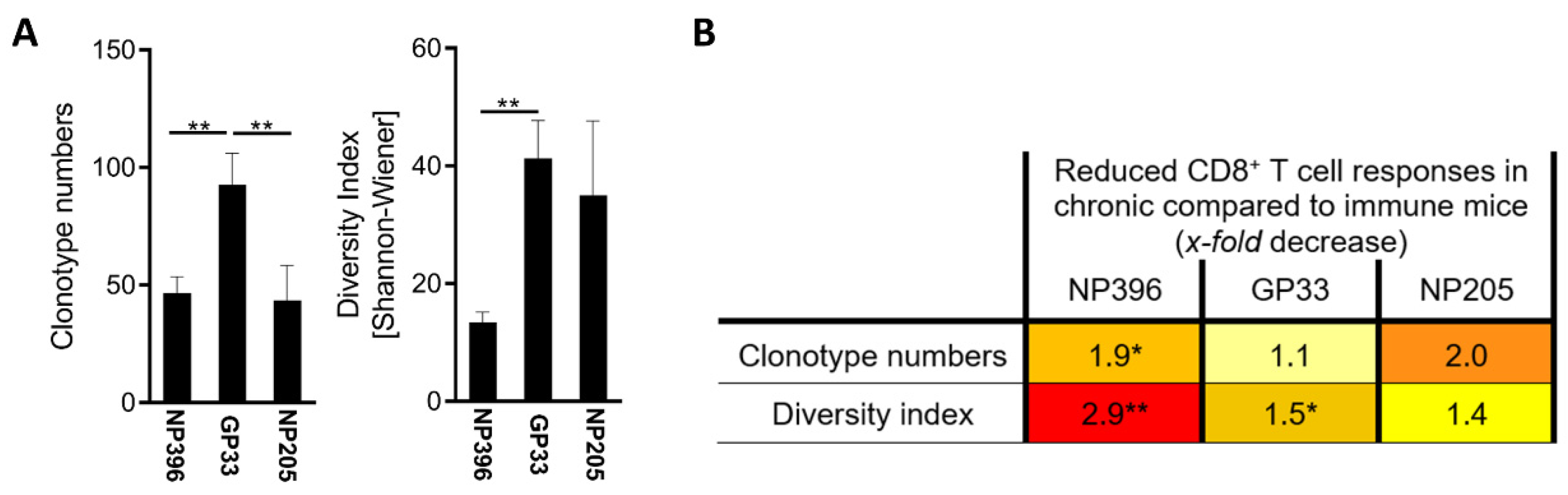
3.2. NP396-, GP33- and NP205-Specific T Cell Responses Displayed Differences in the TCR Repertoire in Chronic LCMV Infection
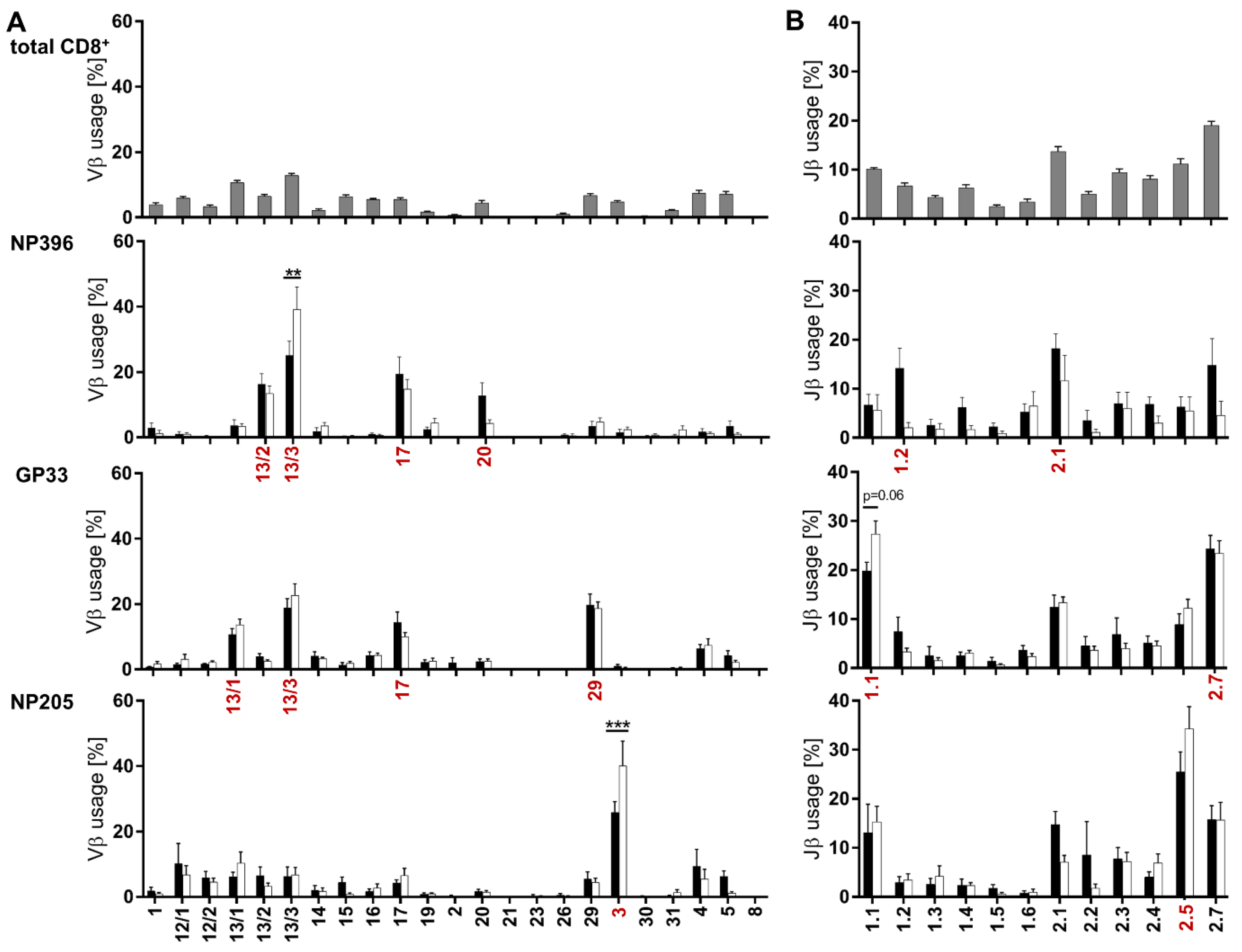
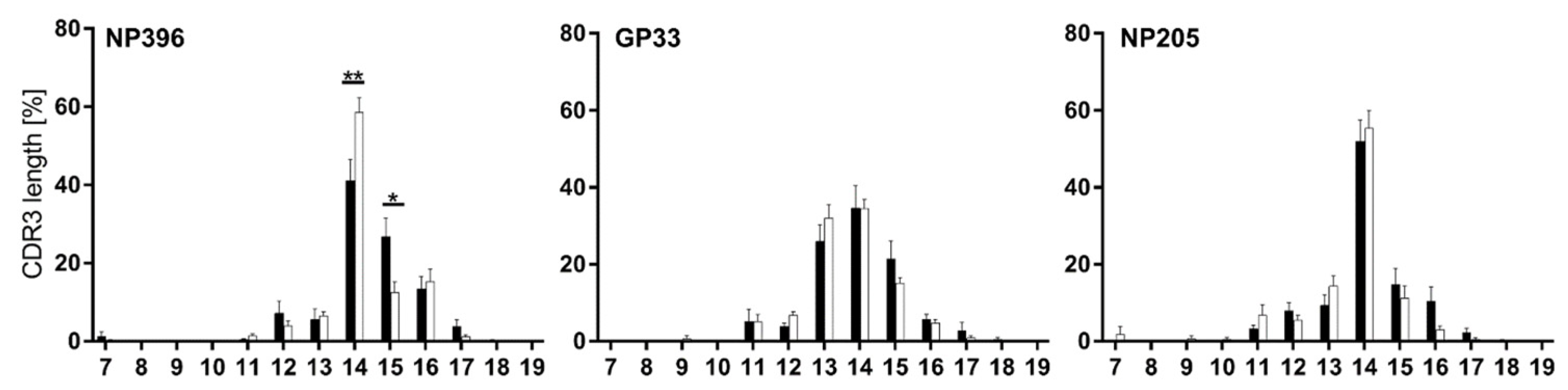
3.3. Distinct Vβ/Jβ Patterns Appeared in Individual Epitope-Specific TCR Repertoires
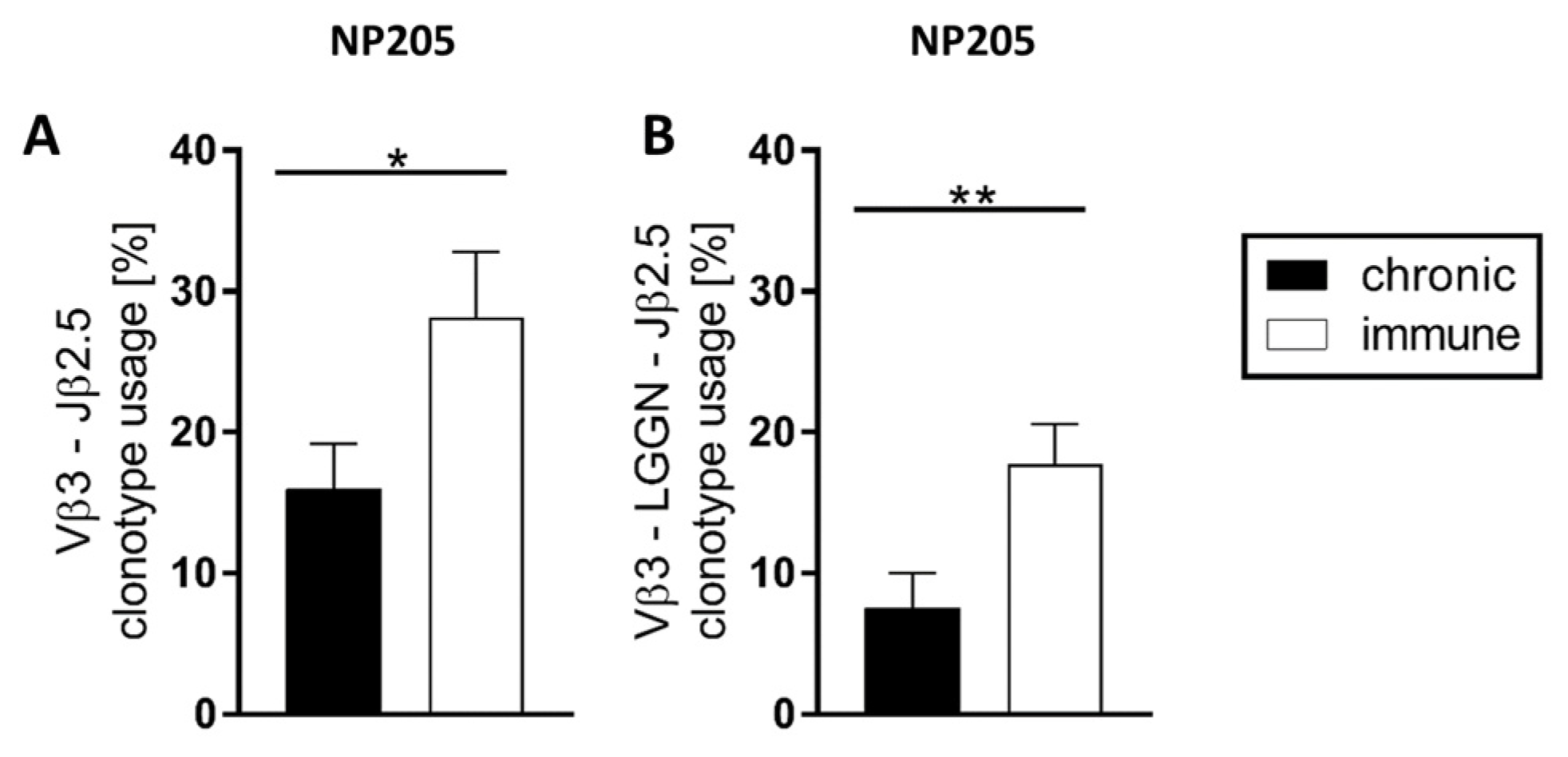
3.4. The TCR Repertoires to NP396 and GP33 Were Highly Private, Whereas NP205-Responses Revealed a Public TCR Repertoire
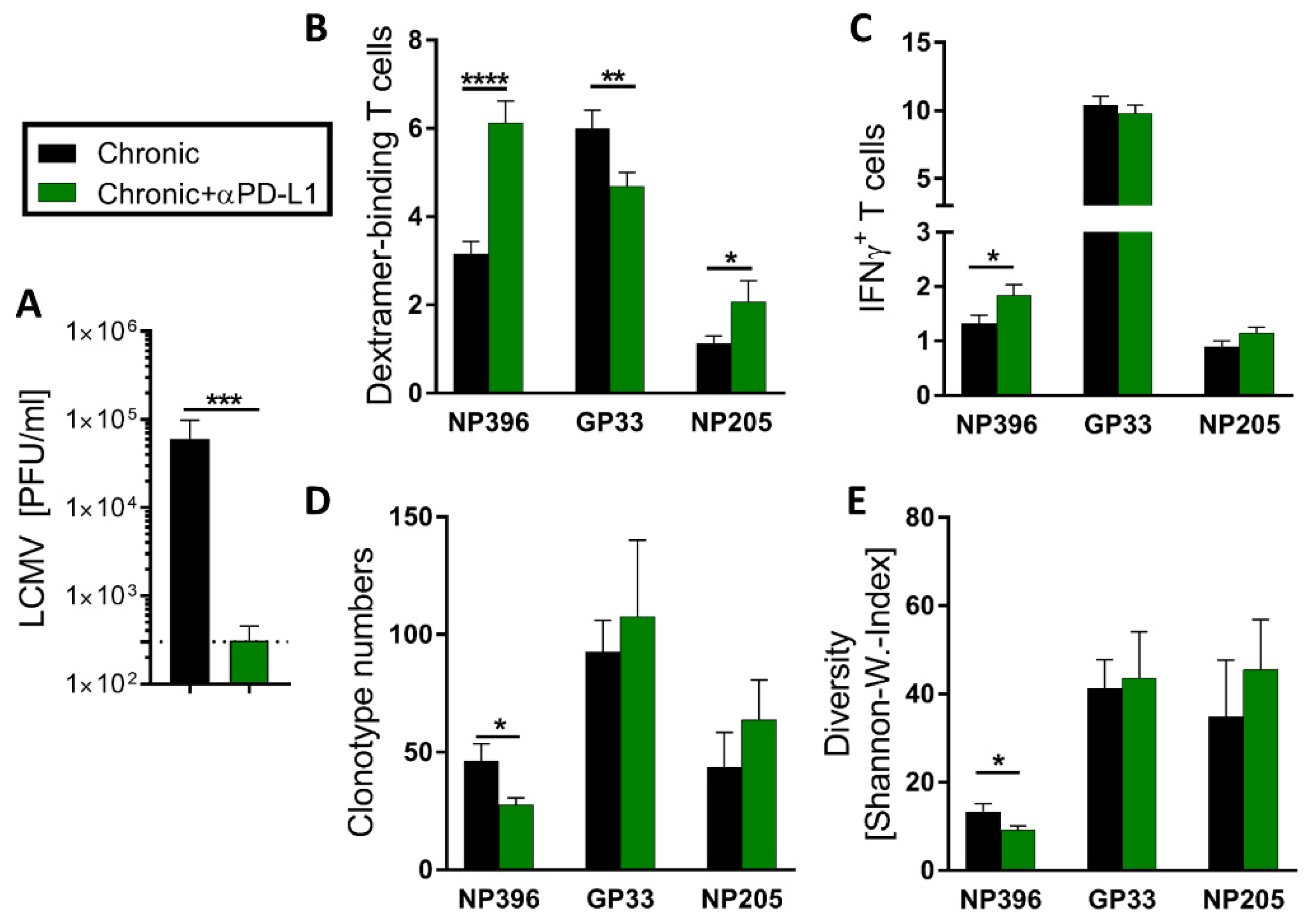
3.5. Checkpoint Inhibitor Treatment Shaped Predominantly the NP396- and NP205-Specific CD8+ TCR Repertoire, but Had Only Minor Effects on the GP33-Specific One
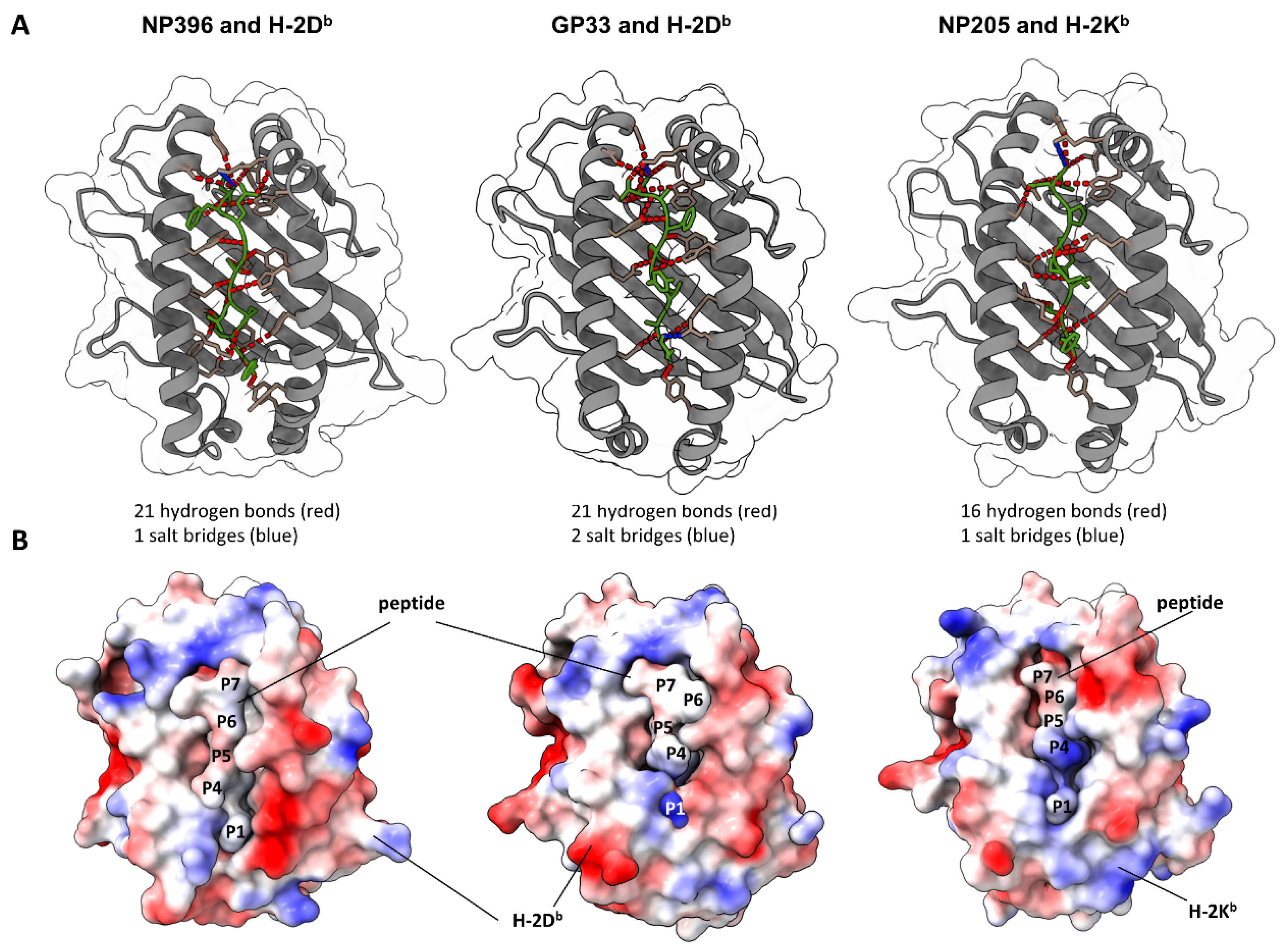
3.6. Crystallographic Structures of Peptide–MHC Complexes Indicated Differences in Epitope-Specific Antigen Presentation to CD8+ T Cells
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| NP396 | GP33 | NP205 | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Mouse | Treatment | Cells (Input) | Reads | Clonotypes | Diversity | Cells (Input) | Reads | Clonotypes | Diversity | Cells (Input) | Reads | Clonotypes | Diversity |
| 1 | chronic | 9000 | 15,551 | 72 | 24.0 | ||||||||
| 2 | chronic | 9000 | 15,584 | 52 | 28.1 | 830 | 103,183 | 21 | 11.2 | ||||
