
Phylogenomics and Diversification of the Schistosomatidae Based on Targeted Sequence Capture of Ultra-Conserved Elements

 , , , ,
, , , , 
Abstract
1. Introduction
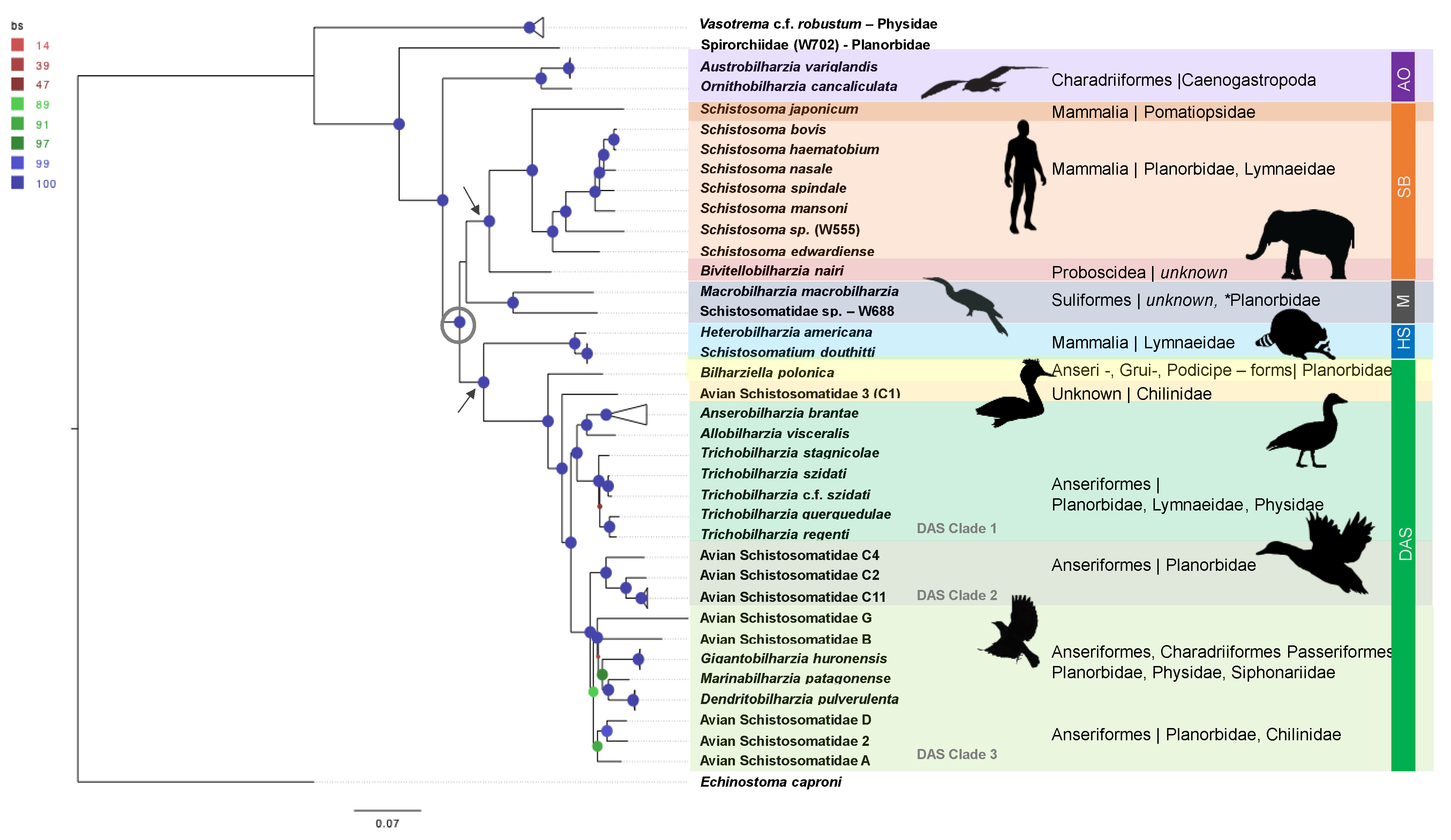
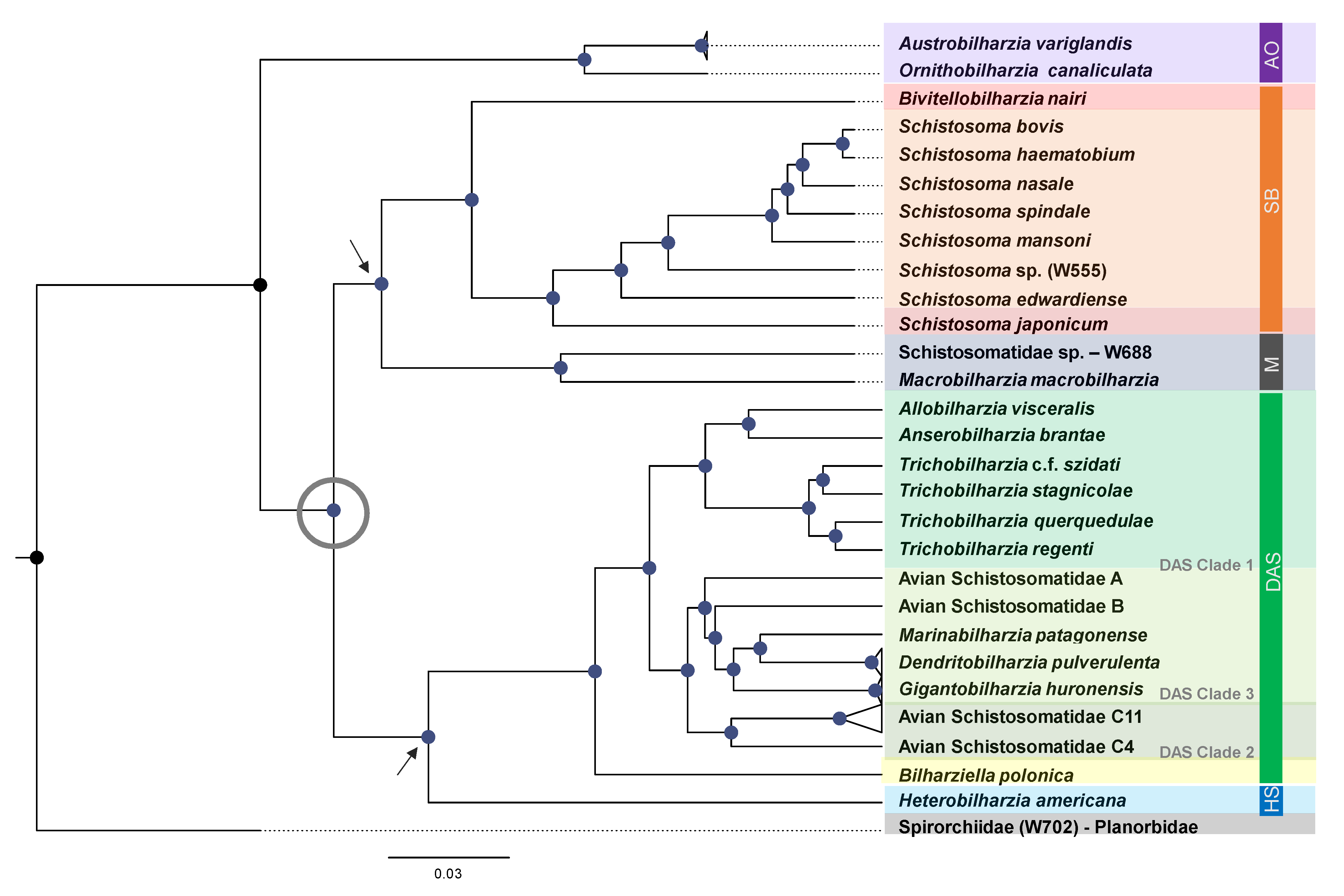
- AO Clade: Marine avian genera, Austrobilharzia and Ornithobilharzia (AO clade), are often recovered as well-supported sister genera [36,39,40], and both are considered globally distributed. In most of the studies over the last 10 years, the AO clade typically acts as a sister group to the remainder of the schistosomatids.
- SB Clade: Species of the Afro-Eurasian mammalian clade (Schistosoma + Bivitellobilharzia, or the SB clade) are found in tropical and sub-tropical latitudes. Schistosoma and Bivitellobilharzia are considered probable sister genera, but the use of traditional loci often does not statistically support this grouping. A better understanding of relationships within the SB clade and its placement within Schistosomatidae is essential to understanding the evolution of human schistosomatids.
- Macrobilharzia:Macrobilharzia is a monotypic genus with species that infect Anhinga anhinga in the Americas and has failed to group consistently with other schistosomatid lineages. Some rRNA phylogenies suggest an (unsupported) affinity with the SB clade, suggesting the possibility of the SB clade having had Afro-Eurasian avian-infecting ancestors. Cercariae (Schistosomatidae-W688) recovered from the freshwater snail Indoplanorbis exustus in Nepal [41] represent an otherwise undescribed, but well-supported, sister lineage to M. macrobilharzia.
- DAS Clade: The derived avian–schistosomatid (DAS) clade includes the majority of avian-infecting genera (Anserobilharzia, Allobilharzia, Bilharziella, Dendritobilharzia, Gigantobilharzia, Marinabilharzia, Nasusbilharzia, Riverabilharzia, Trichobilharzia) and several yet-to-be described genera. The monophyly of DAS is consistently supported (Table 1, [39,40]), though its placement as a sister to the SB clade or North American mammalian schistosomatids is unclear. The relationships within the DAS clade are largely unresolved.
- HS Clade:Heterobilharzia and Schistosomatium (HS clade) are both monotypic genera whose species infect North American mammals and form a well-supported clade in most studies (Table 1). Phylogenies based on oft-used loci have provided weak support for the placement of the HS clade as a sister to the DAS clade (see Table 1). Phylogenetic placement of the HS clade has significant implications for understanding the role of host shifts and biogeography of the Schistosomatidae.
