
Role of Microglia in Herpesvirus-Related Neuroinflammation and Neurodegeneration



{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Herpetic Infections of CNS
3. Neuroinflammation
4. Microglia in Viral Encephalitis
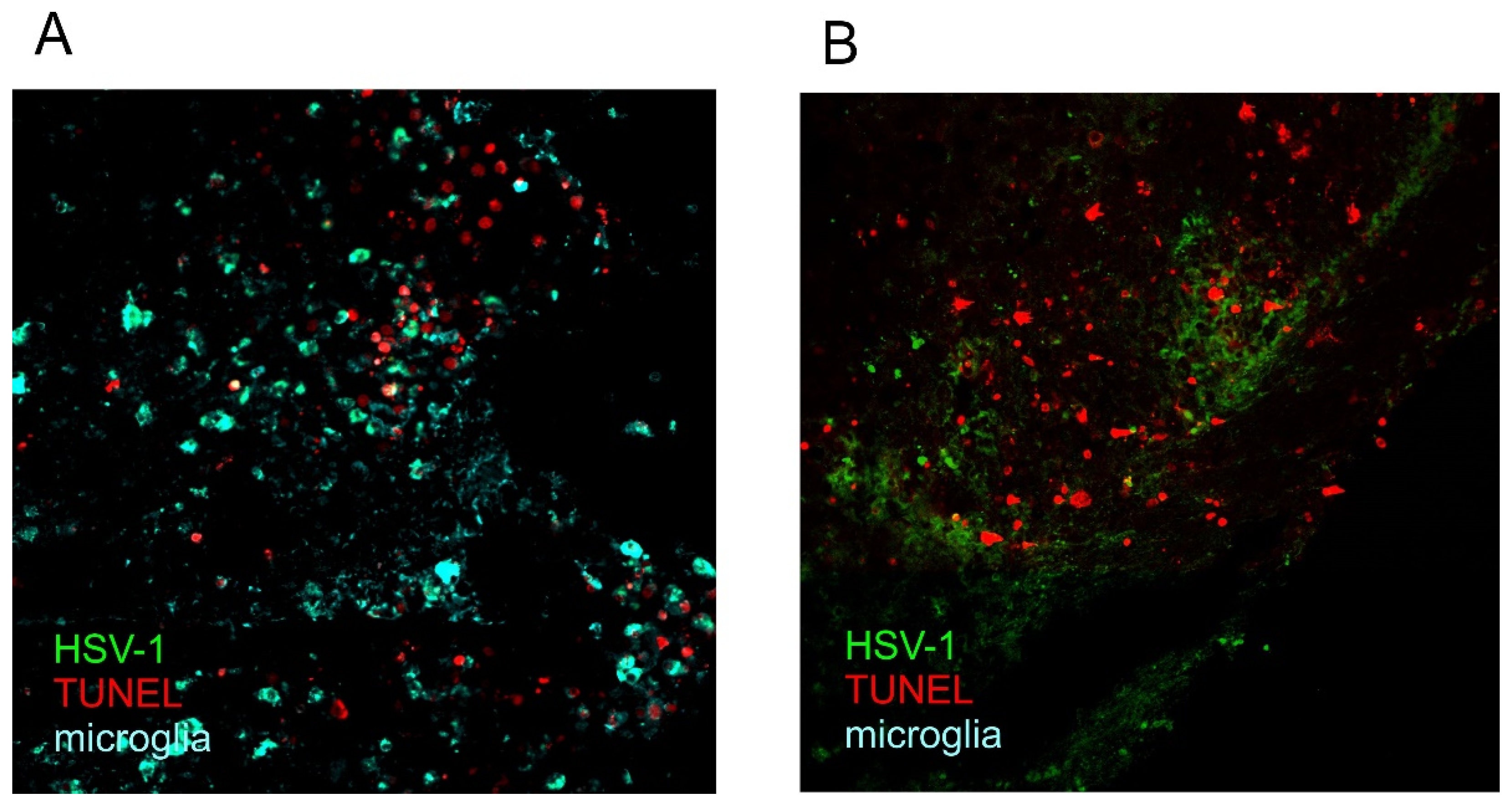
5. Cell Death and Neuroinflammation
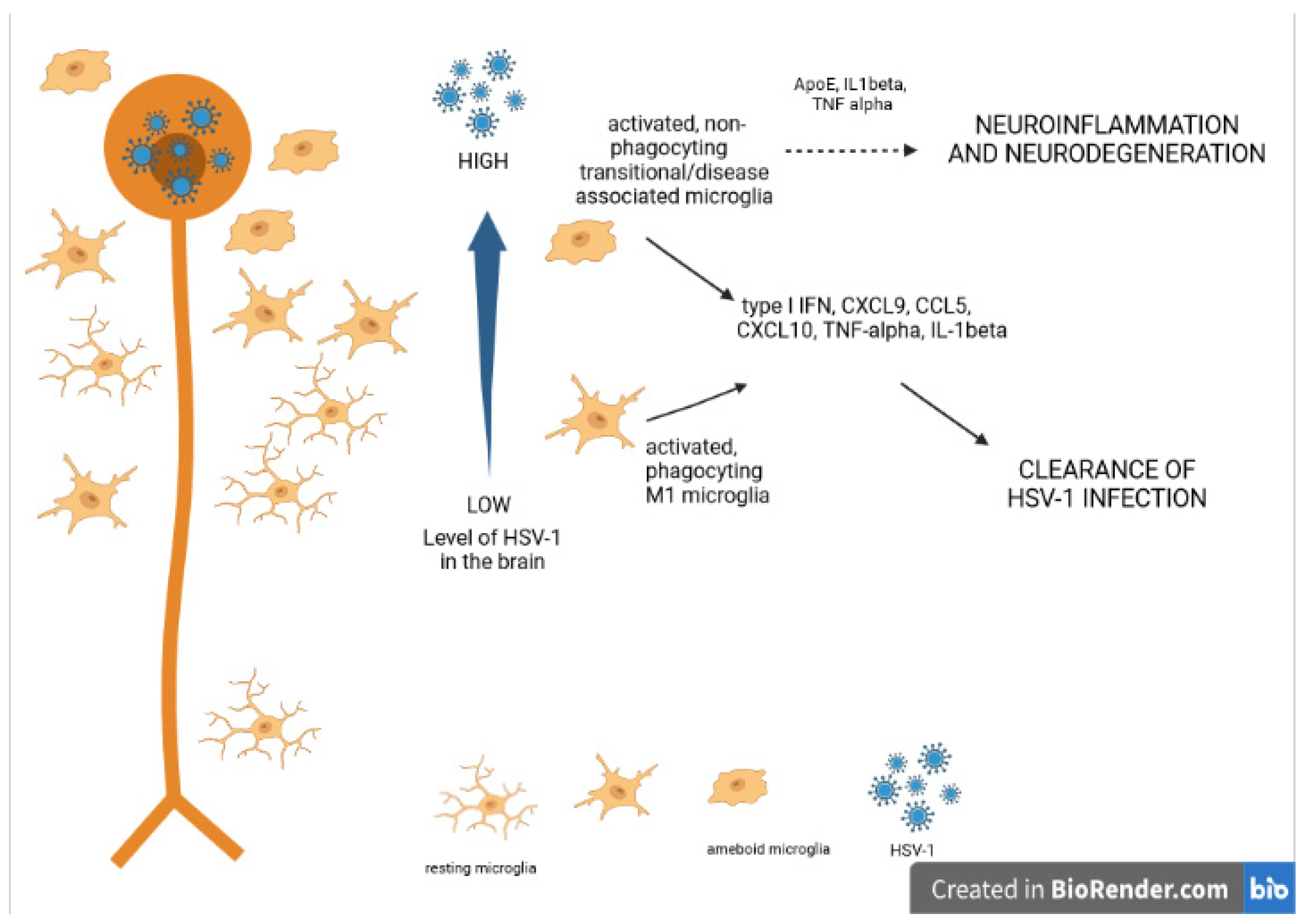
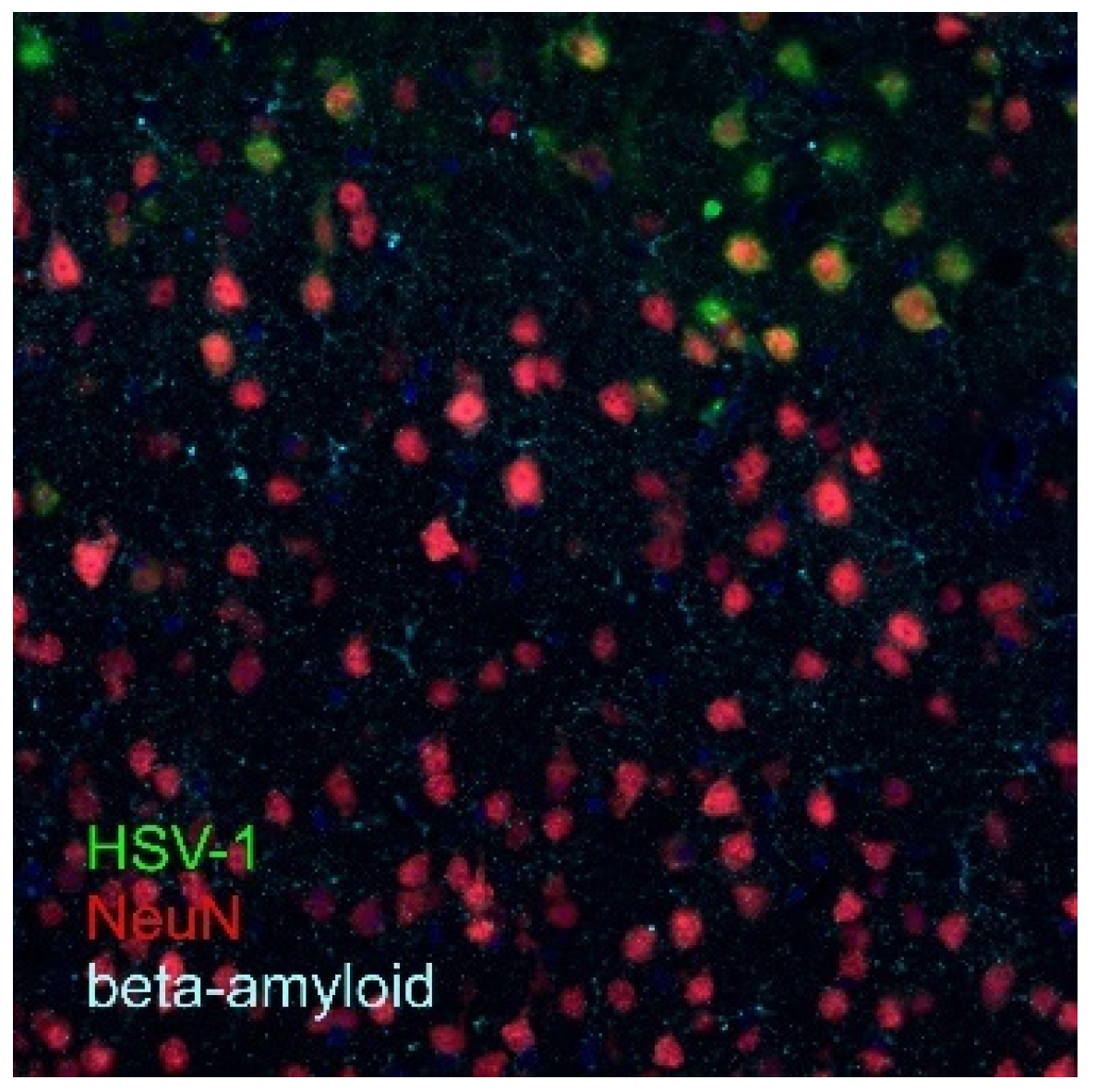
6. Neuroinflammation and Neurodegeneration
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Louveau, A.; Smirnov, I.; Keyes, T.J.; Eccles, J.D.; Rouhani, S.J.; Peske, J.D.; Derecki, N.C.; Castle, D.; Mandell, J.W.; Lee, K.S.; et al. Structural and functional features of central nervous system lymphatic vessels. Nature 2015, 523, 337–341. [Google Scholar] [CrossRef] [PubMed]
- Forrester, V.; McMenamin, P.G.; Dando, S.J. CNS infection and immune privilege. Nat. Rev. Neurosci. 2018, 19, 655–671. [Google Scholar] [CrossRef] [PubMed]
- Marques, C.P.; Cheeran, M.C.; Palmquist, J.M.; Hu, S.; Urban, S.L.; Lokensgard, J.R. Prolonged microglial cell activation and lymphocyte infiltration following experimental herpes encephalitis. J. Immunol. 2008, 181, 6417–6426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lundberg, P.; Ramakrishna, C.; Brown, J.; Tyszka, J.M.; Hamamura, M.; Hinton, D.R.; Kovats, S.; Nalcioglu, O.; Weinberg, K.; Openshaw, H.; et al. The immune response to herpes simplex virus type 1 infection in susceptible mice is a major cause of central nervous system pathology resulting in fatal encephalitis. J. Virol. 2008, 82, 7078–7088. [Google Scholar] [CrossRef] [Green Version]
- Soares, B.P.; Provenzale, J.M. Imaging of Herpesvirus infections of the CNS. Am. J. Roentgenol. 2015, 206, 39–48. [Google Scholar] [CrossRef]
- Roizman, B.; Knipe, D.M. Herpes simplex viruses and their replication. In Fields Virology, 4th ed.; Knipe, D.M., Howley, P.M., Griffen, D.E., Eds.; Lippincott Williams: Philadelphia, PA, USA, 2001; pp. 2399–2459. [Google Scholar]
- Marcocci, M.E.; Napoletani, G.; Protto, V.; Kolesova, O.; Piacentini, R.; Li Puma, D.D.; Lomonte, P.; Grassi, C.; Palamara, A.T.; De Chiara, G. Herpes simplex virus-1 in the brain: The dark side of a sneaky infection. Trends Microbiol. 2020, 28, 808–820. [Google Scholar] [CrossRef]
- Umbach, J.L.; Kramer, M.F.; Jurak, I.; Karnowski, H.W.; Coen, D.M.; Cullen, B.R. MicroRNAs expressed by herpes simplex virus 1 during latent infection regulate viral mRNAs. Nature 2008, 454, 780–783. [Google Scholar] [CrossRef] [Green Version]
- Knipe, D.M.; Cliffe, A. Chromatin control of herpes simplex virus lytic and latent infection. Nat. Rev. 2008, 6, 211–221. [Google Scholar] [CrossRef]
- Gupta, R.; Warren, T.; Wald, A. Genital herpes. Lancet 2007, 370, 2127–2137. [Google Scholar] [CrossRef]
- Halioua, B.; Malkin, J.E. Epidemiology of genital herpes-recent advances. Eur. J. Dermatol. 1999, 9, 177–184. [Google Scholar]