| 3 | chronic | 11,000 | 28,628 | 62 | 23.8 | ||||||||
| 4 | chronic | 9800 | 28,242 | 30 | 10.9 | 11,800 | 24,887 | 151 | 78.4 | ||||
| 5 | chronic | 1840 | 18,410 | 38 | 21.3 | 12,100 | 28,764 | 88 | 41.5 | ||||
| 6 | chronic | 2540 | 19,488 | 16 | 9.1 | 8800 | 18,303 | 70 | 34.9 | ||||
| 7 | chronic | 10,100 | 30,510 | 37 | 11.2 | 12,200 | 35,831 | 100 | 40.2 | ||||
| 8 | chronic | 8400 | 27,096 | 33 | 11.0 | ||||||||
| 9 | chronic | 12,300 | 30,587 | 54 | 9.2 | ||||||||
| 10 | chronic | 10,000 | 20,962 | 30 | 14.9 | ||||||||
| 11 | chronic | 11,300 | 20,164 | 33 | 15.7 | ||||||||
| 12 | chronic | 10,000 | 20,152 | 26 | 5.1 | ||||||||
| 13 | chronic | 8800 | 21,742 | 35 | 9.9 | ||||||||
| 14 | chronic | 6200 | 28,333 | 47 | 8.9 | ||||||||
| 15 | chronic | 6800 | 26,948 | 51 | 23.0 | ||||||||
| 16 | chronic | 6100 | 21,298 | 147 | 36.2 | ||||||||
| 17 | chronic | 5900 | 27,859 | 53 | 12.8 | ||||||||
| 18 | chronic | 10,400 | 3675 | 49 | 5.7 | ||||||||
| 19 | chronic | 9800 | 17,082 | 86 | 8.2 | ||||||||
| 20 | chronic | 10,000 | 24,239 | 54 | 17.6 | ||||||||
| 21 | chronic | 11,100 | 41,995 | 182 | 77.8 | 4022 | 9511 | 23 | 19.2 | ||||
| 22 | chronic | 15,100 | 37,435 | 59 | 27.2 | 1441 | 12,797 | 22 | 18.9 | ||||
| 23 | chronic | 10,800 | 8893 | 91 | 37.3 | 2540 | 14,480 | 124 | 103.5 | ||||