2. Results
2.1. UCE Enrichment and Sequencing
2.2. Phylogenetic Reconstruction
2.2.1. Supermatrix Alignment

2.2.2. Phylogenies
2.3. UCE Loci Sequence Similarity among Blood Flukes
3. Discussion
3.1. Resolution of Pivotal Nodes and Host Divergences
3.1.1. AO Clade
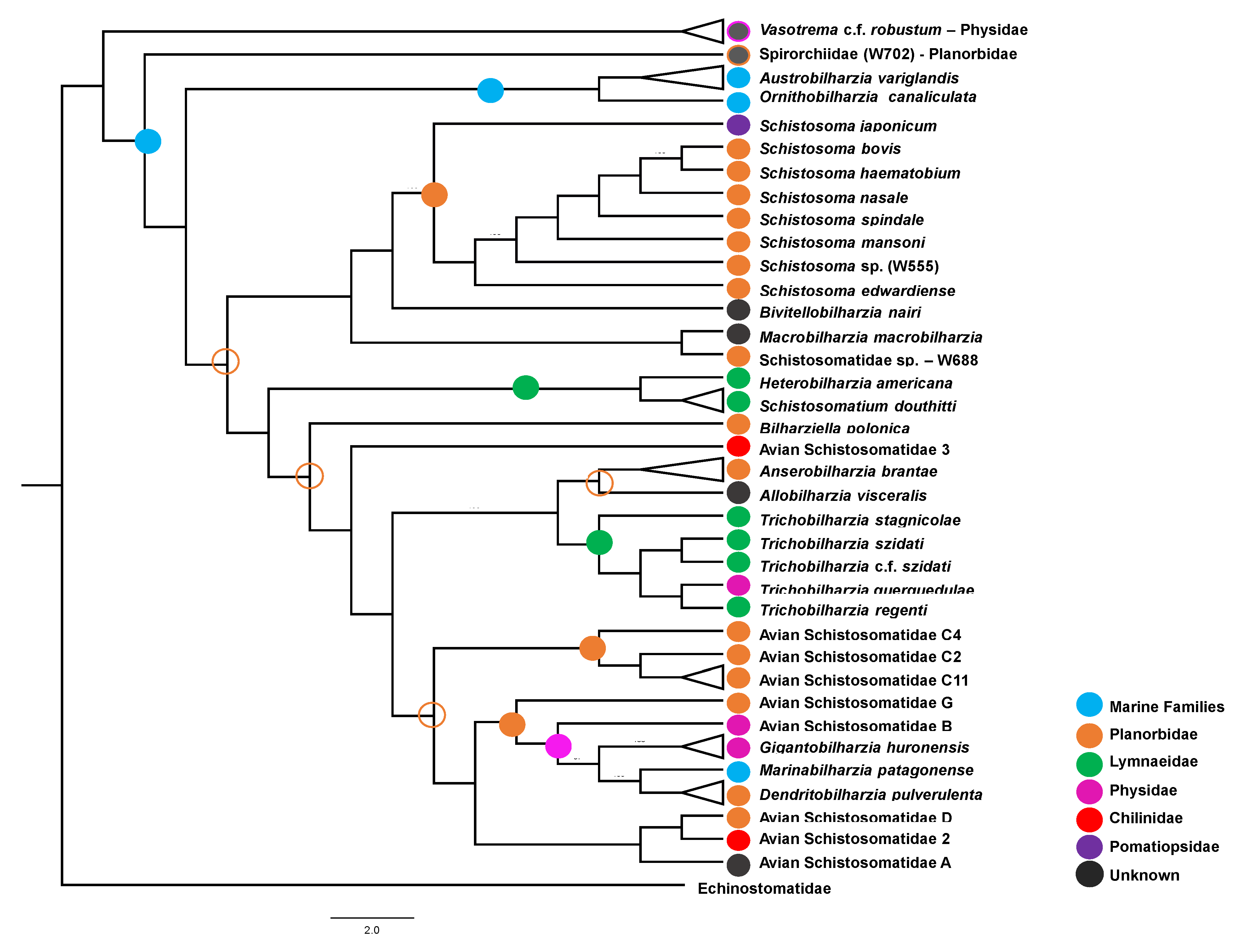
3.1.2. Hypothesized Early Gastropod Host Use Prior to the (M + SB) and (HS +DAS) Split
3.1.3. SB Clade
3.1.4. Macrobilharzia (M Clade)
3.1.5. DAS Clade
- Clade 1: Trichobilharzia, Allobilharzia and Anserobilharzia form a well-supported sub-clade within DAS, reported exclusively from Anseriformes, predominantly ducks. Published single loci studies do not consistently resolve the node connecting Trichobilharzia and Allobilharzia + Anserobilharzia [28,82,83,84]. This is a logical grouping as this diverse radiation appears to have occurred in Anseriformes [28,83,84], and Trichobilharzia demonstrates a strong association with anatids. The diversification of modern Anatidae has been estimated to have occurred between 25–5 mya [85], which might be used as a hard bound for the radiation of Trichobilharzia.
- Clade 2: A second sub-clade within DAS is composed of several lineages retaining the names used from previous studies, Avian Schistosomatidae C (C2, C4, and C11 were sampled, [31]). Avian Schistosomatidae C is a complex of several species distributed globally (Americas, Europe, and Africa), none of which have yet been formally named [31].