- Corey, L.; Wald, A. Maternal and neonatal herpes simplex virus infections. N. Engl. J. Med. 2009, 361, 1376–1385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gershon, A.A.; Breuer, J.; Cohen, J.I.; Cohrs, R.J.; Gershon, M.D.; Gilden, D.; Grose, C.; Hambleton, S.; Kennedy, P.G.; Oxman, M.N.; et al. Varicella zoster virus infection. Nat. Rev. Dis. Primers 2015, 1, 15016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagel, M.A.; Gilden, D.H. The protean neurologic manifestations of varicella-zoster virus. Clevel. Clin. J. Med. 2007, 74, 489–504. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, P.G.E. Neurological complications of varicella-zoster virus infections. In Infections of the Nervous System; Kennedy, P.G.E., Johnson, R.T., Eds.; Butterworth: London, UK, 1987; pp. 177–208. [Google Scholar]
- Balfour, H.H., Jr.; Odumade, O.A.; Schmeling, D.O.; Mullan, B.D.; Ed, J.A.; Knight, J.A.; Vezina, H.E.; Thomas, W.; Hogquist, K.A. Behavioral, Virologic, and Immunologic Factors Associated with Acquisition and Severity of Primary Epstein-Barr Virus Infection in University Students. J. Infect. Dis. 2013, 207, 80–88. [Google Scholar] [CrossRef] [Green Version]
- Hadinoto, V.; Shapiro, M.; Sun, C.C.; Thorley-Lawson, D.A. The Dynamics of EBV Shedding Implicate a Central Role for Epithelial Cells in Amplifying Viral Output. PLoS Pathog. 2009, 5, e1000496. [Google Scholar] [CrossRef] [Green Version]
- Boppana, S.B.; Ross, S.A.; Fowler, K.B. Congenital cytomegalovirus infection: Clinical outcome. Clin. Infect. Dis 2013, 57, 178–181. [Google Scholar] [CrossRef] [Green Version]
- Ludlow, M.; Kortekaas, J.; Herden, C.; Hoffmann, B.; Tappe, D.; Trebst, C.; Griffin, D.E.; Brindle, H.E.; Solomon, T.; Brown, A.S.; et al. Neurotropic virus infections as the cause of immediate and delayed neuropathology. Acta Neuropathol. 2015, 131, 159–184. [Google Scholar] [CrossRef] [Green Version]
- Soldan, S.S.; Leist, T.P.; Juhng, K.N.; McFarland, H.F.; Jacobson, S. Increased Lymphoproliferative Response to Human Herpesvirus Type 6A Variant in Multiple Sclerosis Patients. Ann. Neurol. 2000, 47, 306–313. [Google Scholar] [CrossRef]
- Ortega-Madueño, I.; Garcia-Montojo, M.; Dominguez-Mozo, M.I.; Garcia-Martinez, A.; Arias-Leal, A.M.; Casanova, I.; Arroyo, R.; Alvarez-Lafuente, R. Anti-Human Herpesvirus 6A/B IgG Correlates with Relapses and Progression in Multiple Sclerosis. PLoS ONE 2014, 9, e104836. [Google Scholar] [CrossRef]
- Hall, C.B.; Long, C.E.; Schnabel, K.C.; Caserta, M.T.; McIntyre, K.M.; Costanzo, M.A.; Knott, A.; Dewhurst, S.; Insel, R.A.; Epstein, L.G. Human Herpesvirus-6 Infection in Children. A Prospective Study of Complications and Reactivation. N. Engl. J. Med. 1994, 331, 432–438. [Google Scholar] [CrossRef]
- Caselli, E.; Di Luca, D. Molecular biology and clinical associations of Roseoloviruses human herpesvirus 6 and human herpesvirus 7. New Microbiol. 2007, 30, 173–187. [Google Scholar] [PubMed]
