
Evaluation of Next-Generation Sequencing Applied to Cryptosporidium parvum and Cryptosporidium hominis Epidemiological Study

 , , , , and
, , , , and
Abstract
:1. Introduction
2. Results
2.1. NGS Protocol Gives Reliable Subtyping Results and Allows Detecting DNA Mixtures of Cryptosporidium sp.
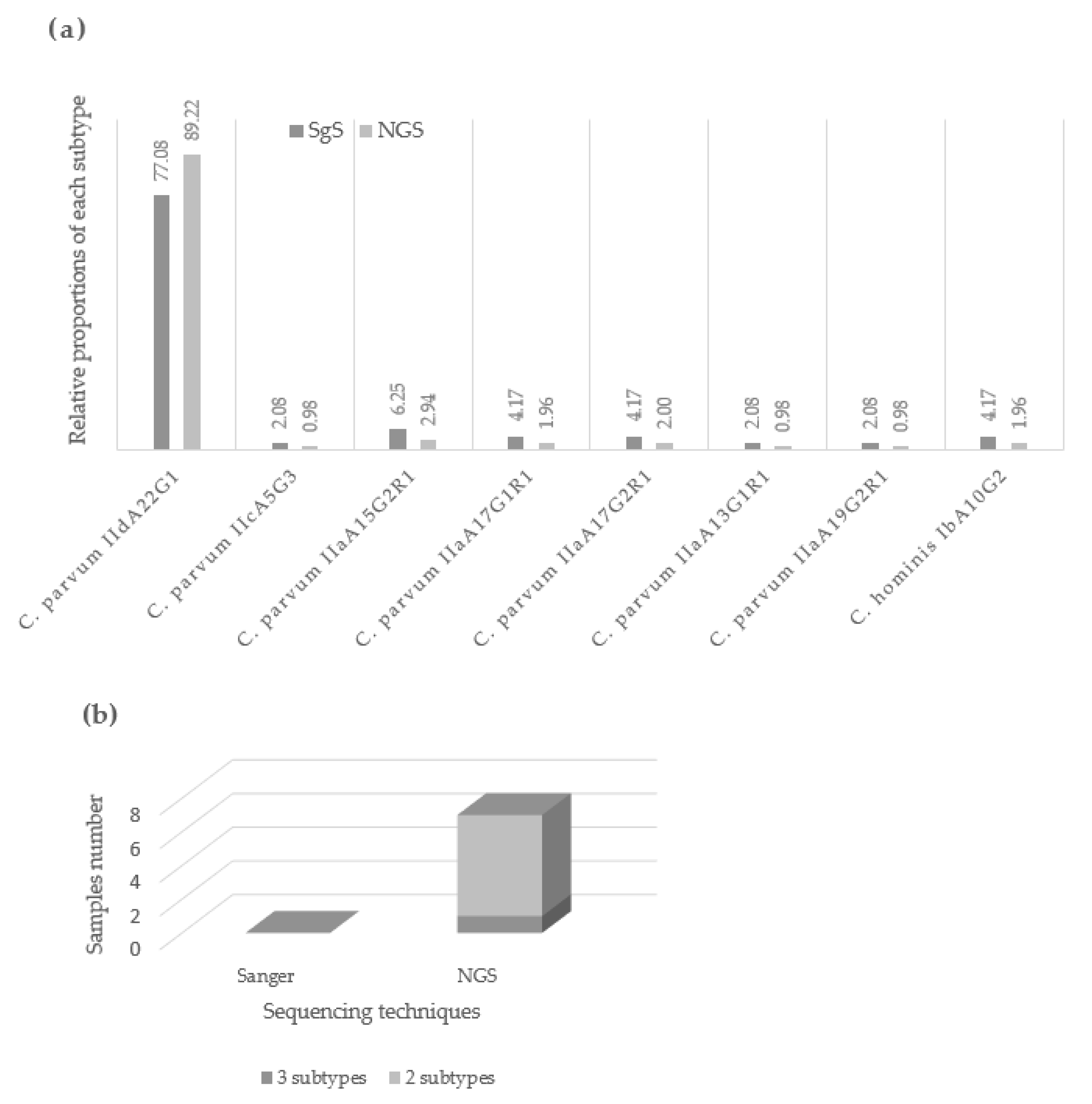
2.2. Application of NGS for Cryptosporidium sp. Subtyping Outbreaks Isolates
3. Discussion
4. Materials and Methods
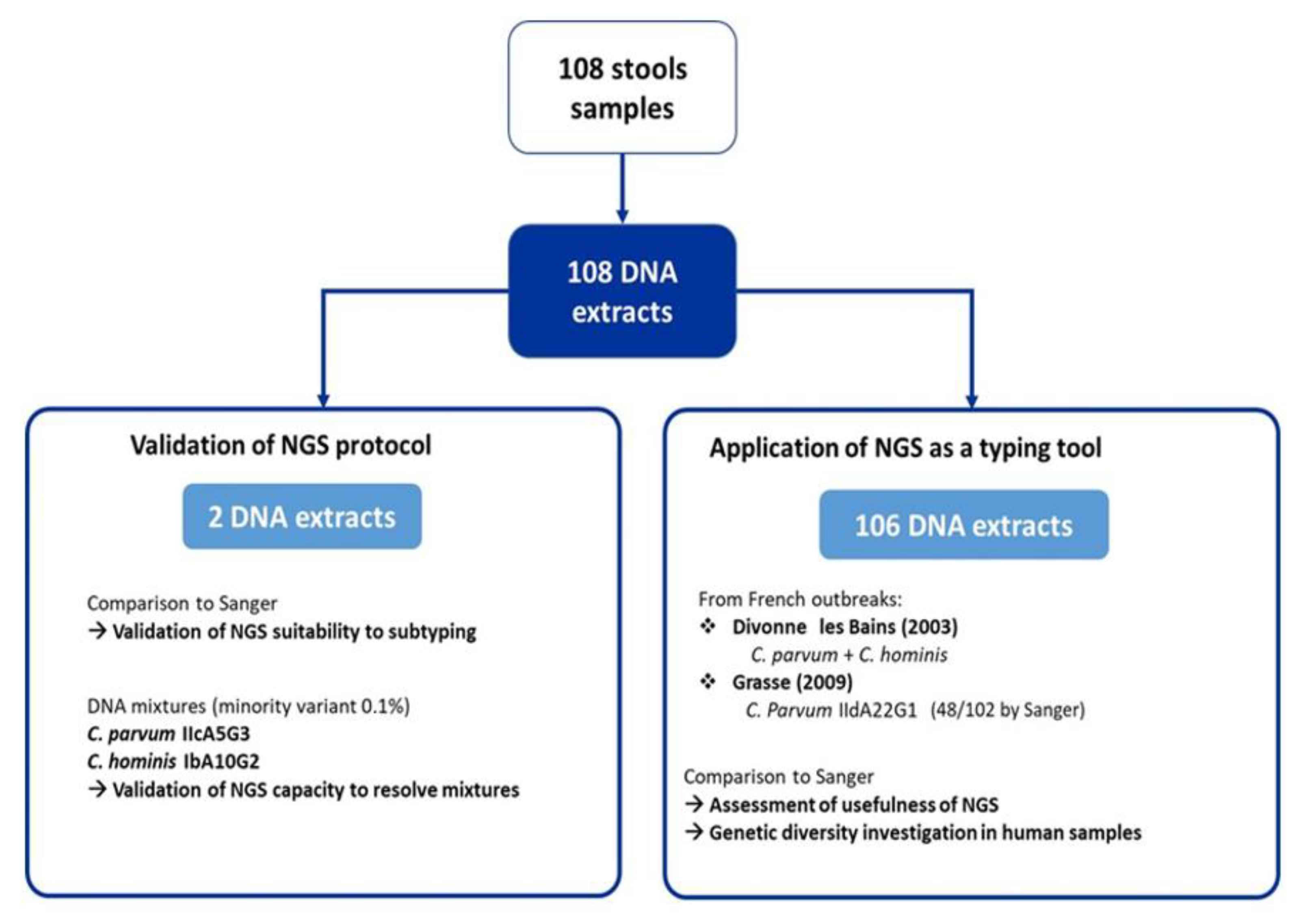
4.1. Sample Collection
4.2. Next-Generation Sequencing (NGS) Protocol
4.3. Bioinformatics Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Leitch, G.J.; He, Q. Cryptosporidiosis-an overview. J. Biomed. Res. 2012, 25, 1–16. [Google Scholar] [CrossRef]
- Chalmers, R.M.; Davies, A.P. Minireview: Clinical cryptosporidiosis. Exp. Parasitol. 2010, 124, 138–146. [Google Scholar] [CrossRef] [PubMed]
