
First Report of a Complete Genome Sequence of a Variant African Swine Fever Virus in the Mekong Delta, Vietnam

 , , and
, , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
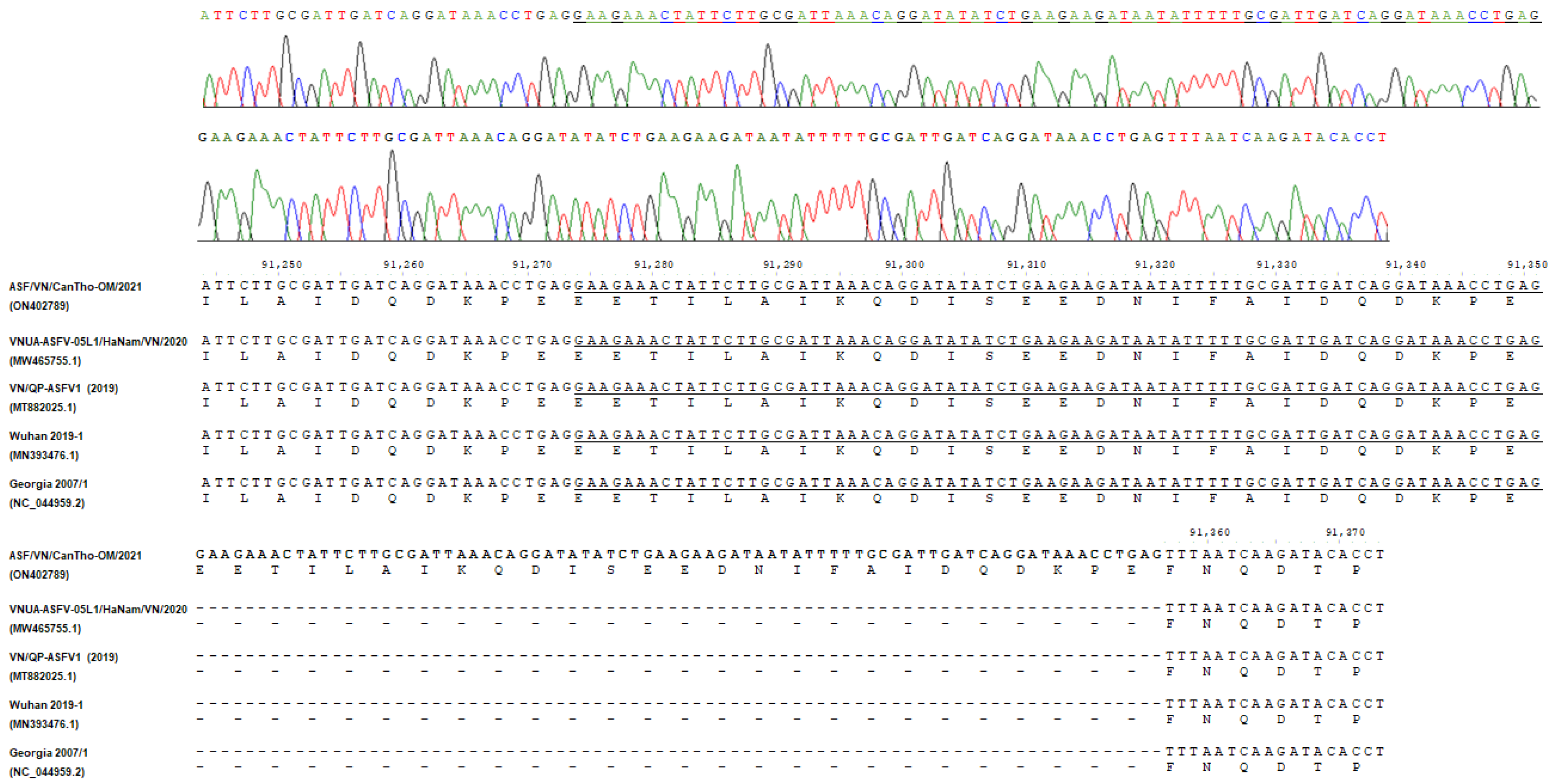
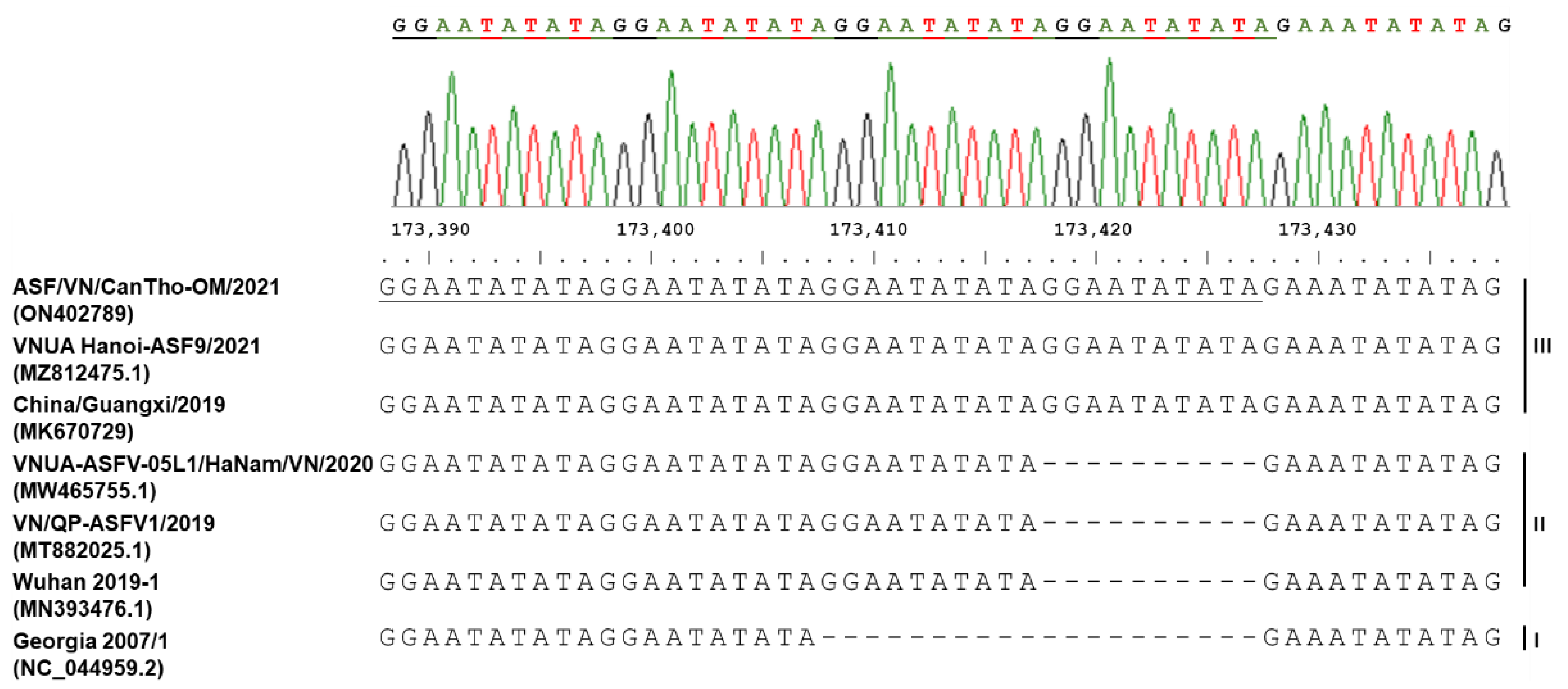
2. Results
3. Discussion
4. Materials and Methods
4.1. Sample Collection and Sampling Site
4.2. Whole-Genome Sequencing
4.3. DNA Alignment and Phylogenetic Analysis
4.4. PCR Amplification and Sanger Sequencing
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Costard, S.; Wieland, B.; de Glanville, W.; Jori, F.; Rowlands, R.; Vosloo, W.; Roger, F.; Pfeiffer, D.U.; Dixon, L.K. African swine fever: How can global spread be prevented? Philos. Trans. R. Soc. Lond. B Biol. Sci. 2009, 364, 2683–2696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sánchez-Vizcaíno, J.M.; Mur, L.; Martínez-López, B. African swine fever: An epidemiological update. Transbound. Emerg. Dis. 2012, 59, 27–35. [Google Scholar] [CrossRef] [PubMed]
- Dixon, L.K.; Chapman, D.A.; Netherton, C.L.; Upton, C. African swine fever virus replication and genomics. Virus Res. 2013, 173, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Alejo, A.; Matamoros, T.; Guerra, M.; Andrés, G. A proteomic atlas of the African swine fever virus particle. J. Virol. 2018, 92, e01293-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bastos, A.D.; Penrith, M.L.; Crucière, C.; Edrich, J.L.; Hutchings, G.; Roger, F.; Couacy-Hymann, E.; Thomson, G.R. Genotyping field strains of African swine fever virus by partial p72 gene characterisation. Arch. Virol. 2003, 148, 693–706. [Google Scholar] [CrossRef] [PubMed]
- Montgomery, R.E. On a form of swine fever occurring in British East Africa (Kenya Colony). J. Comp. Pathol. Ther. 1921, 34, 159–191. [Google Scholar] [CrossRef] [Green Version]