| 24 | chronic | 5547 | 37,082 | 60 | 46.4 | ||||||||
| 25 | chronic | 2190 | 34,981 | 45 | 39.6 | ||||||||
| 26 | chronic | 11,900 | 28,049 | 10 | 6.2 | ||||||||
| 27 | chronic | 6500 | 25,837 | 53 | 12.8 | ||||||||
| 28 | chr.+αPD-L1 | 9600 | 27,130 | 33 | 10.5 | 8000 | 25,475 | 102 | 44.2 | ||||
| 29 | chr.+αPD-L1 | 10,000 | 28,383 | 12 | 5.5 | 9000 | 21,762 | 134 | 63.4 | ||||
| 30 | chr.+αPD-L1 | 9460 | 32,553 | 29 | 7.9 | 14,200 | 31,020 | 63 | 23.6 | ||||
| 31 | chr.+αPD-L1 | 11,300 | 27,835 | 29 | 10.8 | 9400 | 28,884 | 116 | 40.0 | ||||
| 32 | chr.+αPD-L1 | 11,000 | 16,900 | 35 | 14.6 | ||||||||
| 33 | chr.+αPD-L1 | 11,000 | 17,275 | 29 | 10.4 | ||||||||
| 34 | chr.+αPD-L1 | 10,000 | 12,981 | 8 | 5.2 | ||||||||
| 35 | chr.+αPD-L1 | 10,000 | 17,407 | 24 | 13.3 | ||||||||
| 36 | chr.+αPD-L1 | 13,000 | 15,704 | 36 | 13.8 | ||||||||
| 37 | chr.+αPD-L1 | 12,000 | 20,618 | 28 | 8.7 | ||||||||
| 38 | chr.+αPD-L1 | 10,200 | 40,833 | 276 | 92.4 | 2972 | 14,982 | 77 | 56.4 | ||||
| 39 | chr.+αPD-L1 | 10,200 | 37,557 | 10 | 7.3 | 2861 | 13,683 | 74 | 52.1 | ||||
| 40 | chr.+αPD-L1 | 11,100 | 42,348 | 54 | 34.4 | 2536 | 8823 | 105 | 63.7 | ||||
| 41 | chr.+αPD-L1 | 10,000 | 13,757 | 20 | 8.6 | 5711 | 40,250 | 101 | 77.8 | ||||
| 42 | chr.+αPD-L1 | 3722 | 46,224 | 12 | 10.4 | ||||||||