- Clade 3: Gigantobilharzia, Dendritobilharzia, and six undescribed genera form Clade 3. Branch lengths within this clade, specifically the distance between tips, are large, which may suggest missing taxa. Remarkably, the taxa in Clade 3 (Figure 1) are hosted by at least four different snail families (including one marine family) and three orders of birds (Passeriformes, Charadriiformes, and Anseriformes) recovered from Argentina, Kenya, and across North America.
3.1.6. HS Clade
3.2. Diversification via Extensive Intermediate Host-Switching
3.3. Phylogenomic Considerations and Future Directions
3.3.1. Phylogenomics of Schistosomatids
3.3.2. Guiding Future Collection Efforts
4. Materials and Methods
4.1. Taxon Sampling
4.2. Sample Preparation
4.3. Sequence Capture of Ultra-Conserved Elements
4.4. Processing and Alignment of UCE Data
4.5. Alignment Building and Phylogenetic Reconstruction
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hoberg, E.P.; Brooks, D.R. A macroevolutionary mosaic: Episodic host-switching, geographical colonization and diversification in complex host-parasite systems. J. Biogeogr. 2008, 35, 1533–1550. [Google Scholar] [CrossRef]
- Hay, E.M.; Poulin, R.; Jorge, F. Macroevolutionary dynamics of parasite diversification: A reality check. J. Evol. Biol. 2020, 33, 1758–1769. [Google Scholar] [CrossRef] [PubMed]
- Glodosky, C.M.; Sandland, G.J. Assessing host competency between native and invasive snail species exposed to the native parasite Echinostoma revolutum. Aquat. Invasions 2014, 9, 1. [Google Scholar] [CrossRef]
- Ricklefs, R.E.; Fallon, S.M.; Bermingham, E. Evolutionary relationships, cospeciation, and host switching in avian malaria parasites. Syst. Biol. 2004, 53, 111–119. [Google Scholar] [CrossRef]
- Malcicka, M.; Agosta, S.J.; Harvey, J.A. Multi-level ecological fitting: Indirect life cycles are not a barrier to host switching and invasion. Glob. Chang. Biol. 2015, 21, 3210–3218. [Google Scholar] [CrossRef] [PubMed]
- Martin, S.B.; Sasal, P.; Cutmore, S.C.; Ward, S.; Aeby, G.S.; Cribb, T.H. Intermediate host switches drive diversification among the largest trematode family: Evidence from the Polypipapiliotrematinae n. subf. (Opecoelidae), parasites transmitted to butterflyfishes via predation of coral polyps. Int. J. Parasitol. 2018, 48, 1107–1126. [Google Scholar] [CrossRef] [PubMed]
- Jiménez, A.F.; Notarnicola, J.; Gardner, S.L. Host-switching events in Litomosoides Chandler, 1931 (Filarioidea: Onchocercidae) are not rampant but clade dependent. J. Parasitol. 2021, 107, 320–335. [Google Scholar] [CrossRef]
- Jouet, D.; Skírnisson, K.; Kolářová, L.; Ferté, H. Molecular diversity of Trichobilharzia franki in two intermediate hosts (Radix auricularia and Radix peregra): A complex of species. Inf. Gene. Evol. 2010, 10, 1218–1227. [Google Scholar] [CrossRef]
- Detwiler, J.T.; Bos, D.H.; Minchella, D.J. Revealing the secret lives of cryptic species: Examining the phylogenetic relationships of echinostome parasites in North America. Mol. Phylogenetics Evol. 2010, 55, 611–620. [Google Scholar] [CrossRef]
- Ebbs, E.T.; Loker, E.S.; Davis, N.E.; Flores, V.R.; Veleizan, A.; Brant, S.V. Schistosomes with wings: How host phylogeny and ecology shape the global distribution of Trichobilharzia querquedulae (Schistosomatidae). Int. J. Parasitol. 2016, 46, 669–677. [Google Scholar] [CrossRef]
- Kasl, E.L.; Font, W.F.; Criscione, C.D. Resolving evolutionary changes in parasite life cycle complexity: Molecular phylogeny of the trematode genus Alloglossidium indicates more than one origin of precociousness. Mol. Phylogenetics Evol. 2018, 126, 371–381. [Google Scholar] [CrossRef] [PubMed]
- Locke, S.A.; Al-Nasiri, F.S.; Caffara, M.; Drago, F.; Kalbe, M.; Lapierre, A.R.; McLaughlin, J.D.; Nie, P.; Overstreet, R.M.; Souza, G.T.; et al. Diversity, specificity, and speciation in larval Diplostomidae (Platyhelminthes: Digenea) in the eyes of freshwater fish, as revealed by DNA barcodes. Int. J. Parasitol. 2015, 45, 841–855. [Google Scholar] [CrossRef] [PubMed]
- Van Steenberge, M.; Pariselle, A.; Huyse, T.; Volckaert, F.A.; Snoeks, J.; Vanhove, M.P. Morphology, molecules, and monogenean parasites: An example of an integrative approach to cichlid biodiversity. PLoS ONE 2015, 10, 4. [Google Scholar] [CrossRef] [PubMed]
