
Application of CRISPR-Cas9 Gene Editing for HIV Host Factor Discovery and Validation


Abstract
1. Introduction
2. Genetic Screens and the Identification of HIV-1 Host Factors
2.1. RNAi HIV-1 Host Factor Screens
2.2. CRISPR-Cas9 Gene Editing Technologies
2.3. CRISPR-Cas9 Pooled Screens for Identifying HIV Host Factors
2.4. CRISPR Screens Exploring HIV-1 Latency
2.5. Primary Cell CRISPR-Cas9 Screens
2.6. Limitations of CRISPR-Cas9 Screens
2.7. Future Directions for CRISPR-Cas9 Host Factor Screens
3. Application of CRISPR-Cas9 Gene Editing in HIV-1 Research and Therapy
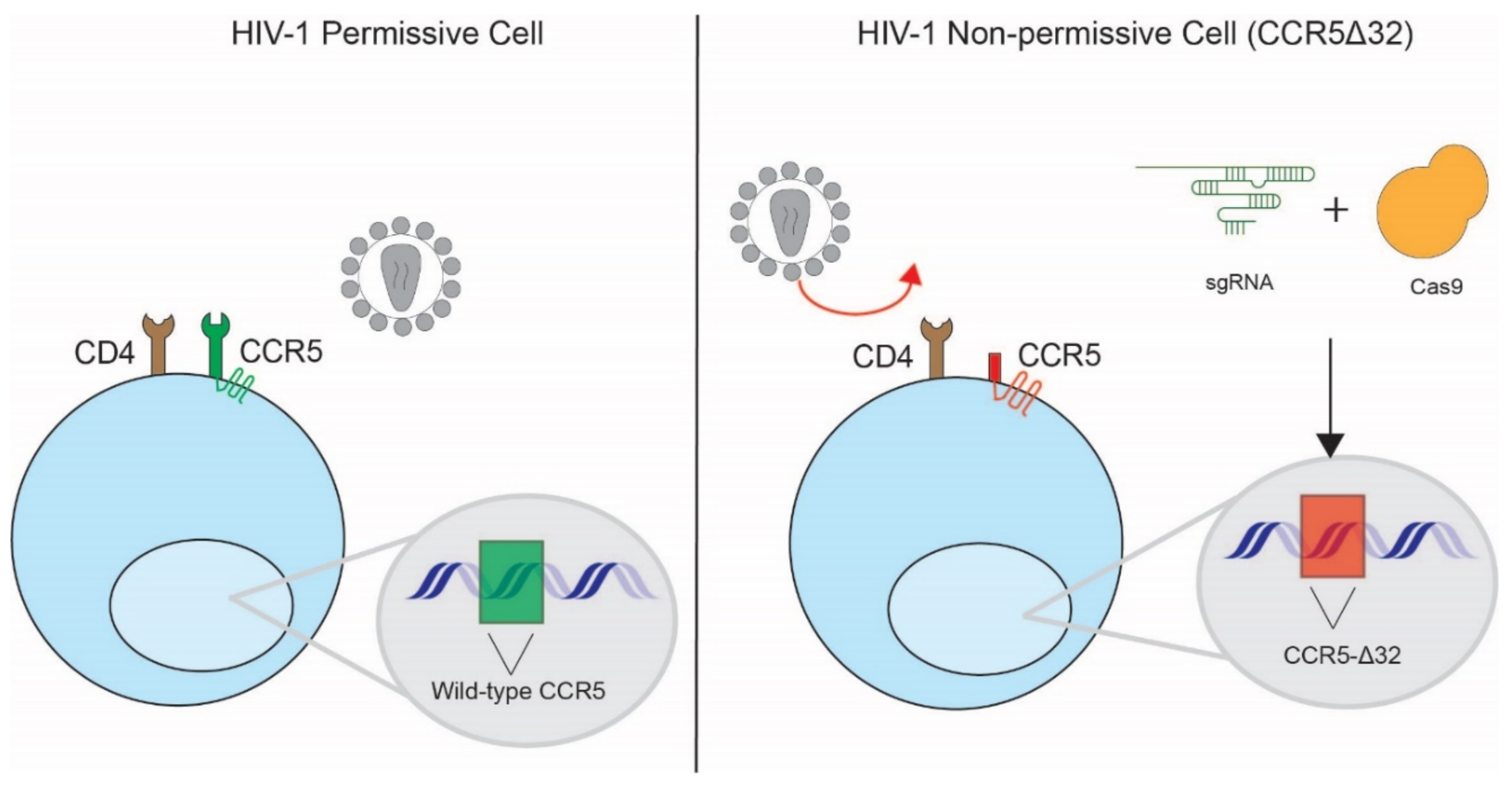
3.1. Disruption of HIV-1 Co-Receptors
3.2. Modification of HIV-1 Dependency Factors
3.3. Modification of Host Restriction Factors
3.4. Modification of Host Immune Factors
3.5. HIV-1 Latency
3.6. Considerations and Future Directions of CRISPR Therapies
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Goff, S.P. Host factors exploited by retroviruses. Nat. Rev. Microbiol. 2007, 5, 253–263. [Google Scholar] [CrossRef] [PubMed]
- Friedrich, B.M.; Dziuba, N.; Li, G.; Endsley, M.A.; Murray, J.L.; Ferguson, M.R. Host factors mediating HIV-1 replication. Virus Res. 2011, 161, 101–114. [Google Scholar] [CrossRef] [PubMed]
- Malim, M.H.; Bieniasz, P.D. HIV Restriction Factors and Mechanisms of Evasion. Cold Spring Harb. Perspect. Med. 2012, 2, a006940. [Google Scholar] [CrossRef] [PubMed]
- Santa-Marta, M.; de Brito, P.M.; Godinho-Santos, A.; Goncalves, J. Host Factors and HIV-1 Replication: Clinical Evidence and Potential Therapeutic Approaches. Front. Immunol. 2013, 4, 343. [Google Scholar] [CrossRef]
- Wood, A.; Armour, D. The discovery of the CCR5 receptor antagonist, UK-427,857, a new agent for the treatment of HIV infection and AIDS. Prog. Med. Chem. 2005, 43, 239–271. [Google Scholar] [CrossRef]
- Dorr, P.; Westby, M.; Dobbs, S.; Griffin, P.; Irvine, B.; Macartney, M.; Mori, J.; Rickett, G.; Smith-Burchnell, C.; Napier, C.; et al. Maraviroc (UK-427,857), a potent, orally bioavailable, and selective small-molecule inhibitor of chemokine receptor CCR5 with broad-spectrum anti-human immunodeficiency virus type 1 activity. Antimicrob. Agents Chemother. 2005, 49, 4721–4732. [Google Scholar] [CrossRef]
- Dufour, C.; Gantner, P.; Fromentin, R.; Chomont, N. The multifaceted nature of HIV latency. J. Clin. Investig. 2020, 130, 3381–3390. [Google Scholar] [CrossRef]
- Ruelas, D.S.; Greene, W.C. An integrated overview of HIV-1 latency. Cell 2013, 155, 519–529. [Google Scholar] [CrossRef]
- Hsu, P.D.; Lander, E.S.; Zhang, F. Development and applications of CRISPR-Cas9 for genome engineering. Cell 2014, 157, 1262–1278. [Google Scholar] [CrossRef]
- Ran, F.A.; Hsu, P.D.; Wright, J.; Agarwala, V.; Scott, D.A.; Zhang, F. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 2013, 8, 2281–2308. [Google Scholar] [CrossRef]
- Hsu, T.H.; Spindler, K.R. Identifying host factors that regulate viral infection. PLoS Pathog. 2012, 8, e1002772. [Google Scholar] [CrossRef]
- Puschnik, A.S.; Majzoub, K.; Ooi, Y.S.; Carette, J.E. A CRISPR toolbox to study virus-host interactions. Nat. Rev. Microbiol. 2017, 15, 351–364. [Google Scholar] [CrossRef]
- Echeverri, C.J.; Perrimon, N. High-throughput RNAi screening in cultured cells: A user’s guide. Nat. Rev. Genet. 2006, 7, 373–384. [Google Scholar] [CrossRef]
- Bock, C.; Datlinger, P.; Chardon, F.; Coelho, M.A.; Dong, M.B.; Lawson, K.A.; Lu, T.; Maroc, L.; Norman, T.M.; Song, B.; et al. High-content CRISPR screening. Nat. Rev. Methods Primers 2022, 2, 8. [Google Scholar] [CrossRef]
- Pache, L.; König, R.; Chanda, S.K. Identifying HIV-1 host cell factors by genome-scale RNAi screening. Methods 2011, 53, 3–12. [Google Scholar] [CrossRef]
- Shalem, O.; Sanjana, N.E.; Zhang, F. High-throughput functional genomics using CRISPR-Cas9. Nat. Rev. Genet. 2015, 16, 299–311. [Google Scholar] [CrossRef]
- Schuster, A.; Erasimus, H.; Fritah, S.; Nazarov, P.V.; van Dyck, E.; Niclou, S.P.; Golebiewska, A. RNAi/CRISPR Screens: From a Pool to a Valid Hit. Trends Biotechnol. 2019, 37, 38–55. [Google Scholar] [CrossRef]
- Yeung, M.L.; Houzet, L.; Yedavalli, V.S.; Jeang, K.T. A genome-wide short hairpin RNA screening of jurkat T-cells for human proteins contributing to productive HIV-1 replication. J. Biol. Chem. 2009, 284, 19463–19473. [Google Scholar] [CrossRef]