- Chalmers, R.M.; Robinson, G.; Elwin, K.; Elson, R. Analysis of the Cryptosporidium spp. and gp60 subtypes linked to human outbreaks of cryptosporidiosis in England and Wales, 2009 to 2017. Parasites Vectors 2019, 12, 95. [Google Scholar] [CrossRef] [PubMed]
- Mac Kenzie, W.R.; Hoxie, N.J.; Proctor, M.E.; Gradus, M.S.; Blair, K.A.; Peterson, D.E.; Kazmierczak, J.J.; Addiss, D.G.; Fox, K.R.; Rose, J.B. A massive outbreak in Milwaukee of cryptosporidium infection transmitted through the public water supply. N. Engl. J. Med. 1994, 331, 161–167. [Google Scholar] [CrossRef]
- Costa, D.; Razakandrainibe, R.; Valot, S.; Vannier, M.; Sautour, M.; Basmaciyan, L.; Gargala, G.; Viller, V.; Lemeteil, D.; Ballet, J.-J.; et al. Epidemiology of Cryptosporidiosis in France from 2017 to 2019. Microorganisms 2020, 8, 1358. [Google Scholar] [CrossRef]
- Mosnier, E.; Martin, N.; Razakandrainibe, R.; Dalle, F.; Roux, G.; Buteux, A.; Favennec, L.; Brousse, P.; Guarmit, B.; Blanchet, D.; et al. Cryptosporidiosis Outbreak in Immunocompetent Children from a Remote Area of French Guiana. Am. J. Trop. Med. Hyg. 2018, 98, 1727–1732. [Google Scholar] [CrossRef]
- Dalle, F.; Roz, P.; Dautin, G.; Di-Palma, M.; Kohli, E.; Sire-Bidault, C.; Fleischmann, M.G.; Gallay, A.; Carbonel, S.; Bon, F.; et al. Molecular characterization of isolates of waterborne Cryptosporidium spp. collected during an outbreak of gastroenteritis in South Burgundy, France. J. Clin. Microbiol. 2003, 41, 2690–2693. [Google Scholar] [CrossRef]
- Santé Publique France. Épidémie de cryptosporidiose dans un collège de l’ouest de la France, novembre 2017. 2018. Available online: http://beh.santepubliquefrance.fr/beh/2019/16/pdf/2019_16_2.pdf (accessed on 1 July 2022).