- Tchuem Tchuenté, L.-A.; Rollinson, D.; Stothard, J.R.; Molyneux, D. Moving from control to elimination of schistosomiasis in sub-Saharan Africa: Time to change and adapt strategies. Infect. Dis. Poverty 2017, 6, 42. [Google Scholar] [CrossRef]
- Klohe, K.; Koudou, B.G.; Fenwick, A.; Fleming, F.; Garba, A.; Gouvras, A.; Harding-Esch, E.M.; Knopp, S.; Molyneux, D.; D’Souza, S.; et al. A systematic literature review of schistosomiasis in urban and peri-urban settings. PLoS Neg. Trop. Dis. 2021, 15, e0008995. [Google Scholar] [CrossRef]
- Horák, P.; Mikeš, L.; Lichtenbergová, L.; Skála, V.; Soldánová, M.; Brant, S.V. Avian schistosomes and outbreaks of cercarial dermatitis. Clin. Microbiol. Rev. 2015, 28, 165–190. [Google Scholar] [CrossRef]
- Lashaki, E.K.; Teshnizi, S.H.; Gholami, S.; Fakhar, M.; Brant, S.V.; Dodangeh, S. Global prevalence status of avian schistosomes: A systematic review with meta-analysis. Parasit. Epidem. Control. 2020, 9, e00142. [Google Scholar] [CrossRef]
- Platt, T.R.; Brooks, D.R. Evolution of schistosomes (Digenea: Schistosomatoidea): The origin of dioecy and colonization of the venous system. J. Parasitol. 1997, 83, 1035–1044. [Google Scholar] [CrossRef][Green Version]
- Loker, E.S.; Brant, S.V. Diversification, dioecy and dimorphism in schistosomes. Trend. Parasitol. 2006, 22, 521–528. [Google Scholar] [CrossRef]
- Morand, S.; Müller-Graf, C.D.M. Muscles or testes? Comparative evidence for sexual competition among dioecious blood parasites (Schistosomatidae) of vertebrates. Parasitology 2000, 120, 45–56. [Google Scholar] [CrossRef]
- Brant, S.V.; Loker, E.S. Discovery-based studies of schistosome diversity stimulate new hypotheses about parasite biology. Trend. Parasitol. 2013, 29, 449–459. [Google Scholar] [CrossRef]
- Farley, J.A. review of the family Schistosomatidae: Excluding the genus Schistosoma from mammals. J. Helm. 1971, 45, 289–320. [Google Scholar] [CrossRef] [PubMed]
- Carmichael, A.C. Phylogeny and Historical Biogeography of the Schistosomatidae; Michigan State University, Department of Zoology: East Lansing, MI, USA, 1984. [Google Scholar]
- Khalil, L.F. Family Schistosomatidae Stiles & Hassall, 1898. In Keys to the Trematoda; CABI Publishing: Cambridge, MA, USA, 2002; Volume 1, pp. 419–432. [Google Scholar]
- Snyder, S.D.; Loker, E.S. Evolutionary relationships among the Schistosomatidae (Platyhelminthes: Digenea) and an Asian origin for Schistosoma. J. Parasitol. 2000, 86, 283–288. [Google Scholar] [CrossRef]
- Lockyer, A.E.; Olson, P.D.; Østergaard, P.; Rollinson, D.; Johnston, D.A.; Attwood, S.W.; Southgate, V.R.; Horák, P.; Snyder, S.D.; Le, T.H.; et al. The phylogeny of the Schistosomatidae based on three genes with emphasis on the interrelationships of Schistosoma Weinland, 1858. Parasitology 2003, 126, 203–224. [Google Scholar] [CrossRef] [PubMed]
- Brant, S.V.; Morgan, J.A.T.; Mkoji, G.M.; Snyder, S.D.; Rajapakse, R.P.V.J.; Loker, E.S. An approach to revealing blood fluke life cycles, taxonomy, and diversity: Provision of key reference data including DNA sequence from single life cycle stages. J. Parasitol. 2006, 92, 77–88. [Google Scholar] [CrossRef] [PubMed]
- Brant, S.V.; Loker, E.S. Molecular systematics of the avian schistosome genus Trichobilharzia (Trematoda: Schistosomatidae) in North America. J. Parasitol. 2009, 95, 941–963. [Google Scholar] [CrossRef]
- Flores, V.R.; Brant, S.V.; Loker, E.S. Avian schistosomes from the South American endemic gastropod genus Chilina (Pulmonata: Chilinidae), with a brief review of South American schistosome species. J. Parasitol. 2015, 101, 565–576. [Google Scholar] [CrossRef]
- Brant, S.V.; Loker, E.S.; Casalins, L.; Flores, V.R. Phylogenetic placement of a schistosome from an unusual marine snail host, the false limpet (Siphonaria lessoni) and gulls (Larus dominicanus) from Argentina with a brief review of marine schistosomes from snails. J. Parasitol. 2017, 103, 75–82. [Google Scholar] [CrossRef]
- Pinto, H.A.; Pulido-Murillo, E.A.; de Melo, A.L.; Brant, S.V. Putative new genera and species of avian schistosomes potentially involved in human cercarial dermatitis in the Americas, Europe and Africa. Acta Trop. 2017, 176, 415–420. [Google Scholar] [CrossRef]