| 43 | chr.+αPD-L1 | 14,700 | 13,123 | 10 | 4.6 | 13,500 | 29,309 | 14 | 12.9 | ||||
| 44 | immune | 13,600 | 36,281 | 156 | 89.8 | 5088 | 20,027 | 124 | 71.2 | ||||
| 45 | immune | 11,500 | 17,831 | 33 | 17.0 | 7865 | 38,368 | 15 | 13.6 | ||||
| 46 | immune | 13,600 | 21,382 | 89 | 37.1 | 13,600 | 32,486 | 86 | 48.3 | ||||
| 47 | immune | 13,600 | 17,748 | 44 | 15.1 | 13,400 | 40,100 | 23 | 16.2 | ||||
| 48 | immune | 15,500 | 23,349 | 141 | 70.9 | ||||||||
| 49 | immune | 10,000 | 22,435 | 124 | 56.8 | ||||||||
| 50 | immune | 8900 | 20,648 | 104 | 39.0 | ||||||||
| 51 | immune | 10,000 | 19,338 | 97 | 45.6 | 6003 | 16,6205 | 177 | 67.4 | ||||
| 52 | immune | 10,000 | 23,712 | 97 | 60.9 | 6068 | 35,970 | 9 | 8.2 | ||||
| 53 | immune | 10,000 | 32,581 | 108 | 60.0 | 5709 | 216,712 | 362 | 215.7 | ||||
| 54 | immune | 9500 | 24,943 | 76 | 48.1 | 11,020 | 64,975 | 9 | 8.3 | ||||
| 55 | immune | 10,000 | 22,220 | 101 | 65.4 | 5140 | 168,059 | 73 | 44.6 | ||||
| 56 | immune | 10,000 | 24,534 | 125 | 76.6 | 15,393 | 108,220 | 23 | 20.3 | ||||
| 57 | immune | 10,000 | 20,314 | 88 | 62.0 | 3408 | 135,541 | 45 | 25.7 | ||||
References
- WHO Organization. World Health Organization Fact Sheets Hepatitis B, Hepatitis C and HIV 2022. Available online: https://www.who.int/news-room/fact-sheets/detail/ (accessed on 25 April 2023).