- Brass, A.L.; Dykxhoorn, D.M.; Benita, Y.; Yan, N.; Engelman, A.; Xavier, R.J.; Lieberman, J.; Elledge, S.J. Identification of host proteins required for HIV infection through a functional genomic screen. Science 2008, 319, 921–926. [Google Scholar] [CrossRef]
- König, R.; Zhou, Y.; Elleder, D.; Diamond, T.L.; Bonamy, G.M.; Irelan, J.T.; Chiang, C.Y.; Tu, B.P.; De Jesus, P.D.; Lilley, C.E.; et al. Global analysis of host-pathogen interactions that regulate early-stage HIV-1 replication. Cell 2008, 135, 49–60. [Google Scholar] [CrossRef]
- Zhou, H.; Xu, M.; Huang, Q.; Gates, A.T.; Zhang, X.D.; Castle, J.C.; Stec, E.; Ferrer, M.; Strulovici, B.; Hazuda, D.J.; et al. Genome-scale RNAi screen for host factors required for HIV replication. Cell Host Microbe 2008, 4, 495–504. [Google Scholar] [CrossRef]
- Park, R.J.; Wang, T.; Koundakjian, D.; Hultquist, J.F.; Lamothe-Molina, P.; Monel, B.; Schumann, K.; Yu, H.; Krupzcak, K.M.; Garcia-Beltran, W.; et al. A genome-wide CRISPR screen identifies a restricted set of HIV host dependency factors. Nat. Genet. 2017, 49, 193–203. [Google Scholar] [CrossRef]
- Huang, H.; Kong, W.; Jean, M.; Fiches, G.; Zhou, D.; Hayashi, T.; Que, J.; Santoso, N.; Zhu, J. A CRISPR/Cas9 screen identifies the histone demethylase MINA53 as a novel HIV-1 latency-promoting gene (LPG). Nucleic Acids Res. 2019, 47, 7333–7347. [Google Scholar] [CrossRef] [PubMed]
- Krasnopolsky, S.; Kuzmina, A.; Taube, R. Genome-wide CRISPR knockout screen identifies ZNF304 as a silencer of HIV transcription that promotes viral latency. PLoS Pathog. 2020, 16, e1008834. [Google Scholar] [CrossRef]
- Li, Z.; Wu, J.; Chavez, L.; Hoh, R.; Deeks, S.G.; Pillai, S.K.; Zhou, Q. Reiterative Enrichment and Authentication of CRISPRi Targets (REACT) identifies the proteasome as a key contributor to HIV-1 latency. PLoS Pathog. 2019, 15, e1007498. [Google Scholar] [CrossRef]
- Li, Z.; Hajian, C.; Greene, W.C. Identification of unrecognized host factors promoting HIV-1 latency. PLoS Pathog. 2020, 16, e1009055. [Google Scholar] [CrossRef]
- Pedersen, S.F.; Collora, J.A.; Kim, R.N.; Yang, K.; Razmi, A.; Catalano, A.A.; Yeh, Y.J.; Mounzer, K.; Tebas, P.; Montaner, L.J.; et al. Inhibition of a Chromatin and Transcription Modulator, SLTM, Increases HIV-1 Reactivation Identified by a CRISPR Inhibition Screen. J. Virol. 2022, 96, e0057722. [Google Scholar] [CrossRef]
- OhAinle, M.; Helms, L.; Vermeire, J.; Roesch, F.; Humes, D.; Basom, R.; Delrow, J.J.; Overbaugh, J.; Emerman, M. A virus-packageable CRISPR screen identifies host factors mediating interferon inhibition of HIV. eLife 2018, 7, e35977. [Google Scholar] [CrossRef]
- Ohainle, M.; Kim, K.; Komurlu Keceli, S.; Felton, A.; Campbell, E.; Luban, J.; Emerman, M. TRIM34 restricts HIV-1 and SIV capsids in a TRIM5α-dependent manner. PLoS Pathog. 2020, 16, e1008507. [Google Scholar] [CrossRef]
- Hultquist, J.F.; Schumann, K.; Woo, J.M.; Manganaro, L.; McGregor, M.J.; Doudna, J.; Simon, V.; Krogan, N.J.; Marson, A. A Cas9 Ribonucleoprotein Platform for Functional Genetic Studies of HIV-Host Interactions in Primary Human T Cells. Cell Rep. 2016, 17, 1438–1452. [Google Scholar] [CrossRef]
- Hiatt, J.; Hultquist, J.F.; McGregor, M.J.; Bouhaddou, M.; Leenay, R.T.; Simons, L.M.; Young, J.M.; Haas, P.; Roth, T.L.; Tobin, V.; et al. A functional map of HIV-host interactions in primary human T cells. Nat. Commun. 2022, 13, 1752. [Google Scholar] [CrossRef] [PubMed]
- Elbashir, S.M.; Harborth, J.; Lendeckel, W.; Yalcin, A.; Weber, K.; Tuschl, T. Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature 2001, 411, 494–498. [Google Scholar] [CrossRef] [PubMed]
- Bushman, F.D.; Malani, N.; Fernandes, J.; D’Orso, I.; Cagney, G.; Diamond, T.L.; Zhou, H.; Hazuda, D.J.; Espeseth, A.S.; König, R.; et al. Host cell factors in HIV replication: Meta-analysis of genome-wide studies. PLoS Pathog. 2009, 5, e1000437. [Google Scholar] [CrossRef]
- Zhu, J.; Davoli, T.; Perriera, J.M.; Chin, C.R.; Gaiha, G.D.; John, S.P.; Sigiollot, F.D.; Gao, G.; Xu, Q.; Qu, H.; et al. Comprehensive identification of host modulators of HIV-1 replication using multiple orthologous RNAi reagents. Cell Rep. 2014, 9, 752–766. [Google Scholar] [CrossRef]
- Liu, L.; Oliveira, N.M.; Cheney, K.M.; Pade, C.; Dreja, H.; Bergin, A.M.; Borgdorff, V.; Beach, D.H.; Bishop, C.L.; Dittmar, M.T.; et al. A whole genome screen for HIV restriction factors. Retrovirology 2011, 8, 94. [Google Scholar] [CrossRef]