- Gordy, M.A.; Hanington, P.C. A fine-scale phylogenetic assessment of digenean trematodes in central Alberta reveals we have yet to uncover their total diversity. Ecol. Evol. 2019, 9, 3153–3238. [Google Scholar] [CrossRef]
- Flores, V.R.; Viozzi, G.; Casalins, L.; Loker, E.S.; Brant, S.V. A new schistosome (Digenea: Schistosomatidae) from the nasal tissue of South America black-necked swans, Cygnus melancoryphus (Anatidae) and the endemic pulmonate snail Chilina gibbosa. Zootaxa 2021, 4948, 404–418. [Google Scholar] [CrossRef] [PubMed]
- McPhail, B.A.; Rudko, S.P.; Turnbull, A.; Gordy, M.A.; Reimink, R.L.; Clyde, D.; Froelich, K.; Brant, S.V.; Hannington, P.C. Evidence of a putative novel species of avian schistosome infecting Planorbella trivolvis. J. Parasitol. 2021, 107, 89–97. [Google Scholar] [CrossRef] [PubMed]
- Lorenti, E.; Brant, S.V.; Gilardoni, C.; Diaz, J.I.; Cremonte, F. Two new genera and species of avian schistosomes from Argentina with proposed recommendations and discussion of the polyphyletic genus Gigantobilharzia (Trematoda, Schistosomatidae). Parasitology 2022, 149, 1–20. [Google Scholar] [CrossRef]
- Brant, S.V.; Loker, E.S. Can specialized pathogens colonize distantly related hosts? Schistosome evolution as a case study. PLoS Pathog. 2005, 1, e38. [Google Scholar] [CrossRef] [PubMed]
- Webster, B.L.; Littlewood, D.T.J. Mitochondrial gene order change in Schistosoma (Platyhelminthes: Digenea: Schistosomatidae). Int. J. Parasitol. 2012, 42, 313–321. [Google Scholar] [CrossRef]
- Schuster, R.K.; Aldhoun, J.A.; O’Donovan, D. Gigantobilharzia melanoidis n. sp. (Trematoda: Schistosomatidae) from Melanoides tuberculata (Gastropoda: Thiaridae) in the United Arab Emirates. Parasitol. Res. 2014, 113, 959–972. [Google Scholar] [CrossRef]
- Khosravi, M.; Thieltges, D.W.; Shamseddin, J.; Georgieva, S. Schistosomes from the Persian Gulf: Phylogenetic Relationships, Host Associations and Life-Cycle Elucidation of Ornithobilharzia canaliculata (Rudolphi, 1819) Odhner, 1912. 2022. Available online: https://assets.researchsquare.com/files/rs-1387572/v2/5451effd-2741-4666-8ee5-7cb9fb843712.pdf?c=1647956870 (accessed on 30 June 2022).
- Oyarzún-Ruiz, P.; Thomas, R.; Santodomingo, A.; Collado, G.; Muñoz, P.; Moreno, L. Morphological, behavioral, and molecular characterization of avian schistosomes (Digenea: Schistosomatidae) in the native snail Chilina dombeyana (Chilinidae) from Southern Chile. Pathogens 2022, 11, 332. [Google Scholar] [CrossRef]
- Devkota, R.; Brant, S.V.; Thapa, S.; Loker, E.S. Two avian schistosome cercariae from Nepal, including a Macrobilharzia-like species from Indoplanorbis exustus. Parasitol. Int. 2014, 63, 374–380. [Google Scholar] [CrossRef]
- Maddison, W.P.; Knowles, L.L. Inferring phylogeny despite incomplete lineage sorting. Syst. Bio. 2006, 55, 21–30. [Google Scholar] [CrossRef]
- Pease, J.B.; Hahn, M.W. More accurate phylogenies inferred from low-recombination regions in the presence of incomplete lineage sorting. Evolution 2013, 67, 2376–2384. [Google Scholar] [CrossRef]
- Suh, A.; Smeds, L.; Ellegren, H. The Dynamics of incomplete lineage sorting across the ancient adaptive radiation of neoavian birds. PLoS Biol. 2015, 13, e1002224. [Google Scholar] [CrossRef] [PubMed]
- Jones, B.P.; Norman, B.F.; Borrett, H.E.; Attwood, S.W.; Mondal, M.M.; Walker, A.J.; Webster, J.P.; Rajapakse, R.P.V.; Lawton, S.P. Divergence across mitochondrial genomes of sympatric members of the Schistosoma indicum group and clues into the evolution of Schistosoma spindale. Sci. Rep. 2020, 10, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Brabec, J.; Kostadinova, A.; Scholz, T.; Littlewood, D.T.J. Complete mitochondrial genomes and nuclear ribosomal RNA operons of two species of Diplostomum (Platyhelminthes: Trematoda): A molecular resource for taxonomy and molecular epidemiology of important fish pathogens. Parasit. Vec. 2015, 8, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Locke, S.A.; Van Dam, A.; Caffara, M.; Pinto, H.A.; Lopez-Hernandez, D.; Blanar, C.A. Validity of the Diplostomoidea and Diplostomida (Digenea, Platyhelminthes) upheld in phylogenomic analysis. Int. J. Parasitol. 2018, 48, 1043–1059. [Google Scholar] [CrossRef] [PubMed]