- Wherry, E.J.; Kurachi, M. Molecular and Cellular Insights into T Cell Exhaustion. Nat. Rev. Immunol. 2015, 15, 486–499. [Google Scholar] [CrossRef]
- Wherry, E.J. T Cell Exhaustion. Nat. Immunol. 2011, 12, 492–499. [Google Scholar] [CrossRef]
- Walunas, T.L.; Bakker, C.Y.; Bluestone, J.A. CTLA-4 Ligation Blocks CD28-Dependent T Cell Activation. J. Exp. Med. 1996, 183, 2541–2550. [Google Scholar] [CrossRef]
- Parry, R.V.; Chemnitz, J.M.; Frauwirth, K.A.; Lanfranco, A.R.; Braunstein, I.; Kobayashi, S.V.; Linsley, P.S.; Thompson, C.B.; Riley, J.L. CTLA-4 and PD-1 Receptors Inhibit T-Cell Activation by Distinct Mechanisms. Mol. Cell Biol. 2005, 25, 9543–9553. [Google Scholar] [CrossRef]
- Li, K.; Yuan, Z.; Lyu, J.; Ahn, E.; Davis, S.J.; Ahmed, R.; Zhu, C. PD-1 Suppresses TCR-CD8 Cooperativity during T-Cell Antigen Recognition. Nat. Commun. 2021, 12, 2746. [Google Scholar] [CrossRef]
- Bucks, C.M.; Norton, J.A.; Boesteanu, A.C.; Mueller, Y.M.; Katsikis, P.D. Chronic Antigen Stimulation Alone is Sufficient to Drive CD8+ T Cell Exhaustion. J. Immunol. 2009, 182, 6697–6708. [Google Scholar] [CrossRef]
- Mueller, S.N.; Ahmed, R. High Antigen Levels are the Cause of T Cell Exhaustion during Chronic Viral Infection. Proc. Natl. Acad. Sci. USA 2009, 106, 8623–8628. [Google Scholar] [CrossRef]
- Angelosanto, J.M.; Blackburn, S.D.; Crawford, A.; Wherry, E.J. Progressive Loss of Memory T Cell Potential and Commitment to Exhaustion during Chronic Viral Infection. J. Virol. 2012, 86, 8161–8170. [Google Scholar] [CrossRef]
- Wherry, E.J.; Blattman, J.N.; Murali-Krishna, K.; van der Most, R.; Ahmed, R. Viral Persistence Alters CD8 T-Cell Immunodominance and Tissue Distribution and Results in Distinct Stages of Functional Impairment. J. Virol. 2003, 77, 4911–4927. [Google Scholar] [CrossRef]
- Kemball, C.C.; Lee, E.D.H.; Vezys, V.; Pearson, T.C.; Larsen, C.P.; Lukacher, A.E. Late Priming and Variability of Epitope-Specific CD8+ T Cell Responses during a Persistent Virus Infection. J. Immunol. 2005, 174, 7950–7960. [Google Scholar] [CrossRef]
- Barber, D.L.; Wherry, E.J.; Masopust, D.; Zhu, B.; Allison, J.P.; Sharpe, A.H.; Freeman, G.J.; Ahmed, R. Restoring Function in Exhausted CD8 T Cells during Chronic Viral Infection. Nature 2006, 439, 682–687. [Google Scholar] [CrossRef]
- Bengsch, B.; Martin, B.; Thimme, R. Restoration of HBV-Specific CD8+ T Cell Function by PD-1 Blockade in Inactive Carrier Patients is Linked to T Cell Differentiation. J. Hepatol. 2014, 61, 1212–1219. [Google Scholar] [CrossRef]
- Wu, T.; Ji, Y.; Moseman, E.A.; Xu, H.C.; Manglani, M.; Kirby, M.; Anderson, S.M.; Handon, R.; Kenyon, E.; Elkahloun, A.; et al. The TCF1-Bcl6 Axis Counteracts Type I Interferon to Repress Exhaustion and Maintain T Cell Stemness. Sci. Immunol. 2016, 1, eaai8593. [Google Scholar] [CrossRef]
- Chen, Z.; Ji, Z.; Ngiow, S.F.; Manne, S.; Cai, Z.; Huang, A.C.; Johnson, J.; Staupe, R.P.; Bengsch, B.; Xu, C.; et al. TCF-1-Centered Transcriptional Network Drives an Effector Versus Exhausted CD8 T Cell-Fate Decision. Immunity 2019, 51, 840–855.e5. [Google Scholar] [CrossRef]
- Welten, S.P.M.; Yermanos, A.; Baumann, N.S.; Wagen, F.; Oetiker, N.; Sandu, I.; Pedrioli, A.; Oduro, J.D.; Reddy, S.T.; Cicin-Sain, L.; et al. Tcf1+ Cells are Required to Maintain the Inflationary T Cell Pool upon MCMV Infection. Nat. Commun. 2020, 11, 2295. [Google Scholar] [CrossRef]
- Martinez, G.J.; Pereira, R.M.; Aijo, T.; Kim, E.Y.; Marangoni, F.; Pipkin, M.E.; Togher, S.; Heissmeyer, V.; Zhang, Y.C.; Crotty, S.; et al. The Transcription Factor NFAT Promotes Exhaustion of Activated CD8+ T Cells. Immunity 2015, 42, 265–278. [Google Scholar] [CrossRef]
- Wang, Y.; Hu, J.; Li, Y.; Xiao, M.; Wang, H.; Tian, Q.; Li, Z.; Tang, J.; Hu, L.; Tan, Y.; et al. The Transcription Factor TCF1 Preserves the Effector Function of Exhausted CD8 T Cells during Chronic Viral Infection. Front. Immunol. 2019, 10, 169. [Google Scholar] [CrossRef]
- Renkema, K.R.; Huggins, M.A.; Borges da Silva, H.; Knutson, T.P.; Henzler, C.M.; Hamilton, S.E. KLRG1+ Memory CD8 T Cells Combine Properties of Short-Lived Effectors and Long-Lived Memory. J. Immunol. 2020, 205, 1059–1069. [Google Scholar] [CrossRef]
- Borst, L.; Sluijter, M.; Sturm, G.; Charoentong, P.; Santegoets, S.J.; van Gulijk, M.; van Elsas, M.J.; Groeneveldt, C.; van Montfoort, N.; Finotello, F.; et al. NKG2A is a Late Immune Checkpoint on CD8 T Cells and Marks Repeated Stimulation and Cell Division. Int. J. Cancer 2022, 150, 688–704. [Google Scholar] [CrossRef]
- Jadhav, R.R.; Im, S.J.; Hu, B.; Hashimoto, M.; Li, P.; Lin, J.X.; Leonard, W.J.; Greenleaf, W.J.; Ahmed, R.; Goronzy, J.J. Epigenetic Signature of PD-1+ TCF1+ CD8 T Cells that Act as Resource Cells during Chronic Viral Infection and Respond to PD-1 Blockade. Proc. Natl. Acad. Sci. USA 2019, 116, 14113–14118. [Google Scholar] [CrossRef]
- Siddiqui, I.; Schaeuble, K.; Chennupati, V.; Fuertes Marraco, S.A.; Calderon-Copete, S.; Pais Ferreira, D.; Carmona, S.J.; Scarpellino, L.; Gfeller, D.; Pradervand, S.; et al. Intratumoral Tcf1+ PD-1+ CD8+ T Cells with Stem-Like Properties Promote Tumor Control in Response to Vaccination and Checkpoint Blockade Immunotherapy. Immunity 2019, 50, 195–211.e10. [Google Scholar] [CrossRef]
- Fisicaro, P.; Valdatta, C.; Massari, M.; Loggi, E.; Biasini, E.; Sacchelli, L.; Cavallo, M.C.; Silini, E.M.; Andreone, P.; Missale, G.; et al. Antiviral Intrahepatic T-Cell Responses can be Restored by Blocking Programmed Death-1 Pathway in Chronic Hepatitis B. Gastroenterology 2010, 138, 682–693. [Google Scholar] [CrossRef]