- Zhu, J.; Gaiha, G.D.; John, S.P.; Pertel, T.; Chin, C.R.; Gao, G.; Qu, H.; Walker, B.D.; Elledge, S.J.; Brass, A.L. Reactivation of latent HIV-1 by inhibition of BRD4. Cell Rep. 2012, 2, 807–816. [Google Scholar] [CrossRef]
- Takeuchi, H.; Saito, H.; Noda, T.; Miyamoto, T.; Yoshinaga, T.; Terahara, K.; Ishii, H.; Tsunetsugu-Yokota, Y.; Yamaoka, S. Phosphorylation of the HIV-1 capsid by MELK triggers uncoating to promote viral cDNA synthesis. PLoS Pathog. 2017, 13, e1006441. [Google Scholar] [CrossRef]
- Ndzinu, J.K.; Takeuchi, H.; Saito, H.; Yoshida, T.; Yamaoka, S. eIF4A2 is a host factor required for efficient HIV-1 replication. Microbes Infect. 2018, 20, 346–352. [Google Scholar] [CrossRef]
- Jackson, A.L.; Linsley, P.S. Recognizing and avoiding siRNA off-target effects for target identification and therapeutic application. Nat. Rev. Drug Discov. 2010, 9, 57–67. [Google Scholar] [CrossRef]
- Shalem, O.; Sanjana, N.E.; Hartenian, E.; Shi, X.; Scott, D.A.; Mikkelson, T.; Heckl, D.; Ebert, B.L.; Root, D.E.; Doench, J.G.; et al. Genome-scale CRISPR-Cas9 knockout screening in human cells. Science 2014, 343, 84–87. [Google Scholar] [CrossRef]
- Wang, T.; Wei, J.J.; Sabatini, D.M.; Lander, E.S. Genetic screens in human cells using the CRISPR-Cas9 system. Science 2014, 343, 80–84. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Zhu, S.; Cai, C.; Yuan, P.; Li, C.; Huang, Y.; Wei, W. High-throughput screening of a CRISPR/Cas9 library for functional genomics in human cells. Nature 2014, 509, 487–491. [Google Scholar] [CrossRef]
- Cong, L.; Ran, F.A.; Cox, D.; Lin, S.; Barretto, R.; Habib, N.; Hsu, P.D.; Wu, X.; Jiang, W.; Marraffini, L.A.; et al. Multiplex genome engineering using CRISPR/Cas systems. Science 2013, 339, 819–823. [Google Scholar] [CrossRef] [PubMed]
- Mali, P.; Yang, L.; Esvelt, K.M.; Aach, J.; Guell, M.; DiCarlo, J.E.; Norville, J.E.; Church, G.M. RNA-guided human genome engineering via Cas9. Science 2013, 339, 823–826. [Google Scholar] [CrossRef]
- Schumann, K.; Lin, S.; Boyer, E.; Simeonov, D.R.; Subramaniam, M.; Gate, R.E.; Haliburton, G.E.; Ye, C.J.; Bluestone, J.A.; Doudna, J.A.; et al. Generation of knock-in primary human T cells using Cas9 ribonucleoproteins. Proc. Natl. Acad. Sci. USA 2015, 112, 10437–10442. [Google Scholar] [CrossRef]
- Qi, L.S.; Larson, M.H.; Gilbert, L.A.; Doudna, J.A.; Weissman, J.S.; Arkin, A.P.; Lim, W.A. Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression. Cell 2013, 152, 1173–1183. [Google Scholar] [CrossRef]
- Larson, M.H.; Gilbert, L.A.; Wang, X.; Lim, W.A.; Weissman, J.S.; Qi, L.S. CRISPR interference (CRISPRi) for sequence-specific control of gene expression. Nat. Protoc. 2013, 8, 2180–2196. [Google Scholar] [CrossRef]
- Rosenbluh, J.; Xu, H.; Harrington, W.; Gill, S.; Wang, X.; Vazquez, F.; Root, D.E.; Tsherniak, A.; Hahn, W.C. Complementary information derived from CRISPR Cas9 mediated gene deletion and suppression. Nat. Commun. 2017, 8, 15403. [Google Scholar] [CrossRef]
- Kampmann, M. CRISPRi and CRISPRa Screens in Mammalian Cells for Precision Biology and Medicine. ACS Chem. Biol. 2018, 13, 406–416. [Google Scholar] [CrossRef]
- Maeder, M.L.; Linder, S.J.; Cascio, V.M.; Fu, Y.; Ho, Q.H.; Joung, J.K. CRISPR RNA-guided activation of endogenous human genes. Nat. Methods 2013, 10, 977–979. [Google Scholar] [CrossRef]
- Roesch, F.; OhAinle, M. HIV-CRISPR: A CRISPR/Cas9 Screening Method to Identify Genes Affecting HIV Replication. Bio-Protocol 2020, 10, e3614. [Google Scholar] [CrossRef] [PubMed]
- Margolis, D.M.; Garcia, J.V.; Hazuda, D.J.; Haynes, B.F. Latency reversal and viral clearance to cure HIV-1. Science 2016, 353, aaf6517. [Google Scholar] [CrossRef] [PubMed]
- Siliciano, R.F.; Greene, W.C. HIV latency. Cold Spring Harb. Perspect. Med. 2011, 1, a007096. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.C.; Xing, S.; Shan, L.; O’Connell, K.; Dinoso, J.; Shen, A.; Zhou, Y.; Shrum, C.K.; Han, Y.; Liu, J.O.; et al. Small-molecule screening using a human primary cell model of HIV latency identifies compounds that reverse latency without cellular activation. J. Clin. Investig. 2009, 119, 3473–3486. [Google Scholar] [CrossRef]
- Liu, R.; Yeh, Y.J.; Varabyou, A.; Collora, J.A.; Sherrill-Mix, S.; Talbot, C.C., Jr.; Mehta, S.; Albrecht, K.; Hao, H.; Zhang, H.; et al. Single-cell transcriptional landscapes reveal HIV-1-driven aberrant host gene transcription as a potential therapeutic target. Sci. Transl. Med. 2020, 12, eaaz0802. [Google Scholar] [CrossRef]