- ANOFEL Cryptosporidium National Network. Laboratory-Based Surveillance for Cryptosporidium in France, 2006–2009. Eurosurveillance 2010, 15, 19642. [Google Scholar]
- Chaud, P.; Ramalli, L.; Raibaut, J.; Ortmans, C.; Joubert, S.; Chereau, F.; Dalle, F.; Valot, S.; Costa, D.; François, A. Épidémie de cryptosporidiose d’origine hydrique dans les Alpes Maritimes–novembre 2019. Médecine Mal. Infect. 2020, 50, S167. [Google Scholar] [CrossRef]
- Xiao, L. Molecular epidemiology of cryptosporidiosis: An update. Exp. Parasitol. 2010, 124, 80–89. [Google Scholar] [CrossRef]
- Khan, A.; Shaik, J.S.; Grigg, M.E. Genomics and molecular epidemiology of Cryptosporidium species. Acta Trop. 2018, 184, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Sulaiman, I.M.; Hira, P.R.; Zhou, L.; Al-Ali, F.M.; Al-Shelahi, F.A.; Shweiki, H.M.; Iqbal, J.; Khalid, N.; Xiao, L. Unique endemicity of cryptosporidiosis in children in Kuwait. J. Clin. Microbiol. 2005, 43, 2805–2809. [Google Scholar] [CrossRef]
- Li, N.; Xiao, L.; Cama, V.A.; Ortega, Y.; Gilman, R.H.; Guo, M.; Feng, Y. Genetic recombination and Cryptosporidium hominis virulent subtype IbA10G2. Emerg. Infect. Dis. 2013, 19, 1573–1582. [Google Scholar] [CrossRef] [PubMed]
- Zahedi, A.; Gofton, A.W.; Jian, F.; Paparini, A.; Oskam, C.; Ball, A.; Robertson, I.; Ryan, U. Next Generation Sequencing uncovers within-host differences in the genetic diversity of Cryptosporidium gp60 subtypes. Int. J. Parasitol. 2017, 47, 601–607. [Google Scholar] [CrossRef] [PubMed]
- Paparini, A.; Yang, R.; Chen, L.; Tong, K.; Gibson-Kueh, S.; Lymbery, A.; Ryan, U.M. Cryptosporidium in fish: Alternative sequencing approaches and analyses at multiple loci to resolve mixed infections. Parasitology 2017, 144, 1811–1820. [Google Scholar] [CrossRef]
- Rotondo, J.C.; Martini, F.; Maritati, M.; Caselli, E.; Gallenga, C.E.; Guarino, M.; De Giorgio, R.; Mazziotta, C.; Tramarin, M.L.; Badiale, G.; et al. Advanced Molecular and Immunological Diagnostic Methods to Detect SARS-CoV-2 Infection. Microorganisms 2022, 10, 1193. [Google Scholar] [CrossRef]
- Grinberg, A.; Widmer, G. Cryptosporidium within-host genetic diversity: Systematic bibliographical search and narrative overview. Int. J. Parasitol. 2016, 46, 465–471. [Google Scholar] [CrossRef]
- Zahedi, A.; Monis, P.; Gofton, A.W.; Oskam, C.L.; Ball, A.; Bath, A.; Bartkow, M.; Robertson, I.; Ryan, U. Cryptosporidium species and subtypes in animals inhabiting drinking water catchments in three states across Australia. Water Res. 2018, 134, 327–340. [Google Scholar] [CrossRef]
- Mphephu, M.G.; Ekwanzala, M.D.; Momba, M.N.B. Cryptosporidium species and subtypes in river water and riverbed sediment using next-generation sequencing. Int. J. Parasitol. 2021, 51, 339–351. [Google Scholar] [CrossRef]
- Paparini, A.; Gofton, A.; Yang, R.; White, N.; Bunce, M.; Ryan, U.M. Comparison of Sanger and next generation sequencing performance for genotyping Cryptosporidium isolates at the 18S rRNA and actin loci. Exp. Parasitol. 2015, 151–152, 21–27. [Google Scholar] [CrossRef]
- Davis, N.M.; Proctor, D.M.; Holmes, S.P.; Relman, D.A.; Callahan, B.J. Simple statistical identification and removal of contaminant sequences in marker-gene and metagenomics data. Microbiome 2018, 6, 226. [Google Scholar] [CrossRef]
- SPF Epidémie de gastro-entérites liée à la pollution du réseau de distribution d’eau potable de la commune de Divonne-les-Bains, Ain (01). Août-septembre 2003. Available online: https://www.santepubliquefrance.fr/determinants-de-sante/pollution-et-sante/eau/documents/rapport-synthese/epidemie-de-gastro-enterites-liee-a-la-pollution-du-reseau-de-distribution-d-eau-potable-de-la-commune-de-divonne-les-bains-ain-01-.-aout-septemb (accessed on 1 July 2022).