- Urbani, S.; Amadei, B.; Tola, D.; Pedrazzi, G.; Sacchelli, L.; Cavallo, M.C.; Orlandini, A.; Missale, G.; Ferrari, C. Restoration of HCV-Specific T Cell Functions by PD-1/PD-L1 Blockade in HCV Infection: Effect of Viremia Levels and Antiviral Treatment. J. Hepatol. 2008, 48, 548–558. [Google Scholar] [CrossRef]
- de Miguel, M.; Calvo, E. Clinical Challenges of Immune Checkpoint Inhibitors. Cancer Cell 2020, 38, 326–333. [Google Scholar] [CrossRef]
- Robert, C. A Decade of Immune-Checkpoint Inhibitors in Cancer Therapy. Nat. Commun. 2020, 11, 3801. [Google Scholar] [CrossRef]
- Buggert, M.; Tauriainen, J.; Yamamoto, T.; Frederiksen, J.; Ivarsson, M.A.; Michaelsson, J.; Lund, O.; Hejdeman, B.; Jansson, M.; Sonnerborg, A.; et al. T-Bet and Eomes are Differentially Linked to the Exhausted Phenotype of CD8+ T Cells in HIV Infection. PLoS Pathog. 2014, 10, e1004251. [Google Scholar] [CrossRef]
- Im, S.J.; Hashimoto, M.; Gerner, M.Y.; Lee, J.; Kissick, H.T.; Burger, M.C.; Shan, Q.; Hale, J.S.; Lee, J.; Nasti, T.H.; et al. Defining CD8+ T Cells that Provide the Proliferative Burst After PD-1 Therapy. Nature 2016, 537, 417–421. [Google Scholar] [CrossRef]
- Snell, L.M.; MacLeod, B.L.; Law, J.C.; Osokine, I.; Elsaesser, H.J.; Hezaveh, K.; Dickson, R.J.; Gavin, M.A.; Guidos, C.J.; McGaha, T.L.; et al. CD8+ T Cell Priming in Established Chronic Viral Infection Preferentially Directs Differentiation of Memory-Like Cells for Sustained Immunity. Immunity 2018, 49, 678–694.e5. [Google Scholar] [CrossRef]
- Velu, V.; Titanji, K.; Zhu, B.; Husain, S.; Pladevega, A.; Lai, L.; Vanderford, T.H.; Chennareddi, L.; Silvestri, G.; Freeman, G.J.; et al. Enhancing SIV-Specific Immunity in Vivo by PD-1 Blockade. Nature 2009, 458, 206–210. [Google Scholar] [CrossRef]
- Sandu, I.; Cerletti, D.; Claassen, M.; Oxenius, A. Exhausted CD8+ T Cells Exhibit Low and Strongly Inhibited TCR Signaling during Chronic LCMV Infection. Nat. Commun. 2020, 11, 4454. [Google Scholar] [CrossRef]
- Colombetti, S.; Basso, V.; Mueller, D.L.; Mondino, A. Prolonged TCR/CD28 Engagement Drives IL-2-Independent T Cell Clonal Expansion through Signaling Mediated by the Mammalian Target of Rapamycin. J. Immunol. 2006, 176, 2730–2738. [Google Scholar] [CrossRef]
- Hofmann, M.; Radsak, M.; Rechtsteiner, G.; Wiemann, K.; Gunder, M.; Bien-Grater, U.; Offringa, R.; Toes, R.E.M.; Rammensee, H.; Schild, H. T Cell Avidity Determines the Level of CTL Activation. Eur. J. Immunol. 2004, 34, 1798–1806. [Google Scholar] [CrossRef]
- La Gruta, N.L.; Turner, S.J.; Doherty, P.C. Hierarchies in Cytokine Expression Profiles for Acute and Resolving Influenza Virus-Specific CD8+ T Cell Responses: Correlation of Cytokine Profile and TCR Avidity. J. Immunol. 2004, 172, 5553–5560. [Google Scholar] [CrossRef]
- Zhong, S.; Malecek, K.; Johnson, L.A.; Yu, Z.; Vega-Saenz de Miera, E.; Darvishian, F.; McGary, K.; Huang, K.; Boyer, J.; Corse, E.; et al. T-Cell Receptor Affinity and Avidity Defines Antitumor Response and Autoimmunity in T-Cell Immunotherapy. Proc. Natl. Acad. Sci. USA 2013, 110, 6973–6978. [Google Scholar] [CrossRef]
- Ploquin, M.J.; Eksmond, U.; Kassiotis, G. B Cells and TCR Avidity Determine Distinct Functions of CD4+ T Cells in Retroviral Infection. J. Immunol. 2011, 187, 3321–3330. [Google Scholar] [CrossRef]
- Cukalac, T.; Chadderton, J.; Zeng, W.; Cullen, J.G.; Kan, W.T.; Doherty, P.C.; Jackson, D.C.; Turner, S.J.; La Gruta, N.L. The Influenza Virus-Specific CTL Immunodominance Hierarchy in Mice is Determined by the Relative Frequency of High-Avidity T Cells. J. Immunol. 2014, 192, 4061–4068. [Google Scholar] [CrossRef]
- Tian, S.; Maile, R.; Collins, E.J.; Frelinger, J.A. CD8+ T Cell Activation is Governed by TCR-Peptide/MHC Affinity, Not Dissociation Rate. J. Immunol. 2007, 179, 2952–2960. [Google Scholar] [CrossRef]
- Nauerth, M.; Weissbrich, B.; Knall, R.; Franz, T.; Dossinger, G.; Bet, J.; Paszkiewicz, P.J.; Pfeifer, L.; Bunse, M.; Uckert, W.; et al. TCR-Ligand Koff Rate Correlates with the Protective Capacity of Antigen-Specific CD8+ T Cells for Adoptive Transfer. Sci. Transl. Med. 2013, 5, 192ra87. [Google Scholar] [CrossRef]
- Sandu, I.; Cerletti, D.; Oetiker, N.; Borsa, M.; Wagen, F.; Spadafora, I.; Welten, S.P.M.; Stolz, U.; Oxenius, A.; Claassen, M. Landscape of Exhausted Virus-Specific CD8 T Cells in Chronic LCMV Infection. Cell. Rep. 2020, 32, 108078. [Google Scholar] [CrossRef]
- Yermanos, A.; Sandu, I.; Pedrioli, A.; Borsa, M.; Wagen, F.; Oetiker, N.; Welten, S.P.M.; Pallmer, K.; Reddy, S.T.; Oxenius, A. Profiling Virus-Specific Tcf1+ T Cell Repertoires during Acute and Chronic Viral Infection. Front. Immunol. 2020, 11, 986. [Google Scholar] [CrossRef]
- Kotturi, M.F.; Scott, I.; Wolfe, T.; Peters, B.; Sidney, J.; Cheroutre, H.; von Herrath, M.G.; Buchmeier, M.J.; Grey, H.; Sette, A. Naive Precursor Frequencies and MHC Binding rather than the Degree of Epitope Diversity Shape CD8+ T Cell Immunodominance. J. Immunol. 2008, 181, 2124–2133. [Google Scholar] [CrossRef]
- Turner, S.J.; Kedzierska, K.; Komodromou, H.; La Gruta, N.L.; Dunstone, M.A.; Webb, A.I.; Webby, R.; Walden, H.; Xie, W.; McCluskey, J.; et al. Lack of Prominent Peptide-Major Histocompatibility Complex Features Limits Repertoire Diversity in Virus-Specific CD8+ T Cell Populations. Nat. Immunol. 2005, 6, 382–389. [Google Scholar] [CrossRef]