- Pan, C.; Kumar, C.; Bohl, S.; Klingmueller, U.; Mann, M. Comparative proteomic phenotyping of cell lines and primary cells to assess preservation of cell type-specific functions. Mol. Cell Proteom. 2009, 8, 443–450. [Google Scholar] [CrossRef]
- Hultquist, J.F.; Hiatt, J.; Schumann, K.; McGregor, M.J.; Roth, T.L.; Haas, P.; Doudna, J.A.; Marson, A.; Krogan, N.J. CRISPR-Cas9 genome engineering of primary CD4(+) T cells for the interrogation of HIV-host factor interactions. Nat. Protoc. 2019, 14, 1–27. [Google Scholar] [CrossRef]
- Hiatt, J.; Cavero, D.A.; McGregor, M.J.; Zheng, W.; Budzik, J.M.; Roth, T.L.; Haas, K.M.; Wu, D.; Rathore, U.; Meyer-Franke, A.; et al. Efficient generation of isogenic primary human myeloid cells using CRISPR-Cas9 ribonucleoproteins. Cell Rep. 2021, 35, 109105. [Google Scholar] [CrossRef]
- Chechik, L.; Martin, O.; Soutoglou, E. Genome Editing Fidelity in the Context of DNA Sequence and Chromatin Structure. Front. Cell Dev. Biol. 2020, 8, 319. [Google Scholar] [CrossRef]
- Rodrigues, V.; Ruffin, N.; San-Roman, M.; Benaroch, P. Myeloid Cell Interaction with HIV: A Complex Relationship. Front. Immunol. 2017, 8, 1698. [Google Scholar] [CrossRef]
- Kwong, P.D.; Wyatt, R.; Robinson, J.; Sweet, R.W.; Sodroski, J.; Hendrickson, W.A. Structure of an HIV gp120 envelope glycoprotein in complex with the CD4 receptor and a neutralizing human antibody. Nature 1998, 393, 648–659. [Google Scholar] [CrossRef]
- Berger, E.A.; Murphy, P.M.; Farber, J.M. Chemokine receptors as HIV-1 coreceptors: Roles in viral entry, tropism, and disease. Annu. Rev. Immunol. 1999, 17, 657–700. [Google Scholar] [CrossRef]
- Melikyan, G.B. Common principles and intermediates of viral protein-mediated fusion: The HIV-1 paradigm. Retrovirology 2008, 5, 111. [Google Scholar] [CrossRef]
- Briz, V.; Poveda, E.; Soriano, V. HIV entry inhibitors: Mechanisms of action and resistance pathways. J. Antimicrob. Chemother. 2006, 57, 619–627. [Google Scholar] [CrossRef]
- Wang, W.; Ye, C.; Liu, J.; Zhang, D.; Kimata, J.T.; Zhou, P. CCR5 gene disruption via lentiviral vectors expressing Cas9 and single guided RNA renders cells resistant to HIV-1 infection. PLoS ONE 2014, 9, e115987. [Google Scholar] [CrossRef]
- Kang, H.; Minder, P.; Park, M.A.; Mesquitta, W.T.; Torbett, B.E.; Slukvin, I.I. CCR5 Disruption in Induced Pluripotent Stem Cells Using CRISPR/Cas9 Provides Selective Resistance of Immune Cells to CCR5-tropic HIV-1 Virus. Mol. Nucleic Acids 2015, 4, e268. [Google Scholar] [CrossRef]
- Yu, S.; Yao, Y.; Xiao, H.; Li, J.; Liu, Q.; Yang, Y.; Adah, D.; Lu, J.; Zhao, S.; Qin, L.; et al. Simultaneous Knockout of CXCR4 and CCR5 Genes in CD4+ T Cells via CRISPR/Cas9 Confers Resistance to Both X4- and R5-Tropic Human Immunodeficiency Virus Type 1 Infection. Hum. Gene 2018, 29, 51–67. [Google Scholar] [CrossRef]
- Dean, M.; Carrington, M.; Winkler, C.; Huttley, G.A.; Smith, M.W.; Allikmets, R.; Goedert, J.J.; Buchbinder, S.P.; Vittinghoff, E.; Gomperts, E.; et al. Genetic restriction of HIV-1 infection and progression to AIDS by a deletion allele of the CKR5 structural gene. Hemophilia Growth and Development Study, Multicenter AIDS Cohort Study, Multicenter Hemophilia Cohort Study, San Francisco City Cohort, ALIVE Study. Science 1996, 273, 1856–1862. [Google Scholar] [CrossRef]
- Liu, R.; Paxton, W.A.; Choe, S.; Ceradini, D.; Martin, S.R.; Horuk, R.; MacDonald, M.E.; Stuhlmann, H.; Koup, R.A.; Landau, N.R. Homozygous defect in HIV-1 coreceptor accounts for resistance of some multiply-exposed individuals to HIV-1 infection. Cell 1996, 86, 367–377. [Google Scholar] [CrossRef]
- Samson, M.; Libert, F.; Doranz, B.J.; Rucker, J.; Liesnard, C.; Farber, C.M.; Saragosti, S.; Lapoumeroulie, C.; Cognaux, J.; Forceille, C.; et al. Resistance to HIV-1 infection in caucasian individuals bearing mutant alleles of the CCR-5 chemokine receptor gene. Nature 1996, 382, 722–725. [Google Scholar] [CrossRef]
- Hütter, G.; Nowak, D.; Mossner, M.; Ganepola, S.; Müssig, A.; Allers, K.; Schneider, T.; Hofmann, J.; Kücherer, C.; Blau, O.; et al. Long-term control of HIV by CCR5 Delta32/Delta32 stem-cell transplantation. N. Engl. J. Med. 2009, 360, 692–698. [Google Scholar] [CrossRef] [PubMed]