- Martikainen, M.H.; Grönroos, J.O.; Vuorinen, T. Detection of human herpesvirus 7 DNA from the CSF in association with neurosarcoidosis. J. Med. Virol. 2013, 11, 1935–1939. [Google Scholar] [CrossRef] [PubMed]
- Ward, K.N. The natural history and laboratory diagnosis of human herpesviruses-6 and -7 infections in the immunocompetent. J. Clin. Virol. 2005, 32, 183–193. [Google Scholar] [CrossRef]
- Okin, D.; Medzhitov, R. Evolution of inflammatory diseases. Curr. Biol. 2012, 22, 733–740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takeuchi, O.; Akira, S. Pattern recognition receptors and inflammation. Cell 2010, 140, 805–820. [Google Scholar] [CrossRef] [Green Version]
- Estes, M.; McAllister, K. Alterations in immune cells and mediators in the brain: It’s not always neuroinflammation! Brain Pathol. 2014, 24, 623–630. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.; Zhong, D.; Li, G. The role of microglia in viral encephalitis: A review. J. Neuroinflamm. 2019, 16, 76. [Google Scholar] [CrossRef]
- Ginhoux, F.; Garel, S. The mysterious origins of microglia. Nat. Neurosci. 2018, 21, 897–899. [Google Scholar] [CrossRef]
- Bar, E.; Barak, B. Microglia roles in synaptic plasticity and myelination in homeostatic conditions and neurodevelopmental disorders. Glia 2019, 67, 2125–2141. [Google Scholar] [CrossRef]
- Tang, Y.; Le, W. Differential Roles of M1 and M2 Microglia in Neurodegenerative Diseases. Mol. Neurobiol. 2016, 53, 1181–1194. [Google Scholar] [CrossRef]
- Wang, J.; Xing, H.; Wan, L.; Jiang, X.; Wang, C.; Wu, Y. Treatment targets for M2 microglia polarization in ischemic stroke. Biomed. Pharmacother. 2018, 105, 518–525. [Google Scholar] [CrossRef]
- Liddelow, S.A.; Guttenplan, K.A.; Clarke, L.E.; Bennett, F.C.; Bohlen, C.J.; Schirmer, L.; Bennett, M.L.; Münch, A.E.; Chung, W.S.; Peterson, T.C.; et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 2017, 541, 481–487. [Google Scholar] [CrossRef] [PubMed]
- Piret, J.; Boivin, G. Innate immune response during herpes simplex virus encephalitis and development of immunomodulatory strategies. Rev. Med. Virol. 2015, 25, 300–319. [Google Scholar] [CrossRef] [PubMed]
- Krzyzowska, M.; Kowalczyk, A.; Skulska, K.; Thörn, K.; Eriksson, K. Fas/FasL Contributes to HSV-1 Brain Infection and Neuroinflammation. Front. Immunol. 2021, 12, 714821. [Google Scholar] [CrossRef] [PubMed]
- Lafaille, F.G.; Pessach, I.M.; Zhang, S.Y.; Ciancanelli, M.J.; Herman, M.; Abhyankar, A.; Ying, S.W.; Keros, S.; Goldstein, P.A.; Mostoslavsky, G.; et al. Impaired intrinsic immunity to HSV-1 in human iPSC-derived TLR3-deficient CNS cells. Nature 2012, 491, 769–773. [Google Scholar] [CrossRef]
- Holm, T.H.; Draeby, D.; Owens, T. Microglia are required for astroglial Toll-like receptor 4 response and for optimal TLR2 and TLR3 response. Glia 2012, 60, 630–638. [Google Scholar] [CrossRef]
- Rosenberger, K.; Derkow, K.; Dembny, P.; Kruger, C.; Schott, E.; Lehnardt, S. The impact of single and pairwise Toll-like receptor activation on neuroinflammation and neurodegeneration. J. Neuroinflamm. 2014, 11, 166. [Google Scholar] [CrossRef] [Green Version]