- Cornberg, M.; Chen, A.T.; Wilkinson, L.A.; Brehm, M.A.; Kim, S.K.; Calcagno, C.; Ghersi, D.; Puzone, R.; Celada, F.; Welsh, R.M.; et al. Narrowed TCR Repertoire and Viral Escape as a Consequence of Heterologous Immunity. J. Clin. Investig. 2006, 116, 1443–1456. [Google Scholar] [CrossRef]
- Yager, E.J.; Ahmed, M.; Lanzer, K.; Randall, T.D.; Woodland, D.L.; Blackman, M.A. Age-Associated Decline in T Cell Repertoire Diversity Leads to Holes in the Repertoire and Impaired Immunity to Influenza Virus. J. Exp. Med. 2008, 205, 711–723. [Google Scholar] [CrossRef]
- Klein, S.; Ghersi, D.; Manns, M.P.; Prinz, I.; Cornberg, M.; Kraft, A.R.M. PD-L1 Checkpoint Inhibition Narrows the Antigen-Specific T Cell Receptor Repertoire in Chronic LCMV Infection. J. Virol. 2020, 94, e00795-20. [Google Scholar] [CrossRef]
- Chang, Y.M.; Wieland, A.; Li, Z.; Im, S.J.; McGuire, D.J.; Kissick, H.T.; Antia, R.; Ahmed, R. T Cell Receptor Diversity and Lineage Relationship between Virus-Specific CD8 T Cell Subsets during Chronic Lymphocytic Choriomeningitis Virus Infection. J. Virol. 2020, 94, e00935-20. [Google Scholar] [CrossRef]
- Wherry, E.J.; Ahmed, R. Memory CD8 T-Cell Differentiation during Viral Infection. J. Virol. 2004, 78, 5535–5545. [Google Scholar] [CrossRef]
- Zehn, D.; Wherry, E.J. Immune Memory and Exhaustion: Clinically Relevant Lessons from the LCMV Model. Adv. Exp. Med. Biol. 2015, 850, 137–152. [Google Scholar]
- Mischke, J.; Klein, S.; Seamann, A.; Prinz, I.; Selin, L.; Ghersi, D.; Cornberg, M.; Kraft, A.R.M. Cross-Reactive T Cell Response Exists in Chronic Lymphocytic Choriomeningitis Virus Infection upon Pichinde Virus Challenge. Viruses 2022, 14, 2293. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. Fastp: An Ultra-Fast all-in-One FASTQ Preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Bolotin, D.A.; Poslavsky, S.; Mitrophanov, I.; Shugay, M.; Mamedov, I.Z.; Putintseva, E.V.; Chudakov, D.M. MiXCR: Software for Comprehensive Adaptive Immunity Profiling. Nat. Methods 2015, 12, 380–381. [Google Scholar] [CrossRef]
- Warren, R.L.; Freeman, J.D.; Zeng, T.; Choe, G.; Munro, S.; Moore, R.; Webb, J.R.; Holt, R.A. Exhaustive T-Cell Repertoire Sequencing of Human Peripheral Blood Samples Reveals Signatures of Antigen Selection and a Directly Measured Repertoire Size of at Least 1 Million Clonotypes. Genome Res. 2011, 21, 790–797. [Google Scholar] [CrossRef]
- Shugay, M.; Bagaev, D.V.; Turchaninova, M.A.; Bolotin, D.A.; Britanova, O.V.; Putintseva, E.V.; Pogorelyy, M.V.; Nazarov, V.I.; Zvyagin, I.V.; Kirgizova, V.I.; et al. VDJtools: Unifying Post-Analysis of T Cell Receptor Repertoires. PLoS Comput. Biol. 2015, 11, e1004503. [Google Scholar] [CrossRef]
- Welsh, R.M.; Seedhom, M.O. Lymphocytic Choriomeningitis Virus (LCMV): Propagation, Quantitation, and Storage. Curr. Protoc. Microbiol. 2008, 8, 15A.1.1–15A.1.11. [Google Scholar] [CrossRef]
- Bunztman, A.; Vincent, B.G.; Krovi, H.; Steele, S.; Frelinger, J.A. The LCMV gp33-Specific Memory T Cell Repertoire Narrows with Age. Immun. Ageing 2012, 9, 17. [Google Scholar] [CrossRef]
- Schurich, A.; Pallett, L.J.; Lubowiecki, M.; Singh, H.D.; Gill, U.S.; Kennedy, P.T.; Nastouli, E.; Tanwar, S.; Rosenberg, W.; Maini, M.K. The Third Signal Cytokine IL-12 Rescues the Anti-Viral Function of Exhausted HBV-Specific CD8 T Cells. PLoS Pathog. 2013, 9, e1003208. [Google Scholar] [CrossRef]
- Grusdat, M.; McIlwain, D.R.; Xu, H.C.; Pozdeev, V.I.; Knievel, J.; Crome, S.Q.; Robert-Tissot, C.; Dress, R.J.; Pandyra, A.A.; Speiser, D.E.; et al. IRF4 and BATF are Critical for CD8+ T-Cell Function Following Infection with LCMV. Cell Death Differ. 2014, 21, 1050–1060. [Google Scholar] [CrossRef]
- Schuch, A.; Alizei, E.S.; Heim, K.; Wieland, D.; Kiraithe, M.M.; Kemming, J.; Llewellyn-Lacey, S.; Sogukpinar, Ö.; Ni, Y.; Urban, S.; et al. Phenotypic and Functional Differences of HBV Core-Specific Versus HBV Polymerase-Specific CD8+ T Cells in Chronically HBV-Infected Patients with Low Viral Load. Gut 2019, 68, 905–915. [Google Scholar] [CrossRef]
- Egui, A.; Lopez, M.C.; Gomez, I.; Simon, M.; Segovia, M.; Thomas, M.C. Differential Phenotypic and Functional Profile of Epitope-Specific Cytotoxic CD8+ T Cells in Benznidazole-Treated Chronic Asymptomatic Chagas Disease Patients. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165629. [Google Scholar] [CrossRef]
- Aliabadi, E.; Urbanek-Quaing, M.; Maasoumy, B.; Bremer, B.; Grasshoff, M.; Li, Y.; Niehaus, C.E.; Wedemeyer, H.; Kraft, A.R.M.; Cornberg, M. Impact of HBsAg and HBcrAg Levels on Phenotype and Function of HBV-Specific T Cells in Patients with Chronic Hepatitis B Virus Infection. Gut 2022, 71, 2300–2312. [Google Scholar] [CrossRef]
- Kuhn, R.; Sandu, I.; Agrafiotis, A.; Hong, K.; Shlesinger, D.; Neimeier, D.; Merkler, D.; Oxenius, A.; Reddy, S.T.; Yermanos, A. Clonally Expanded Virus-Specific CD8 T Cells Acquire Diverse Transcriptional Phenotypes during Acute, Chronic, and Latent Infections. Front. Immunol. 2022, 13, 782441. [Google Scholar] [CrossRef] [PubMed]
- Fuller, M.J.; Khanolkar, A.; Tebo, A.E.; Zajac, A.J. Maintenance, Loss, and Resurgence of T Cell Responses during Acute, Protracted, and Chronic Viral Infections. J. Immunol. 2004, 172, 4204–4214. [Google Scholar] [CrossRef]
- Tikochinski, Y.; Elias, D.; Steeg, C.; Marcus, H.; Kantorowitz, M.; Reshef, T.; Ablamunits, V.; Cohen, I.R.; Friedmann, A. A Shared TCR CDR3 Sequence in NOD Mouse Autoimmune Diabetes. Int. Immunol. 1999, 11, 951–956. [Google Scholar] [CrossRef] [PubMed]