- Gupta, R.K.; Abdul-Jawad, S.; McCoy, L.E.; Mok, H.P.; Peppa, D.; Salgado, M.; Martinez-Picado, J.; Nijhuis, M.; Wensing, A.M.J.; Lee, H.; et al. HIV-1 remission following CCR5Δ32/Δ32 haematopoietic stem-cell transplantation. Nature 2019, 568, 244–248. [Google Scholar] [CrossRef]
- Arslan, S.; Litzow, M.R.; Cummins, N.W.; Rizza, S.A.; Badley, A.D.; Navarro, W.; Hashmi, S.K. Risks and Outcomes of Allogeneic Hematopoietic Stem Cell Transplantation for Hematologic Malignancies in Patients with HIV Infection. Biol. Blood Marrow Transplant. 2019, 25, e260–e267. [Google Scholar] [CrossRef]
- Ye, L.; Wang, J.; Beyer, A.I.; Teque, F.; Cradick, T.J.; Qi, Z.; Chang, J.C.; Bao, G.; Muench, M.O.; Yu, J.; et al. Seamless modification of wild-type induced pluripotent stem cells to the natural CCR5Δ32 mutation confers resistance to HIV infection. Proc. Natl. Acad. Sci. USA 2014, 111, 9591–9596. [Google Scholar] [CrossRef]
- Yusa, K.; Zhou, L.; Li Meng, A.; Bradley, A.; Craig Nancy, L. A hyperactive piggyBac transposase for mammalian applications. Proc. Natl. Acad. Sci. USA 2011, 108, 1531–1536. [Google Scholar] [CrossRef]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef]
- Deeks, S.G.; Overbaugh, J.; Phillips, A.; Buchbinder, S. HIV infection. Nat. Rev. Dis. Primers 2015, 1, 15035. [Google Scholar] [CrossRef]
- Glass, W.G.; McDermott, D.H.; Lim, J.K.; Lekhong, S.; Yu, S.F.; Frank, W.A.; Pape, J.; Cheshier, R.C.; Murphy, P.M. CCR5 deficiency increases risk of symptomatic West Nile virus infection. J. Exp. Med. 2006, 203, 35–40. [Google Scholar] [CrossRef]
- Farzan, M.; Mirzabekov, T.; Kolchinsky, P.; Wyatt, R.; Cayabyab, M.; Gerard, N.P.; Gerard, C.; Sodroski, J.; Choe, H. Tyrosine sulfation of the amino terminus of CCR5 facilitates HIV-1 entry. Cell 1999, 96, 667–676. [Google Scholar] [CrossRef]
- Hou, P.; Chen, S.; Wang, S.; Yu, X.; Chen, Y.; Jiang, M.; Zhuang, K.; Ho, W.; Hou, W.; Huang, J.; et al. Genome editing of CXCR4 by CRISPR/cas9 confers cells resistant to HIV-1 infection. Sci. Rep. 2015, 5, 15577. [Google Scholar] [CrossRef] [PubMed]
- Qi, C.; Li, D.; Jiang, X.; Jia, X.; Lu, L.; Wang, Y.; Sun, J.; Shao, Y.; Wei, M. Inducing CCR5Δ32/Δ32 Homozygotes in the Human Jurkat CD4+ Cell Line and Primary CD4+ Cells by CRISPR-Cas9 Genome-Editing Technology. Mol. Ther.-Nucleic Acids 2018, 12, 267–274. [Google Scholar] [CrossRef] [PubMed]
- Grivel, J.-C.; Shattock, R.J.; Margolis, L.B. Selective transmission of R5 HIV-1 variants: Where is the gatekeeper? J. Transl. Med. 2011, 9, S6. [Google Scholar] [CrossRef] [PubMed]
- Sundquist, W.I.; Hill, C.P. How to Assemble a Capsid. Cell 2007, 131, 17–19. [Google Scholar] [CrossRef][Green Version]
- Selyutina, A.; Persaud, M.; Simons, L.M.; Bulnes-Ramos, A.; Buffone, C.; Martinez-Lopez, A.; Scoca, V.; Di Nunzio, F.; Hiatt, J.; Marson, A.; et al. Cyclophilin A Prevents HIV-1 Restriction in Lymphocytes by Blocking Human TRIM5α Binding to the Viral Core. Cell Rep. 2020, 30, 3766–3777. [Google Scholar] [CrossRef]
- Aiken, C.; Rousso, I. The HIV-1 capsid and reverse transcription. Retrovirology 2021, 18, 29. [Google Scholar] [CrossRef]
- Bejarano, D.A.; Peng, K.; Laketa, V.; Börner, K.; Jost, K.L.; Lucic, B.; Glass, B.; Lusic, M.; Müller, B.; Kräusslich, H.-G. HIV-1 nuclear import in macrophages is regulated by CPSF6-capsid interactions at the nuclear pore complex. eLife 2019, 8, e41800. [Google Scholar] [CrossRef]
- Selyutina, A.; Hu, P.; Miller, S.; Simons, L.M.; Yu, H.J.; Hultquist, J.F.; Lee, K.; KewalRamani, V.N.; Diaz-Griffero, F. GS-CA1 and lenacapavir stabilize the HIV-1 core and modulate the core interaction with cellular factors. iScience 2022, 25, 103593. [Google Scholar] [CrossRef]
- Lampi, Y.; Van Looveren, D.; Vranckx, L.S.; Thiry, I.; Bornschein, S.; Debyser, Z.; Gijsbers, R. Targeted editing of the PSIP1 gene encoding LEDGF/p75 protects cells against HIV infection. Sci. Rep. 2019, 9, 2389. [Google Scholar] [CrossRef]
- Zhou, Q.; Chen, D.; Pierstorff, E.; Luo, K. Transcription elongation factor P-TEFb mediates Tat activation of HIV-1 transcription at multiple stages. EMBO J. 1998, 17, 3681–3691. [Google Scholar] [CrossRef]
- Price, D.H. P-TEFb, a cyclin-dependent kinase controlling elongation by RNA polymerase II. Mol. Cell Biol. 2000, 20, 2629–2634. [Google Scholar] [CrossRef]
- Ganser-Pornillos, B.K.; Pornillos, O. Restriction of HIV-1 and other retroviruses by TRIM5. Nat. Rev. Microbiol. 2019, 17, 546–556. [Google Scholar] [CrossRef]