- Lima, G.K.; Zolini, G.P.; Santos Mansur, D.; Lima, B.H.F.; Wischhoff, U.; Astigarraga, R.G.; Dias, M.F.; das Graças Almeida Silva, M.; Ribeiro Béla, S.; do Valle Antonelli, L.R.; et al. Toll-like receptor (TLR) 2 and TLR9 expressed in trigeminal ganglia are critical to viral control during herpes simplex virus 1 infection. Am. J. Pathol. 2010, 177, 2433–2445. [Google Scholar] [CrossRef]
- Wang, J.P.; Bowen, G.N.; Zhou, S.; Cerny, A.; Zacharia, A.; Knipe, D.M.; Finberg, R.W.; Kurt-Jones, E.A. Role of specific innate immune responses in herpes simplex virus infection of the central nervous system. J. Virol. 2012, 86, 2273–2281. [Google Scholar] [CrossRef] [Green Version]
- Zolini, G.P.; Lima, G.K.; Lucinda, N.; Silva, M.A.; Dias, M.F.; Lima Pessoa, N.; Pizziolo Coura, B.; Teixeira Cartelle, C.; Arantes, R.M.; Geessien Kroon, E.; et al. Defense against HSV-1 in a murine model is mediated by iNOS and orchestrated by the activation of TLR2 and TLR9 in trigeminal ganglia. J. Neuroinflamm. 2014, 11, 20. [Google Scholar] [CrossRef] [Green Version]
- Reinert, L.S.; Lopušná, K.; Winther, H.; Sun, C.; Thomsen, M.K.; Nandakumar, R.; Mogensen, T.H.; Meyer, M.; Vægter, C.; Nyengaard, J.R.; et al. Sensing of HSV-1 by the cGAS-STING pathway in microglia orchestrates antiviral defence in the CNS. Nat. Commun. 2016, 7, 13348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paludan, S.R.; Bowie, A.G. Immune sensing of DNA. Immunity 2013, 38, 870–880. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uyar, O.; Laflamme, N.; Piret, J.; Venable, M.C.; Carbonneau, J.; Zarrouk, K.; Rivest, S.; Boivin, G. An early microglial response is needed to efficiently control herpes simplex virus encephalitis. J. Virol. 2020, 94, e01428-20. [Google Scholar] [CrossRef]
- Tsai, M.-S.; Wang, L.-C.; Tsai, H.-Y.; Lin, Y.-J.; Wu, H.-L.; Tzeng, S.-F.; Hsu, S.-M.; Chen, S.-H. Microglia Reduce Herpes Simplex Virus 1 Lethality of Mice with Decreased T Cell and Interferon Responses in Brains. Int. J. Mol. Sci. 2021, 22, 12457. [Google Scholar] [CrossRef] [PubMed]
- Uyar, O.; Dominguez, J.M.; Bordeleau, M.; Lapeyre, L.; Ibáñez, F.G.; Vallières, L.; Tremblay, M.E.; Corbeil, J.; Boivin, G. Single-cell transcriptomics of the ventral posterolateral nucleus-enriched thalamic regions from HSV-1-infected mice reveal a novel microglia/microglia-like transcriptional response. J. Neuroinflamm. 2022, 19, 81. [Google Scholar] [CrossRef]
- Fekete, R.; Cserép, C.; Lénárt, N.; Tóth, K.; Orsolits, B.; Martinecz, B.; Méhes, E.; Szabó, B.; Németh, V.; Gönci, B.; et al. Microglia control the spread of neurotropic virus infection via P2Y12 signalling and recruit monocytes through P2Y12-independent mechanisms. Acta Neuropathol. 2018, 136, 461–482. [Google Scholar] [CrossRef] [Green Version]