- Cwynar, P.; Stojkov, J.; Wlazlak, K. African swine fever status in Europe. Viruses 2019, 11, 310. [Google Scholar] [CrossRef] [Green Version]
- EFSA (European Food Safety Authority); Baños, J.V.; Boklund, A.; Gogin, A.; Gortázar, C.; Guberti, V.; Helyes, G.; Kantere, M.; Korytarova, D.; Linden, A. Epidemiological analyses of African swine fever in the European Union: (September 2020 to August 2021). EFSA J. 2022, 20, e07290. [Google Scholar] [CrossRef]
- Mighell, E.; Ward, M.P. African swine fever spread across Asia, 2018–2019. Transbound. Emerg. Dis. 2021, 68, 2722–2732. [Google Scholar] [CrossRef]
- Van Phan, L.; Dae Gwin, J.; Sun-Woo, Y.; Hye-Min, K.; Thi Bich Ngoc, T.; Thi Lan, N.; Thi To Nga, B.; Jinsik, O.; Joon Bae, K.; Kwang Myun, C.; et al. Outbreak of African swine fever, Vietnam, 2019. Emerg. Infect. Dis. 2019, 25, 1433–1435. [Google Scholar] [CrossRef]
- Mai, N.T.A.; Vu, X.D.; Nguyen, T.T.H.; Nguyen, V.T.; Trinh, T.B.N.; Kim, Y.J.; Kim, H.J.; Cho, K.H.; Nguyen, T.L.; Bui, T.T.N.; et al. Molecular profile of African swine fever virus (ASFV) circulating in Vietnam during 2019-2020 outbreaks. Arch. Virol. 2021, 166, 885–890. [Google Scholar] [CrossRef] [PubMed]
- Tran, H.T.T.; Truong, A.D.; Dang, A.K.; Ly, D.V.; Nguyen, C.T.; Chu, N.T.; Nguyen, H.T.; Dang, H.V. Genetic characterization of African swine fever viruses circulating in North Central region of Vietnam. Transbound. Emerg. Dis. 2021, 68, 1697–1699. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, V.T.; Cho, K.-h.; Mai, N.T.A.; Park, J.-Y.; Trinh, T.B.N.; Jang, M.-K.; Nguyen, T.T.H.; Vu, X.D.; Nguyen, T.L.; Nguyen, V.D. Multiple variants of African swine fever virus circulating in Vietnam. Arch. Virol. 2022, 167, 1137–1140. [Google Scholar] [CrossRef]