- Désaulniers, K.; Ortiz, L.; Dufour, C.; Claudel, A.; Plourde, M.B.; Merindol, N.; Berthoux, L. Editing of the TRIM5 Gene Decreases the Permissiveness of Human T Lymphocytic Cells to HIV-1. Viruses 2020, 13, 24. [Google Scholar] [CrossRef] [PubMed]
- Bogerd, H.P.; Kornepati, A.V.; Marshall, J.B.; Kennedy, E.M.; Cullen, B.R. Specific induction of endogenous viral restriction factors using CRISPR/Cas-derived transcriptional activators. Proc. Natl. Acad. Sci. USA 2015, 112, E7249–E7256. [Google Scholar] [CrossRef]
- Malim, M.H. APOBEC proteins and intrinsic resistance to HIV-1 infection. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2009, 364, 675–687. [Google Scholar] [CrossRef]
- Zhang, Y.; Ozono, S.; Yao, W.; Tobiume, M.; Yamaoka, S.; Kishigami, S.; Fujita, H.; Tokunaga, K. CRISPR-mediated activation of endogenous BST-2/tetherin expression inhibits wild-type HIV-1 production. Sci. Rep. 2019, 9, 3134. [Google Scholar] [CrossRef]
- Jia, X.; Weber, E.; Tokarev, A.; Lewinski, M.; Rizk, M.; Suarez, M.; Guatelli, J.; Xiong, Y. Structural basis of HIV-1 Vpu-mediated BST2 antagonism via hijacking of the clathrin adaptor protein complex 1. eLife 2014, 3, e02362. [Google Scholar] [CrossRef]
- Lahouassa, H.; Daddacha, W.; Hofmann, H.; Ayinde, D.; Louge, E.C.; Dragin, L.; Bloch, N.; Maudet, C.; Bertrand, M.; Gramberg, T.; et al. SAMHD1 restricts the replication of human immunodeficiency virus type 1 by depleting the intracellular pool of deoxynucleoside triphosphates. Nat. Immunol. 2012, 13, 223–228. [Google Scholar] [CrossRef]
- Baldauf, H.M.; Pan, X.; Erikson, E.; Schmidt, S.; Daddacha, W.; Burggraf, M.; Schenkova, K.; Ambiel, I.; Wabnitz, G.; Gramberg, T.; et al. SAMHD1 restricts HIV-1 infection in resting CD4+ T cells. Nat. Med. 2012, 18, 1682–1688. [Google Scholar] [CrossRef]
- Krecka, K.; Hao, C.; Gierszewska, M.; Swanson, S.K.; Kesik-Brodacka, M.; Srivastava, S.; Florens, L.; Washburn, M.P.; Skowronski, J. Vpx relieves inhibition if HIV-1 infection of macrophages mediated by the SAMHD1 protein. Nature 2011, 474, 658–661. [Google Scholar] [CrossRef]
- Hofmann, H.; Logue, E.C.; Bloch, N.; Daddacha, W.; Polsky, S.B.; Schultz, M.L.; Kim, B.; Landau, N.R. The Vpx lentiviral accessory protein targets SAMHD1 for degradation in the nucleus. JVI 2012, 86, 12552–12560. [Google Scholar] [CrossRef]
- Perrillo, R. Benefits and risks of interferon therapy for hepatitis B. Hepatology 2009, 49, S103–S111. [Google Scholar] [CrossRef] [PubMed]
- Mitsuyasu, R.T. Interferon alpha in the treatment of AIDS-related Kaposi’s sarcoma. Br. J. Haematol. 1991, 79 (Suppl. 1), 69–73. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.Q.; Huang, Y.L.; Huang, J.; Zheng, J.L.; Qian, G.X. RIG-I detects HIV-1 infection and mediates type I interferon response in human macrophages from patients with HIV-1-associated neurocognitive disorders. Genet. Mol. Res. 2015, 14, 13799–13811. [Google Scholar] [CrossRef] [PubMed]
- Solis, M.; Nakhaei, P.; Jalalirad, M.; Lacoste, J.; Douville, R.; Arguello, M.; Zhao, T.; Laughrea, M.; Wainberg, M.A.; Hiscott, J. RIG-I-mediated antiviral signaling is inhibited in HIV-1 infection by a protease-mediated sequestration of RIG-I. J. Virol. 2011, 85, 1224–1236. [Google Scholar] [CrossRef]
- Jakobsen, M.R.; Bak, R.O.; Andersen, A.; Berg, R.K.; Jensen, S.B.; Jin, T.; Laustsen, A.; Hansen, K.; Østergaard, L.; Fitzgerald, K.A.; et al. IFI16 senses DNA forms of the lentiviral replication cycle and controls HIV-1 replication. Proc. Natl. Acad. Sci. USA 2013, 110, E4571–E4580. [Google Scholar] [CrossRef]
- Vermeire, J.; Roesch, F.; Sauter, D.; Rua, R.; Hotter, D.; Van Nuffel, A.; Vanderstraeten, H.; Naessens, E.; Iannucci, V.; Landi, A.; et al. HIV Triggers a cGAS-Dependent, Vpu- and Vpr-Regulated Type I Interferon Response in CD4(+) T Cells. Cell Rep. 2016, 17, 413–424. [Google Scholar] [CrossRef]
- Sumner, R.P.; Harrison, L.; Touizer, E.; Peacock, T.P.; Spencer, M.; Zuliani-Alvarez, L.; Towers, G.J. Disrupting HIV-1 capsid formation causes cGAS sensing of viral DNA. EMBO J. 2020, 39, e103958. [Google Scholar] [CrossRef]
- Lahaye, X.; Gentili, M.; Silvin, A.; Conrad, C.; Picard, L.; Jouve, M.; Zueva, E.; Maurin, M.; Nadalin, F.; Knott, G.J.; et al. NONO Detects the Nuclear HIV Capsid to Promote cGAS-Mediated Innate Immune Activation. Cell 2018, 175, 488–501. [Google Scholar] [CrossRef]