- McCormack, J.E.; Harvey, M.G.; Faircloth, B.C.; Crawford, N.G.; Glenn, T.C.; Brumfield, R.T. A phylogeny of birds based on over 1500 loci collected by target enrichment and high-throughput sequencing. PLoS ONE 2013, 8, e54848. [Google Scholar] [CrossRef] [PubMed]
- Grégoir, A.F.; Hablützel, P.I.; Vanhove, M.P.M.; Pariselle, A.; Bamps, J.; Volckaert, F.A.M.; Raeymaekers, J.A.M. A link between host dispersal and parasite diversity in two sympatric cichlids of Lake Tanganyika. Freshw. Biol. 2015, 60, 323–335. [Google Scholar] [CrossRef]
- Faircloth, B.C.; McCormack, J.E.; Crawford, N.G.; Harvey, M.G.; Brumfield, R.T.; Glenn, T.C. Ultraconserved elements anchor thousands of genetic markers spanning multiple evolutionary timescales. Syst. Biol. 2012, 61, 717–726. [Google Scholar] [CrossRef]
- Faircloth, B.C.; Sorenson, L.; Santini, F.; Alfaro, M.E. A Phylogenomic perspective on the radiation of ray-finned fishes based upon targeted sequencing of ultraconserved elements (UCEs). PLoS ONE 2013, 8, e65923. [Google Scholar] [CrossRef]
- Esselstyn, J.A.; Oliveros, C.H.; Swanson, M.T.; Faircloth, B.C. Investigating difficult nodes in the placental mammal tree with expanded taxon sampling and thousands of ultraconserved elements. Genome Biol. Evol. 2017, 9, 2308–2321. [Google Scholar] [CrossRef]
- Prum, R.O.; Berv, J.S.; Dornburg, A.; Field, D.J.; Townsend, J.P.; Lemmon, E.M.; Lemmon, A.R. A comprehensive phylogeny of birds (Aves) using targeted next-generation DNA sequencing. Nature 2015, 526, 569–573. [Google Scholar] [CrossRef]
- Parada, A.; Hanson, J.; D’Eiía, G. Ultraconserved Elements improve the resolution of difficult nodes within the rapid radiation of neotropical sigmodontine rodents (Cricetidae: Sigmodontinae). Syst. Biol. 2021, 70, 1090–1100. [Google Scholar] [CrossRef] [PubMed]
- Blaimer, B.B.; Brady, S.G.; Schultz, T.R.; Lloyd, M.W.; Fisher, B.L.; Ward, P.S. Phylogenomic methods outperform traditional multi-locus approaches in resolving deep evolutionary history: A case study of formicine ants. BMC Evol. Biol. 2015, 15, 271. [Google Scholar] [CrossRef] [PubMed]
- Faircloth, B.C.; Branstetter, M.G.; White, N.D.; Brady, S.G. Target enrichment of ultraconserved elements from arthropods provides a genomic perspective on relationships among Hymenoptera. Mol. Ecol. Res. 2015, 15, 489–501. [Google Scholar] [CrossRef] [PubMed]
- Blaimer, B.B.; LaPolla, J.S.; Branstetter, M.G.; Lloyd, M.W.; Brady, S.G. Phylogenomics, biogeography and diversification of obligate mealybug-tending ants in the genus Acropyga. Mol. Phylo. Evol. 2016, 102, 20–29. [Google Scholar] [CrossRef] [PubMed]
- Branstetter, M.G.; Danforth, B.N.; Pitts, J.P.; Faircloth, B.C.; Ward, P.S.; Buffington, M.L.; Gates, M.W.; Kula, R.R.; Brady, S.G. Phylogenomic analysis of ants, bees, and stinging wasps: Improved taxon sampling enhances understanding of hymenopteran Evolution. bioRxiv 2016, 068957. [Google Scholar] [CrossRef]
- Blaimer, B.B.; Lloyd, M.W.; Guillory, W.X.; Brady, S.G. Sequence capture and phylogenetic utility of genomic ultraconserved elements obtained from pinned insect specimens. PLoS ONE 2016, 11, e0161531. [Google Scholar] [CrossRef]
- Stamatakis, A.; Hoover, P.; Rougemont, J. A rapid bootstrap algorithm for the RAxML web servers. Syst. Biol. 2008, 57, 758–771. [Google Scholar] [CrossRef]
- Bullard, S.A.; Roberts, J.R.; Warren, M.B.; Dutton, H.; Whelan, N.V.; Ruiz, C.F.; Platt, T.R.; Tkach, V.V.; Brant, S.V.; Halanych, K.M. Neotropical turtle blood flukes: Two new genera and species from the Amazon river basin with a key to genera and comments on a marine-derived parasite lineage in South America. J. Parasitol. 2019, 105, 497–523. [Google Scholar] [CrossRef]
- Barker, S.C.; Blair, D. Molecular phylogeny of Schistosoma species supports traditional groupings within the genus. J. Parasitol. 1996, 82, 292. [Google Scholar] [CrossRef]
- Devkota, R.; Brant, S.V.; Loker, E.S. A genetically distinct Schistosoma from Radix luteola from Nepal related to Schistosoma turkestanicum: A phylogenetic study of schistosome and snail host. Acta Trop. 2016, 164, 45–53. [Google Scholar] [CrossRef]