- Braima, K.; Zahedi, A.; Egan, S.; Austen, J.; Xiao, L.; Feng, Y.; Witham, B.; Pingault, N.; Perera, S.; Oskam, C.; et al. Molecular analysis of cryptosporidiosis cases in Western Australia in 2019 and 2020 supports the occurrence of two swimming pool associated outbreaks and reveals the emergence of a rare C. hominis IbA12G3 subtype. Infect. Genet. Evol. 2021, 92, 104859. [Google Scholar] [CrossRef]
- Ramo, A.; Quílez, J.; Del Cacho, E.; Sánchez-Acedo, C. Optimization of a fragment size analysis tool for identification of Cryptosporidium species and Gp60 alleles infecting domestic ruminants. Vet. Parasitol. 2014, 205, 466–471. [Google Scholar] [CrossRef] [PubMed]
- Costa, D.; Soulieux, L.; Razakandrainibe, R.; Basmaciyan, L.; Gargala, G.; Valot, S.; Dalle, F.; Favennec, L. Comparative Performance of Eight PCR Methods to Detect Cryptosporidium Species. Pathogens 2021, 10, 647. [Google Scholar] [CrossRef] [PubMed]
- DeMone, C.; Hwang, M.-H.; Feng, Z.; McClure, J.T.; Greenwood, S.J.; Fung, R.; Kim, M.; Weese, J.S.; Shapiro, K. Application of next generation sequencing for detection of protozoan pathogens in shellfish. Food Waterborne Parasitol. 2020, 21, e00096. [Google Scholar] [CrossRef]
- Stoler, N.; Nekrutenko, A. Sequencing error profiles of Illumina sequencing instruments. NAR Genom. Bioinform. 2021, 3, lqab019. [Google Scholar] [CrossRef] [PubMed]
- Rojas-Lopez, L.; Elwin, K.; Chalmers, R.M.; Enemark, H.L.; Beser, J.; Troell, K. Development of a gp60-subtyping method for Cryptosporidium felis. Parasite Vectors 2020, 13, 39. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.; Roellig, D.M.; Guo, Y.; Li, N.; Feng, Y.; Xiao, L. Development of a Subtyping Tool for Zoonotic Pathogen Cryptosporidium canis. J. Clin. Microbiol. 2021, 59, e02474-20. [Google Scholar] [CrossRef]
- Valeix, N.; Costa, D.; Basmaciyan, L.; Valot, S.; Vincent, A.; Razakandrainibe, R.; Robert-Gangneux, F.; Nourrisson, C.; Pereira, B.; Fréalle, E.; et al. Multicenter Comparative Study of Six Cryptosporidium parvum DNA Extraction Protocols Including Mechanical Pretreatment from Stool Samples. Microorganisms 2020, 8, 1450. [Google Scholar] [CrossRef]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| C. parvum_IIcA5G3 | C. hominis_IbA10G2 | C. parvum_IIaA18G1R1 | C. parvum_IIdA22G1 | Total (Not Assigned) | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Samples | Expected Proportion of C. parvum_IIcA5G3/C. hominis_IbA10G2 | Number of Sequences | % | Number of Sequences | % | Number of Sequences | % | Number of Sequences | % | |
| 1572 | 100%/0% | 240,932 | 99.85 | 93 | 0.04 | 19 | 0.01 | 97 | 0.04 | 241,302 (161) |
| 2055 | 0%/100% | 17 | 0.01 | 200,160 | 99.94 | 13 | 0.01 | 46 | 0.02 | 200,284 (48) |
| mix A | 50%/50% | 117,150 | 74.07 | 40,930 | 25.89 | 0 | 0 | 33 | 0.02 | 158,113 (0) |