- Gringhuis, S.I.; Hertoghs, N.; Kaptein, T.M.; Zijlstra-Willems, E.M.; Sarrami-Forooshani, R.; Sprokholt, J.K.; van Teijlingen, N.H.; Kootstra, N.A.; Booiman, T.; van Dort, K.A.; et al. HIV-1 blocks the signaling adaptor MAVS to evade antiviral host defense after sensing of abortive HIV-1 RNA by the host helicase DDX3. Nat. Immunol. 2017, 18, 225–235. [Google Scholar] [CrossRef]
- Lai, M.; Maori, E.; Quaranta, P.; Matteoli, G.; Maggi, F.; Sgarbanti, M.; Crucitta, S.; Pacini, S.; Turriziani, O.; Antonelli, G.; et al. CRISPR/Cas9 Ablation of Integrated HIV-1 Accumulates Proviral DNA Circles with Reformed Long Terminal Repeats. J. Virol. 2021, 95, e01358-21. [Google Scholar] [CrossRef]
- Dampier, W.; Nonnemacher, M.R.; Sullivan, N.T.; Jacobson, J.M.; Wigdahl, B. HIV Excision Utilizing CRISPR/Cas9 Technology: Attacking the Proviral Quasispecies in Reservoirs to Achieve a Cure. MOJ Immunol. 2014, 1, 00022. [Google Scholar] [CrossRef]
- Herskovitz, J.; Hasan, M.; Patel, M.; Blomberg, W.R.; Cohen, J.D.; Machhi, J.; Shahjin, F.; Mosley, R.L.; McMillan, J.; Kevadiya, B.D.; et al. CRISPR-Cas9 Mediated Exonic Disruption for HIV-1 Elimination. EBioMedicine 2021, 73, 103678. [Google Scholar] [CrossRef]
- Ma, X.; Yang, T.; Luo, Y.; Wu, L.; Jiang, Y.; Song, Z.; Pan, T.; Liu, B.; Liu, G.; Liu, J.; et al. TRIM28 promotes HIV-1 latency by SUMOylating CDK9 and inhibiting P-TEFb. eLife 2019, 8, e42426. [Google Scholar] [CrossRef]
- Spina, C.A.; Anderson, J.; Archin, N.M.; Bosque, A.; Chan, J.; Famiglietti, M.; Greene, W.C.; Kashuba, A.; Lewin, S.R.; Margolis, D.M.; et al. An In-Depth Comparison of Latent HIV-1 Reactivation in Multiple Cell Model Systems and Resting CD4+ T Cells from Aviremic Patients. PLoS Pathog. 2013, 9, e1003834. [Google Scholar] [CrossRef]
- Symons, J.; Chopra, A.; Malatinkova, E.; De Spiegelaere, W.; Leary, S.; Cooper, D.; Abana, C.O.; Rhodes, A.; Rezaei, S.D.; Vandekerckhove, L.; et al. HIV integration sites in latently infected cell lines: Evidence of ongoing replication. Retrovirology 2017, 14, 2. [Google Scholar] [CrossRef]
- Cantero-Pérez, J.; Grau-Expósito, J.; Serra-Peinado, C.; Rosero, D.A.; Luque-Ballesteros, L.; Astorga-Gamaza, A.; Castellví, J.; Sanhueza, T.; Tapia, G.; Lloveras, B.; et al. Resident memory T cells are a cellular reservoir for HIV in the cervical mucosa. Nat. Commun. 2019, 10, 4739. [Google Scholar] [CrossRef]
- Chavez, L.; Calvanese, V.; Verdin, E. HIV Latency Is Established Directly and Early in Both Resting and Activated Primary CD4 T Cells. PLoS Pathog. 2015, 11, e1004955. [Google Scholar] [CrossRef]
- Cohn, L.B.; Chomont, N.; Deeks, S.G. The Biology of the HIV-1 Latent Reservoir and Implications for Cure Strategies. Cell Host Microbe 2020, 27, 519–530. [Google Scholar] [CrossRef]
- Tran, T.A.; de Goër de Herve, M.G.; Hendel-Chavez, H.; Dembele, B.; Le Névot, E.; Abbed, K.; Pallier, C.; Goujard, C.; Gasnault, J.; Delfraissy, J.F.; et al. Resting regulatory CD4 T cells: A site of HIV persistence in patients on long-term effective antiretroviral therapy. PLoS ONE 2008, 3, e3305. [Google Scholar] [CrossRef]
- Lee, E.; Bacchetti, P.; Milush, J.; Shao, W.; Boritz, E.; Douek, D.; Fromentin, R.; Liegler, T.; Hoh, R.; Deeks, S.G.; et al. Memory CD4+ T-Cells Expressing HLA-DR Contribute to HIV Persistence During Prolonged Antiretroviral Therapy. Front. Microbiol 2019, 10, 2214. [Google Scholar] [CrossRef]
- Leoni, C.; Bianchi, N.; Vincenzetti, L.; Monticelli, S. An optimized workflow for CRISPR-Cas9 deletion of surface and intracellular factors in primary human T lymphocytes. PLoS ONE 2021, 16, e0247232. [Google Scholar] [CrossRef] [PubMed]
- Albanese, M.; Ruhle, A.; Mittermaier, J.; Mejías-Pérez, E.; Gapp, M.; Linder, A.; Schmacke, N.A.; Hofmann, K.; Hennrich, A.A.; Levy, D.N.; et al. Rapid, efficient and activation-neutral gene editing of polyclonal primary human resting CD4+ T cells allows complex functional analyses. Nat. Methods 2022, 19, 81–89. [Google Scholar] [CrossRef] [PubMed]
- Kurata, M.; Yamamoto, K.; Moriarity, B.S.; Kitagawa, M.; Largaespada, D.A. CRISPR/Cas9 library screening for drug target discovery. J. Hum. Genet. 2018, 63, 179–186. [Google Scholar] [CrossRef] [PubMed]
- Büning, H.; Schambach, A. A first step toward in vivo gene editing in patients. Nat. Med. 2021, 27, 1515–1517. [Google Scholar] [CrossRef] [PubMed]