- De Chiara, G.; Piacentini, R.; Fabiani, M.; Mastrodonato, A.; Marcocci, M.E.; Limongi, D.; Napoletani, G.; Protto, V.; Coluccio, P.; Celestino, I.; et al. Recurrent herpes simplex virus-1 infection induces hall arks of neurodegeneration and cognitive deficits in mice. PLoS Pathog. 2019, 15, e1007617. [Google Scholar] [CrossRef] [Green Version]
- Lokensgard, J.R.; Hu, S.; Sheng, W.; van Oijen, M.; Cox, D.; Cheeran, M.C.-J.J.; Peterson, P.K. Robust expression of TNF-, IL-1, RANTES, and IP-10 by human microglial cells during nonproductive infection with herpes simplex virus. J. Neurovirol. 2001, 7, 208–219. [Google Scholar]
- Chucair-Elliott, A.J.; Conrady, C.; Zheng, M.; Kroll, C.M.; Lane, T.E.; Carr, D.J.J. Microglia-induced IL-6 protects against neuronal loss following HSV-1 infection of neural progenitor cells. Glia 2014, 62, 1418–1434. [Google Scholar] [CrossRef] [Green Version]
- Cymerys, J.; Kowalczyk, A.; Mikołajewicz, K.; Słońska, A.; Krzyżowska, M. Nitric oxide influences HSV-1-induced neuroinflammation. Oxid. Med. Cell. Longev. 2019, 5, 1–17. [Google Scholar] [CrossRef]
- Kveštak, D.; Juranić Lisnić, V.; Lisnić, B.; Tomac, J.; Golemac, M.; Brizić, I.; Indenbirken, D.; Cokarić Brdovčak, M.; Bernardini, G.; Krstanović, F.; et al. NK/ILC1 cells mediate neuroinflammation and brain pathology following congenital CMV infection. J. Exp. Med. 2021, 218, e20201503. [Google Scholar] [CrossRef] [PubMed]
- Rock, R.B.; Gekker, G.; Hu, S.; Sheng, W.S.; Cheeran, M.; Lokensgard, J.R.; Peterson, P.K. Role of microglia in central nervous system infections. Clin. Microbiol. Rev. 2004, 17, 942–964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koontz, T.; Bralic, M.; Tomac, J.; Pernjak-Pugel, E.; Bantug, G.; Jonjic, S.; Britt, W.J. Altered development of the brain after focal herpesvirus infection of the central nervous system. J. Exp. Med. 2008, 205, 423–435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geginat, J.; Paroni, M.; Pagani, M.; Galimberti, D.; De Francesco, R.; Scarpini, E.; Abrignani, S. The Enigmatic Role of Viruses in Multiple Sclerosis: Molecular Mimicry or Disturbed Immune Surveillance? Trends Immunol. 2017, 38, 498–512. [Google Scholar] [CrossRef] [PubMed]
- Tzartos, J.S.; Khan, G.; Vossenkamper, A.; Cruz-Sadaba, M.; Lonardi, S.; Sefia, E.; Meager, A.; Elia, A.; Middeldorp, J.M.; Clemens, M.; et al. Association of Innate Immune Activation with Latent Epstein-Barr Virus in Active MS Lesions. Neurology 2012, 78, 15–23. [Google Scholar] [CrossRef]
- Baglio, S.R.; van Eijndhoven, M.A.; Koppers-Lalic, D.; Berenguer, J.; Lougheed, S.M.; Gibbs, S.; Léveillé, N.; Rinkel, R.N.P.M.; Hopmans, E.S.; Swaminathan, S.; et al. Sensing of Latent EBV Infection Through Exosomal Transfer of 5’ppprna. Proc. Natl. Acad. Sci. USA 2016, 113, 587–596. [Google Scholar] [CrossRef] [Green Version]
- Hassani, A.; Corboy, J.R.; Al-Salam, S.; Khan, G. Epstein-Barr virus is present in the brain of most cases of multiple sclerosis and may engage more than just B cells. PLoS ONE 2018, 13, e0192109. [Google Scholar] [CrossRef] [Green Version]