- Tran, H.T.T.; Truong, A.D.; Dang, A.K.; Ly, D.V.; Nguyen, C.T.; Chu, N.T.; Hoang, T.V.; Nguyen, H.T.; Dang, H.V. Circulation of two different variants of intergenic region (IGR) located between the I73R and I329L genes of African swine fever virus strains in Vietnam. Transbound. Emerg. Dis. 2021, 68, 2693–2695. [Google Scholar] [CrossRef]
- Chen, S.-N.; Li, C.-L.; Lin, J.-S.; Zhai, S.-L.; Sun, M.-F. Diverse African swine fever viruses in China. New Microbes New Infect. 2022, 46, 100976. [Google Scholar] [CrossRef]
- Malogolovkin, A.; Burmakina, G.; Titov, I.; Sereda, A.; Gogin, A.; Baryshnikova, E.; Kolbasov, D. Comparative analysis of African swine fever virus genotypes and serogroups. Emerg. Infect. Dis. 2015, 21, 312–315. [Google Scholar] [CrossRef] [PubMed]
- Ge, S.; Liu, Y.; Li, L.; Wang, Q.; Li, J.; Ren, W.; Liu, C.; Bao, J.; Wu, X.; Wang, Z. An extra insertion of tandem repeat sequence in African swine fever virus, China, 2019. Virus Genes 2019, 55, 843–847. [Google Scholar] [CrossRef]
- Kim, S.H.; Lee, S.I.; Jeong, H.G.; Yoo, J.; Jeong, H.; Choi, Y.; Son, K.; Jheong, W.H. Rapid emergence of African swine fever virus variants with different numbers of a tandem repeat sequence in South Korea. Transbound. Emerg. Dis. 2021, 68, 1726–1730. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. Fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [Green Version]
- Wick, R.R.; Judd, L.M.; Gorrie, C.L.; Holt, K.E. Unicycler: Resolving bacterial genome assemblies from short and long sequencing reads. PLoS Comput. Biol. 2017, 13, e1005595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paradis, E.; Claude, J.; Strimmer, K. APE: Analyses of phylogenetics and evolution in R language. Bioinformatics 2004, 20, 289–290. [Google Scholar] [CrossRef] [Green Version]