- Kaminski, R.; Chen, Y.; Fischer, T.; Tedaldi, E.; Napoli, A.; Zhang, Y.; Karn, J.; Hu, W.; Khalili, K. Elimination of HIV-1 Genomes from Human T-lymphoid Cells by CRISPR/Cas9 Gene Editing. Sci. Rep. 2016, 6, 22555. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Study | Hits | Genes Targeted | Hit Rate | Gene List Description | Technology | Pooled or Arrayed | Cell Type | Readout | Virus Used | Genes of Interest | Screen Hits |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Yeung [18] | 252 | 54,509 | 0.46% | 54,509 transcripts, 3–5 shRNA per gene | shRNA | Pooled | Jurkat | Cell survival | HIV (NL4-3) | HIV replication | NRF1, STXBP2, NCOA3, PRDM2, EXOSC5 |
| Brass [19] | 386 | 21,121 | 1.83% | Genome wide, 21,121 pools of 4 siRNAs per gene | siRNA | Arrayed | TZM-bl | p24 Immunofluorescence, β-galactosidase | HIV (IIIB) | HIV replication | Med28, TNPO3, Rab6, VPS53 |
| König [20] | 295 | 20,000 | 1.48% | Genome wide, targeting 20,000 genes | siRNA | Arrayed | HEK293T | Luciferase | HIV (VSV-pseudotyped HIV-1-luciferase vector) | HIV replication | TNPO3, NUP153, NUP358 |
| Zhou [21] | 311 | 19,709 | 1.58% | Genome wide, targeting 19,709 genes, pools of 3 siRNAs per gene | siRNA | Arrayed | HeLa P4/R5 | β-galactosidase | HIV (HXB2) | HIV replication | AKT1, PRKAA1, CD97, NEIL3, BMP2K, SERPINB6 |
| Park [22] | 5 | 18,543 | 0.03% | Genome wide, 187,536 sgRNAs targeting 18,543 protein-coding human genes | CRISPR | Pooled | GXCRCas9 T cell line | Cell survival | CCR5-tropic HIV-1 strain JR-CSF | Host dependency factors | TPST2, SLC35B2, ALCAM |
| Huang [23] | 446 | 3733 | 11.9% | sgRNA libraries enriched with human nuclear proteins | CRISPR | Pooled | J–LAT A2 | GFP expression | Latently infected ‘LTR-Tat-IRES-GFP’ HIV-1 minigenome’ proviral sequence | Latency | MINA53 |
| Krasnopolsky [24] | 5 | 19,052 | 0.03% | Genome-scale CRISPR Knock-Out (GeCKO) whole genome library, 19,052 genes, 6 sgRNA constructs per gene | CRISPR | Pooled | Jurkat (2D10) | BFP expression | HIV-LTR-2dGFP proviral sequence | Latency | ZNF304 |
| Li, 2019 [25] | 6 | 20,000 | 0.03% | Whole genome sgRNA library containing a total of ~200,000 sgRNAs at an average of 10 per gene | CRISPRi | Pooled | Jurkat (2D10-CRISPRi) | GFP expression | HIV-LTR-2dGFP proviral sequence | Latency | PSMD1, NFKBIA, CYLD, GON4L, PSMD3, PSMD8 |
| Li, 2020 [26] | 4 | 20,000 | 0.02% | ~100,000 sgRNA sequences, 5 sgRNAs/gene | CRISPRi | Pooled | JiL cell lines | GFP expression | HIV-LTR-2dGFP proviral sequence | Latency | FTSJ3, TMEM178A, NICN1 |
| Pedersen [27] | 18 | 18,905 | 0.095% | Genome-wide CRISPRi library, 5 sgRNA per gene | CRISPRi | Pooled | HIV-1-d6-GFP-Jurkat | GFP expression | HIV-1-d6-GFP proviral sequence | Latency | SLTM, SRRM2, NCBP2 |
| OhAinle, 2018 [28] | 15 | 1905 | 0.79% | 1905 human ISGs, 4 sgRNAs per gene | CRISPR (PIKA-HIV) | Pooled | THP-1 | Newly budded viruses | PIKA-HIV | ISGs, dependency factors | MxB, TRIM5α, IFITM1, Tetherin |
| OhAinle, 2020 [29] | 9 | 1905 | 0.47% | 1905 human ISGs, 4 sgRNAs per gene | CRISPR (PIKA-HIV) | Pooled | THP-1 | Newly budded viruses | HIV capsid mutants P90A, N74D | Capsid targeting restriction factors | TRIM34 |
| Hultquist [30] | 8 | 45 | 17.78% | 146 crRNAs | CRISPR RNPs | Arrayed | Primary human CD4+ T cells | GFP expression | HIV (NL4-3) | Genes associated with HIV integrase | GEMIN2, KPNA1, KPNA5, XRCC6 |
| Hiatt [31] | 47 | 426 | 11.03% | HIV-1 Interactome | CRISPR RNPs | Arrayed | Primary human CD4+ T cells | GFP expression | HIV (NL4-3) | Viral restriction and dependency factors | HUWE1, ELOC, AFF1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cisneros, W.J.; Cornish, D.; Hultquist, J.F. Application of CRISPR-Cas9 Gene Editing for HIV Host Factor Discovery and Validation. Pathogens 2022, 11, 891. https://doi.org/10.3390/pathogens11080891
Cisneros WJ, Cornish D, Hultquist JF. Application of CRISPR-Cas9 Gene Editing for HIV Host Factor Discovery and Validation. Pathogens. 2022; 11(8):891. https://doi.org/10.3390/pathogens11080891
Chicago/Turabian StyleCisneros, William J., Daphne Cornish, and Judd F. Hultquist. 2022. "Application of CRISPR-Cas9 Gene Editing for HIV Host Factor Discovery and Validation" Pathogens 11, no. 8: 891. https://doi.org/10.3390/pathogens11080891
APA StyleCisneros, W. J., Cornish, D., & Hultquist, J. F. (2022). Application of CRISPR-Cas9 Gene Editing for HIV Host Factor Discovery and Validation. Pathogens, 11(8), 891. https://doi.org/10.3390/pathogens11080891