- Bortolotti, D.; Gentili, V.; Rotola, A.; Caselli, E.; Rizzo, R. HHV-6A infection induces amyloid-beta expression and activation of microglial cells. Alzheimers Res. Ther. 2019, 11, 104. [Google Scholar] [CrossRef] [Green Version]
- Galluzzi, L.; Maiuri, M.C.; Vitale, I.; Zischka, H.; Castedo, M.; Zitvogel, L.; Kroemer, G. Cell death modalities: Classification and pathophysiological implications. Cell Death Differ. 2007, 14, 1237–1243. [Google Scholar] [CrossRef]
- Debiasi, R.L.; Kleinschmidt-DeMasters, B.K.; Richardson-burns, S.; Tyler, K.L. Central nervous system apoptosis in human herpes simplex virus and cytomegalovirus encephalitis. J. Infect. Dis. 2002, 186, 1547–1557. [Google Scholar] [CrossRef]
- Shaw, M.M.; Gürr, W.K.; Thackray, A.M.; Watts, P.A.; Littler, E.; Field, H.J. Temporal pattern of herpes simplex virus type 1 infection and cell death in the mouse brain stem: Influence of guanosine nucleoside analogues. J. Virol. Methods 2002, 102, 93–102. [Google Scholar] [CrossRef]
- Zhou, G.; Galvan, V.; Campadelli-fiume, G.; Roizman, B. Glycoprotein D or J delivered in trans blocks apoptosis in SK-NSH cells induced by a herpes simplex virus 1 mutant lacking intact genes expressing both glycoproteins. J. Virol. 2000, 74, 11782–11791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, M.L.; Blaho, J.A. Apoptosis during herpes simplex virus infection. Adv. Virus Res. 2007, 69, 67–97. [Google Scholar] [PubMed]
- Aubert, M.; Blaho, J.A. The herpes simplex virus type 1 regulatory protein ICP27 is required for the prevention of apoptosis in infected human cells. J. Virol. 1999, 73, 2803–2813. [Google Scholar] [CrossRef] [Green Version]
- Jiang, X.; Chentoufi, A.A.; Hsiang, C.; Carpenter, D.; Osorio, N.; Benmohamed, L.; Fraser, N.W.; Jones, C.; Wechsler, S.L. The herpes simplex virus type 1 latency-associated transcript can protect neuron-derived C1300 and Neuro2A cells from granzyme B-induced apoptosis and CD8 T-cell killing. J. Virol. 2011, 85, 2325–2332. [Google Scholar] [CrossRef] [Green Version]
- Reinert, L.S.; Rashidi, A.S.; Tran, D.N.; Katzilieris-Petras, G.; Hvidt, A.K.; Gohr, M.; Fruhwürth, S.; Bodda, C.; Thomsen, M.K.; Vendelbo, M.H.; et al. Brain immune cells undergo cGAS/STING-dependent apoptosis during herpes simplex virus type 1 infection to limit type I IFN production. J. Clin. Investig. 2021, 131, e136824. [Google Scholar] [CrossRef]
- Scott, F.L.; Stec, B.; Pop, C.; Dobaczewska, M.K.; Lee, J.J.; Monosov, E.; Robinson, H.; Salvesen, G.S.; Schwarzenbacher, R.; Riedl, S.J. The Fas-FADD death domain complex structure unravels signalling by receptor clustering. Nature 2009, 457, 1019–1022. [Google Scholar] [CrossRef] [Green Version]
- Dowling, P.; Shang, G.; Raval, S.; Menonna, J.; Cook, S.; Husar, W. Involvement of the CD95 (Apo-1/Fas) receptor/ligand system in multiple sclerosis brain. J. Exp. Med. 1996, 184, 1513–1518. [Google Scholar] [CrossRef] [Green Version]