| mix B | 90%/10% | 178,170 | 94.61 | 10,040 | 5.33 | 27 | 0.01 | 49 | 0.03 | 188,317 (31) |
| mix C | 99%/1% | 155,550 | 99.44 | 777 | 0.50 | 23 | 0.01 | 63 | 0.04 | 156,431 (18) |
| mix D | 99.9%/0.1% | 163,383 | 99.91 | 75 | 0.05 | 22 | 0.01 | 27 | 0.02 | 163,534 (27) |
| mix E | 10%/90% | 81,076 | 31.19 | 178,697 | 68.75 | 0 | 0 | 108 | 0.04 | 259,924 (43) |
| mix F | 1%/99% | 6280 | 2.31 | 265,724 | 97.63 | 33 | 0.01 | 105 | 0.04 | 272,165 (23) |
| mix G | 0.1%/99.9% | 1140 | 0.46 | 247,914 | 99.48 | 0 | 0 | 133 | 0.05 | 249,215 (28) |
| CTRL neg | 0%/0% | 94 | 7.76 | 131 | 10.82 | 2 | 0.16 | 983 | 81.2 | 1210 (0) |
| C. parvum_IIcA5G3 | C. hominis_IbA10G2 | C. parvum_IiaA18G1R1 | C. parvum_IidA22G1 | Total (Not Assigned) | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Samples | Expected Results | Number of Sequences | % | Number of Sequences | % | Number of Sequences | % | Number of Sequences | % | |
| D23 | C. hominis + C. parvum | 127 | 48.46 | 58 | 22.14 | 44 | 16.79 | 33 | 12.6 | 262 (0) |
| D24 | C. hominis | 82 | 0.04 | 206,058 | 99.88 | 59 | 0.03 | 43 | 0.02 | 206,309 (67) |
| D26 | C. hominis + C. parvum | 60,215 | 63.04 | 35,172 | 36.82 | 31 | 0.03 | 16 | 0.02 | 95,512 (78) |
| D45 | C. hominis + C. parvum | 296,688 | 99.91 | 84 | 0.03 | 0 | 0 | 101 | 0.03 | 296,954 (81) |
| CTRL neg | Negative | 94 | 7.76 | 131 | 10.82 | 2 | 0.16 | 983 | 81.2 | 1210 (0) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bailly, E.; Valot, S.; Vincent, A.; Duffourd, Y.; Grangier, N.; Chevarin, M.; Costa, D.; Razakandrainibe, R.; Favennec, L.; Basmaciyan, L.; et al. Evaluation of Next-Generation Sequencing Applied to Cryptosporidium parvum and Cryptosporidium hominis Epidemiological Study. Pathogens 2022, 11, 938. https://doi.org/10.3390/pathogens11080938
Bailly E, Valot S, Vincent A, Duffourd Y, Grangier N, Chevarin M, Costa D, Razakandrainibe R, Favennec L, Basmaciyan L, et al. Evaluation of Next-Generation Sequencing Applied to Cryptosporidium parvum and Cryptosporidium hominis Epidemiological Study. Pathogens. 2022; 11(8):938. https://doi.org/10.3390/pathogens11080938
Chicago/Turabian StyleBailly, Eloïse, Stéphane Valot, Anne Vincent, Yannis Duffourd, Nadège Grangier, Martin Chevarin, Damien Costa, Romy Razakandrainibe, Loïc Favennec, Louise Basmaciyan, and et al. 2022. "Evaluation of Next-Generation Sequencing Applied to Cryptosporidium parvum and Cryptosporidium hominis Epidemiological Study" Pathogens 11, no. 8: 938. https://doi.org/10.3390/pathogens11080938
APA StyleBailly, E., Valot, S., Vincent, A., Duffourd, Y., Grangier, N., Chevarin, M., Costa, D., Razakandrainibe, R., Favennec, L., Basmaciyan, L., & Dalle, F. (2022). Evaluation of Next-Generation Sequencing Applied to Cryptosporidium parvum and Cryptosporidium hominis Epidemiological Study. Pathogens, 11(8), 938. https://doi.org/10.3390/pathogens11080938