- Madi, A.; Shifrut, E.; Reich-Zeliger, S.; Gal, H.; Best, K.; Ndifon, W.; Chain, B.; Cohen, I.R.; Friedman, N. T-Cell Receptor Repertoires Share a Restricted Set of Public and Abundant CDR3 Sequences that are Associated with Self-Related Immunity. Genome Res. 2014, 24, 1603–1612. [Google Scholar] [CrossRef] [PubMed]
- Wolpert, E.Z.; Grufman, P.; Sandberg, J.K.; Tegnesjo, A.; Karre, K. Immunodominance in the CTL Response Against Minor Histocompatibility Antigens: Interference between Responding T Cells, rather than with Presentation of Epitopes. J. Immunol. 1998, 161, 4499–4505. [Google Scholar] [CrossRef] [PubMed]
- Sandberg, J.K.; Grufman, P.; Wolpert, E.Z.; Franksson, L.; Chambers, B.J.; Karre, K. Superdominance among Immunodominant H-2Kb-Restricted Epitopes and Reversal by Dendritic Cell-Mediated Antigen Delivery. J. Immunol. 1998, 160, 3163–3169. [Google Scholar] [CrossRef] [PubMed]
- Butz, E.A.; Bevan, M.J. Massive Expansion of Antigen-Specific CD8+ T Cells during an Acute Virus Infection. Immunity 1998, 8, 167–175. [Google Scholar] [CrossRef]
- Smith, A.L.; Wikstrom, M.E.; Fazekas de St Groth, B. Visualizing T Cell Competition for Peptide/MHC Complexes: A Specific Mechanism to Minimize the Effect of Precursor Frequency. Immunity 2000, 13, 783–794. [Google Scholar] [CrossRef]
- Gane, E.; Verdon, D.J.; Brooks, A.E.; Gaggar, A.; Nguyen, A.H.; Subramanian, G.M.; Schwabe, C.; Dunbar, P.R. Anti-PD-1 Blockade with Nivolumab with and without Therapeutic Vaccination for Virally Suppressed Chronic Hepatitis B: A Pilot Study. J. Hepatol. 2019, 71, 900–907. [Google Scholar] [CrossRef]
- Li, H.; van der Merwe, P.A.; Sivakumar, S. Biomarkers of Response to PD-1 Pathway Blockade. Br. J. Cancer 2022, 126, 1663–1675. [Google Scholar] [CrossRef]
- Abdel-Hakeem, M.S.; Manne, S.; Beltra, J.; Stelekati, E.; Chen, Z.; Nzingha, K.; Ali, M.; Johnson, J.L.; Giles, J.R.; Mathew, D.; et al. Epigenetic Scarring of Exhausted T Cells Hinders Memory Differentiation upon Eliminating Chronic Antigenic Stimulation. Nat. Immunol. 2021, 22, 1008–1019. [Google Scholar] [CrossRef] [PubMed]
- Dahling, S.; Mansilla, A.M.; Knopper, K.; Grafen, A.; Utzschneider, D.T.; Ugur, M.; Whitney, P.G.; Bachem, A.; Arampatzi, P.; Imdahl, F.; et al. Type 1 Conventional Dendritic Cells Maintain and Guide the Differentiation of Precursors of Exhausted T Cells in Distinct Cellular Niches. Immunity 2022, 55, 656–670.e8. [Google Scholar] [CrossRef]
- Hashimoto, M.; Araki, K.; Cardenas, M.A.; Li, P.; Jadhav, R.R.; Kissick, H.T.; Hudson, W.H.; McGuire, D.J.; Obeng, R.C.; Wieland, A.; et al. PD-1 Combination Therapy with IL-2 Modifies CD8+ T Cell Exhaustion Program. Nature 2022, 610, 173–181. [Google Scholar] [CrossRef]
- Madden, D.R.; Garboczi, D.N.; Wiley, D.C. The Antigenic Identity of Peptide-MHC Complexes: A Comparison of the Conformations of Five Viral Peptides Presented by HLA-A2. Cell 1993, 75, 693–708. [Google Scholar] [CrossRef]
- Antunes, D.A.; Vieira, G.F.; Rigo, M.M.; Cibulski, S.P.; Sinigaglia, M.; Chies, J.A.B. Structural Allele-Specific Patterns Adopted by Epitopes in the MHC-I Cleft and Reconstruction of MHC:Peptide Complexes to Cross-Reactivity Assessment. PLoS ONE 2010, 5, e10353. [Google Scholar] [CrossRef] [PubMed]
- Achour, A.; Michaelsson, J.; Harris, R.A.; Odeberg, J.; Grufman, P.; Sandberg, J.K.; Levitsky, V.; Karre, K.; Sandalova, T.; Schneider, G. A Structural Basis for LCMV Immune Evasion: Subversion of H-2D(B) and H-2K(B) Presentation of gp33 Revealed by Comparative Crystal Structure Analyses. Immunity 2002, 17, 757–768. [Google Scholar] [CrossRef]
- Cerletti, D.; Sandu, I.; Gupta, R.; Oxenius, A.; Claassen, M. Fate Trajectories of CD8+ T Cells in Chronic LCMV Infection. bioRxiv 2020. [Google Scholar] [CrossRef]
- Brandle, D.; Brduscha-Riem, K.; Hayday, A.C.; Owen, M.J.; Hengartner, H.; Pircher, H. T Cell Development and Repertoire of Mice Expressing a Single T Cell Receptor Alpha Chain. Eur. J. Immunol. 1995, 25, 2650–2655. [Google Scholar] [CrossRef]
- Yang, L.; Jama, B.; Wang, H.; Labarta-Bajo, L.; Zuniga, E.I.; Morris, G.P. TCRalpha Reporter Mice Reveal Contribution of Dual TCRalpha Expression to T Cell Repertoire and Function. Proc. Natl. Acad. Sci. USA 2020, 117, 32574–32583. [Google Scholar] [CrossRef]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Klein, S.; Mischke, J.; Beruldsen, F.; Prinz, I.; Antunes, D.A.; Cornberg, M.; Kraft, A.R.M. Individual Epitope-Specific CD8+ T Cell Immune Responses Are Shaped Differently during Chronic Viral Infection. Pathogens 2023, 12, 716. https://doi.org/10.3390/pathogens12050716
Klein S, Mischke J, Beruldsen F, Prinz I, Antunes DA, Cornberg M, Kraft ARM. Individual Epitope-Specific CD8+ T Cell Immune Responses Are Shaped Differently during Chronic Viral Infection. Pathogens. 2023; 12(5):716. https://doi.org/10.3390/pathogens12050716
Chicago/Turabian StyleKlein, Sebastian, Jasmin Mischke, Finn Beruldsen, Immo Prinz, Dinler A. Antunes, Markus Cornberg, and Anke R. M. Kraft. 2023. "Individual Epitope-Specific CD8+ T Cell Immune Responses Are Shaped Differently during Chronic Viral Infection" Pathogens 12, no. 5: 716. https://doi.org/10.3390/pathogens12050716
APA StyleKlein, S., Mischke, J., Beruldsen, F., Prinz, I., Antunes, D. A., Cornberg, M., & Kraft, A. R. M. (2023). Individual Epitope-Specific CD8+ T Cell Immune Responses Are Shaped Differently during Chronic Viral Infection. Pathogens, 12(5), 716. https://doi.org/10.3390/pathogens12050716