- Ethell, D.W.; Buhler, L.A. Fas ligand-mediated apoptosis in degenerative disorders of the brain. J. Clin. Immunol. 2003, 23, 363–370. [Google Scholar] [CrossRef]
- Jeffries, A.M.; Suptela, A.J.; Marriott, I. Z-DNA binding protein 1 mediates necroptotic and apoptotic cell death pathways in murine astrocytes following herpes simplex virus-1 infection. J. Neuroinflamm. 2022, 19, 109. [Google Scholar] [CrossRef]
- Alzheimers Asociation Report. Alzheimers disease facts and figures. Alzheimers Dement. 2018, 3, 367–429. [Google Scholar]
- Lane, C.A. Alzheimers disease. Eur. J. Neurol. 2018, 25, 59–70. [Google Scholar] [CrossRef] [PubMed]
- Wozniak, M.A.; Frost, A.L.; Itzhaki, R.F. Alzheimers disease-specific tau phosphorylation is induced by herpes simplex virus type 1. J. Alzheimers Dis. 2009, 16, 341–350. [Google Scholar] [CrossRef] [PubMed]
- Itzhaki, R.F.; Dobson, C.B.; Shipley, S.J.; Wozniak, M.A. The role of viruses and of APOE in dementia. Ann. N. Y. Acad. Sci. 2004, 1019, 15–18. [Google Scholar] [CrossRef] [PubMed]
- Agostini, S.; Mancuso, R.; Baglioa, F.; Cabinioa, M.; Hernisa, A.; SaulCosta, A.; Calabresea, E.; Nemnia, R.; Clericia, M. High avidity HSV-1 antibodies correlate with absence of amnestic Mild Cognitive Impairment conversion to Alzheimers disease. BBI-Health 2016, 58, 254–260. [Google Scholar]
- Carbone, I.; Lazzarotto, T.; Ianni, M.; Porcellini, E.; Forti, P.; Masliah, E.; Gabrielli, L.; Licastro, F. Herpes Virus in Alzheimers Disease: Relation to Progression of the Disease. Neurobiol. Aging 2014, 35, 122–129. [Google Scholar] [CrossRef] [PubMed]
- Readhead, B.; Haure-Mirande, J.V.; Funk, C.C.; Richards, M.A.; Shannon, P.; Haroutunian, V.; Sano, M.; Liang, W.S.; Beckmann, N.D.; Price, N.D.; et al. Multiscale Analysis of Independent Alzheimers Cohorts Finds Disruption of Molecular, Genetic, and Clinical Networks by Human Herpesvirus. Neuron 2018, 99, 64–82. [Google Scholar] [CrossRef] [Green Version]
- De Chiara, G.; Marcocci, M.E.; Livia Civitelli, L.; Argnani, R.; Piacentini, R.; Ripoli, C.; Manservigi, R.; Grassi, C.; Garaci, E.; Palamara, A.T. APP processing induced by herpes simplex virus type 1 (HSV-1) yields several APP fragments in human and rat neuronal cells. PLoS ONE 2010, 5, e13989. [Google Scholar] [CrossRef] [Green Version]
- Eimer, W.A.; Kumar, D.K.V.; Shanmugam, N.K.N.; Rodriguez, A.S.; Mitchell, T.; Washicosky, K.J.; György, B.; Breakefield, X.O.; Tanzi, R.E.; Moir, R.D. Alzheimers Disease-Associated β-Amyloid Is Rapidly Seeded by Herpesviridae to Protect against Brain Infection. Neuron 2018, 99, 56–63. [Google Scholar] [CrossRef] [Green Version]
- Bocharova, O.; Pandit, N.P.; Molesworth, K.; Fisher, A.; Mychko, O.; Makarava, N.; Baskakov, I.V. Alzheimers disease-associated β-amyloid does not protect against herpes simplex virus 1 infection in the mouse brain. J. Biol. Chem. 2021, 297, 100845. [Google Scholar] [CrossRef]