- Heibl, C.; Cusimano, N.; Krah, F.-S.; Heibl, M.C. Package ‘ips’, version 0.0.11. 2019. Available online: http://www.christophheibl.de/Rpackages.html (accessed on 1 May 2022).
- Bodenhofer, U.; Bonatesta, E.; Horejš-Kainrath, C.; Hochreiter, S. msa: An R package for multiple sequence alignment. Bioinformatics 2015, 31, 3997–3999. [Google Scholar] [CrossRef]
- Hall, T. BioEdit: An important software for molecular biology. GERF Bull. Biosci. 2011, 2, 60–61. [Google Scholar]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hien, N.D.; Nguyen, L.T.; Hoang, L.T.; Bich, N.N.; Quyen, T.M.; Isoda, N.; Sakoda, Y. First Report of a Complete Genome Sequence of a Variant African Swine Fever Virus in the Mekong Delta, Vietnam. Pathogens 2022, 11, 797. https://doi.org/10.3390/pathogens11070797
Hien ND, Nguyen LT, Hoang LT, Bich NN, Quyen TM, Isoda N, Sakoda Y. First Report of a Complete Genome Sequence of a Variant African Swine Fever Virus in the Mekong Delta, Vietnam. Pathogens. 2022; 11(7):797. https://doi.org/10.3390/pathogens11070797
Chicago/Turabian StyleHien, Nguyen Duc, Lam Thanh Nguyen, Le Trung Hoang, Nguyen Ngoc Bich, To My Quyen, Norikazu Isoda, and Yoshihiro Sakoda. 2022. "First Report of a Complete Genome Sequence of a Variant African Swine Fever Virus in the Mekong Delta, Vietnam" Pathogens 11, no. 7: 797. https://doi.org/10.3390/pathogens11070797
APA StyleHien, N. D., Nguyen, L. T., Hoang, L. T., Bich, N. N., Quyen, T. M., Isoda, N., & Sakoda, Y. (2022). First Report of a Complete Genome Sequence of a Variant African Swine Fever Virus in the Mekong Delta, Vietnam. Pathogens, 11(7), 797. https://doi.org/10.3390/pathogens11070797