- Davalos, D.; Grutzendler, J.; Yang, G.; Kim, J.V.; Zuo, Y.; Jung, S.; Littman, D.R.; Dustin, M.L.; Gan, W.B. ATP mediates rapid microglial response to local brain injury in vivo. Nat. Neurosci. 2005, 8, 752–758. [Google Scholar] [CrossRef] [PubMed]
- Au, N.P.B.; Ma, C.H.E. Recent Advances in the Study of Bipolar/Rod-Shaped Microglia and their Roles in Neurodegeneration. Front. Aging Neurosci. 2017, 9, 128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, Z.; Hussain, M.D.; Yan, L.J. Microglia, neuroinflammation, and beta-amyloid protein in Alzheimers disease. Int. J. Neurosci. 2014, 124, 307–321. [Google Scholar] [CrossRef] [PubMed]
- Griffin, W.S. Neuroinflammatory cytokine signaling and Alzheimers disease. N. Engl. J. Med. 2013, 368, 770–771. [Google Scholar] [CrossRef] [PubMed]
- Perego, C.; Fumagalli, S.; De Simoni, M.G. Temporal pattern of expression and colocalization of microglia/macrophage phenotype markers following brain ischemic injury in mice. J. Neuroinflamm. 2011, 8, 174. [Google Scholar] [CrossRef] [Green Version]
- Moyse, E.; Krantic, S.; Djellouli, N.; Roger, S.; Angoulvant, D.; Debacq, C.; Leroy, V.; Fougere, B.; Aidoud, A. Neuroinflammation: A Possible Link Between Chronic Vascular Disorders and Neurodegenerative Diseases. Front. Aging Neurosci. 2022, 14, 827263. [Google Scholar] [CrossRef]
- Krasemann, S.; Madore, C.; Cialic, R.; Baufeld, C.; Calcagno, N.; El Fatimy, R.; Beckers, L.; O’Loughlin, E.; Xu, Y.; Fanek, Z.; et al. The TREM2-APOE pathway drives the transcriptional phenotype of dysfunctional microglia in neurodegenerative diseases. Immunity 2017, 47, 566–581. [Google Scholar] [CrossRef] [Green Version]
- Davie, C.A. A review of Parkinson’s disease. Br. Med. Bull. 2008, 86, 109–127. [Google Scholar] [CrossRef] [Green Version]
- Woulfe, J.M.; Gray, M.T.; Gray, D.A.; Munoz, D.G.; and Middeldorp, J.M. Hypothesis: A Role for EBV-Induced Molecular Mimicry in Parkinson’s Disease. Parkinsonism Relat. Disord. 2014, 20, 685–694. [Google Scholar] [CrossRef]
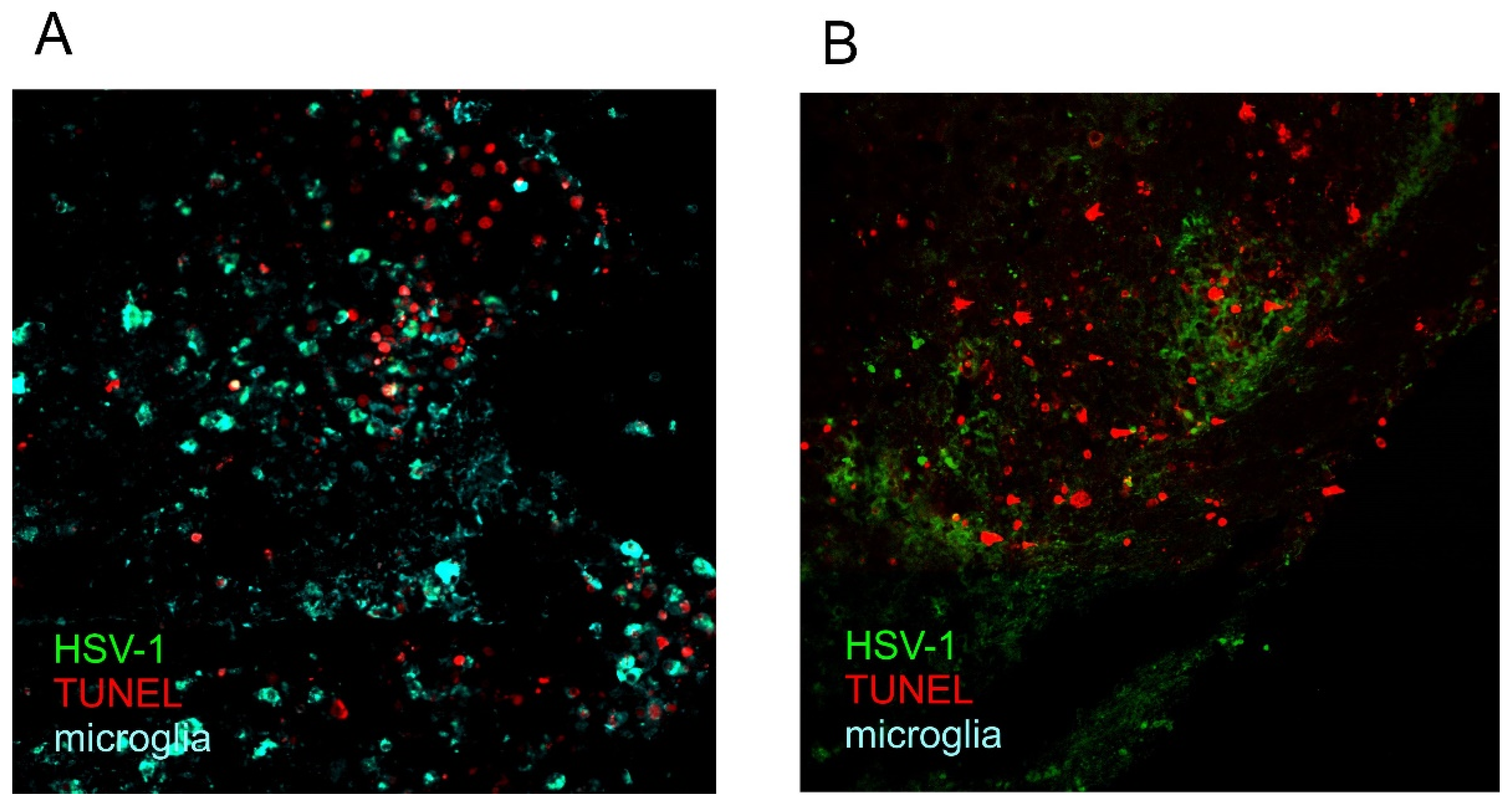
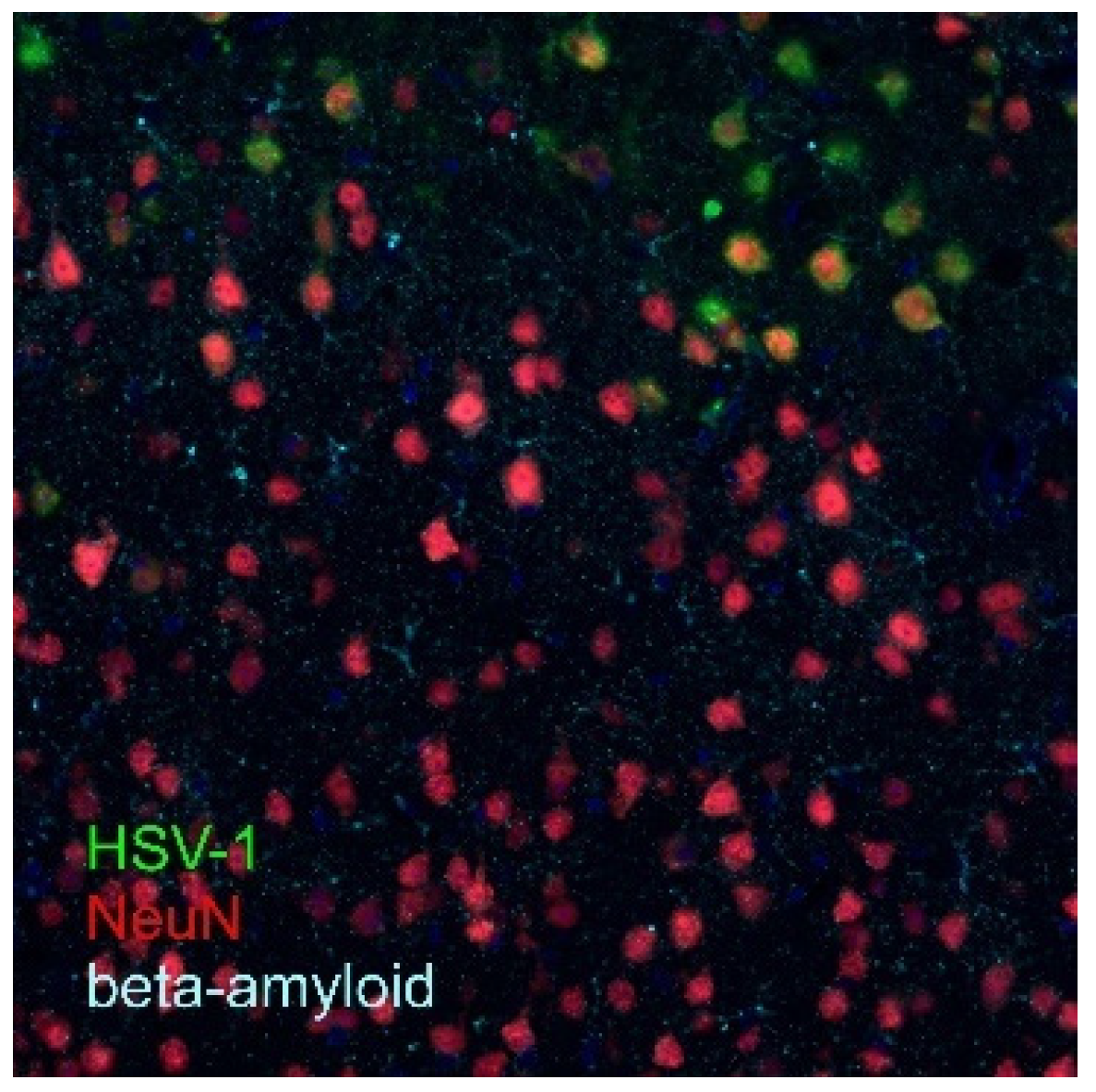
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Patrycy, M.; Chodkowski, M.; Krzyzowska, M. Role of Microglia in Herpesvirus-Related Neuroinflammation and Neurodegeneration. Pathogens 2022, 11, 809. https://doi.org/10.3390/pathogens11070809
Patrycy M, Chodkowski M, Krzyzowska M. Role of Microglia in Herpesvirus-Related Neuroinflammation and Neurodegeneration. Pathogens. 2022; 11(7):809. https://doi.org/10.3390/pathogens11070809
Chicago/Turabian StylePatrycy, Magdalena, Marcin Chodkowski, and Malgorzata Krzyzowska. 2022. "Role of Microglia in Herpesvirus-Related Neuroinflammation and Neurodegeneration" Pathogens 11, no. 7: 809. https://doi.org/10.3390/pathogens11070809
APA StylePatrycy, M., Chodkowski, M., & Krzyzowska, M. (2022). Role of Microglia in Herpesvirus-Related Neuroinflammation and Neurodegeneration. Pathogens, 11(7), 809. https://doi.org/10.3390/pathogens11070809