- Lawton, S.P.; Hirai, H.; Ironside, J.E.; Johnston, D.A.; Rollinson, D. Genomes and geography: Genomic insights into the evolution and phylogeography of the genus Schistosoma. Parasit. Vec. 2010, 4, 131. [Google Scholar] [CrossRef] [PubMed]
- Longo, S.J.; Faircloth, B.C.; Meyer, A.; Westneat, M.W.; Alfaro, M.E.; Wainwright, P.C. Phylogenomic analysis of a rapid radiation of misfit fishes (Syngnathiformes) using ultraconserved elements. Mol. Phylo. Evol. 2017, 113, 33–48. [Google Scholar] [CrossRef] [PubMed]
- Jorge, F.; Perera, A.; Poulin, R.; Roca, V.; Carretero, M.A. Getting there and around: Host range oscillations during colonization of the Canary Islands by the parasitic nematode Spauligodon. Mol. Ecol. 2018, 27, 533–549. [Google Scholar] [CrossRef] [PubMed]
- Huyse, T.; Volckaert, F.A.M. Comparing host and parasite phylogenies: Gyrodactylus flatworms jumping from goby to goby. Syst. Biol. 2005, 54, 710–718. [Google Scholar] [CrossRef]
- Whiteman, N.K.; Kimball, R.T.; Parker, P.G. Co-phylogeography and comparative population genetics of the threatened Galápagos hawk and three ectoparasite species: Ecology shapes population histories within parasite communities. Mol. Ecol. 2007, 16, 4759–4773. [Google Scholar] [CrossRef]
- Hahn, C.; Weiss, S.J.; Stojanovski, S.; Bachmann, L. Co-Speciation of the ectoparasite Gyrodactylus teuchis (Monogenea, Platyhelminthes) and its salmonid hosts. PLoS ONE 2015, 10, e0127340-20. [Google Scholar] [CrossRef]
- Jenkins, T.; Thomas, G.H.; Hellgren, O.; Owens, I.P. Migratory behavior of birds affects their coevolutionary relationship with blood parasites. Evol. Int. J. Org. Evol. 2012, 66, 740–751. [Google Scholar] [CrossRef]
- Ellis, V.A.; Collins, M.D.; Medeiros, M.C.I.; Sari, E.H.R.; Coffey, E.D.; Dickerson, R.C.; Lugarini, C.; Stratford, J.A.; Henry, D.R.; Merrill, L.; et al. Local host specialization, host-switching, and dispersal shape the regional distributions of avian haemosporidian parasites. Proc. Natl. Acad. Sci. USA 2015, 112, 11294–11299. [Google Scholar] [CrossRef]
- Snyder, S.D. Phylogeny and paraphyly among tetrapod blood flukes (Digenea: Schistosomatidae and Spirorchiidae). Int. J. Parasitol. 2004, 34, 1385–1392. [Google Scholar] [CrossRef]
- Shaffer, H.B.; McCartney-Melstad, E.; Near, T.J.; Mount, G.G.; Spinks, P.Q. Phylogenomic analyses of 539 highly informative loci dates a fully resolved time tree for the major clades of living turtles (Testudines). Mol. Phylogenetics Evol. 2017, 115, 7–15. [Google Scholar] [CrossRef]
- Galindo, L.A.; Puillandre, N.; Utge, J.; Lozouet, P.; Bouchet, P. The phylogeny and systematics of the Nassariidae revisited (Gastropoda, Buccinoidea). Mol. Phylogenetics Evol. 2016, 99, 337–353. [Google Scholar] [CrossRef] [PubMed]
- Baker, A.J.; Pereira, S.L.; Paton, T.A. Phylogenetic relationships and divergence times of Charadriiformes genera: Multigene evidence for the Cretaceous origin of at least 14 clades of shorebirds. Biol. Lett. 2007, 3, 205–210. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, M.; Spencer, H.G. Classification of the cormorants of the world. Mol. Phylo. Evol. 2014, 79, 249–257. [Google Scholar] [CrossRef] [PubMed]
- Davis, G.M. Evolution of prosobranch snails transmitting Asian Schistosoma; coevolution with Schistosoma: A review. Prog. Clin. Parasitol. 1993, 3, 145–204. [Google Scholar]
- Devkota, R.; Brant, S.V.; Thapa, A.; Loker, E.S. Sharing schistosomes: The elephant schistosome Bivitellobilharzia nairi also infects the greater one-horned rhinoceros (Rhinoceros unicornis) in Chitwan National Park, Nepal. J. Helm. 2014, 88, 32–40. [Google Scholar] [CrossRef]
- Liu, L.; Huo, G.-N.; He, H.-B.; Zhou, B.; Attwood, S.W. A phylogeny for the pomatiopsidae (Gastropoda: Rissooidea): A resource for taxonomic, parasitological and biodiversity studies. BMC Evol. Biol. 2014, 14, 29. [Google Scholar] [CrossRef]
- Morgan, J.A.T.; Blair, D. Relative merits of nuclear ribosomal internal transcribed spacers and mitochondrial CO1 and ND1 genes for distinguishing among Echinostoma species (Trematoda). Parasitology 1998, 116, 289–297. [Google Scholar] [CrossRef]
- Chan, A.H.E.; Chaisiri, K.; Morand, S.; Saralamba, N.; Thaenkham, U. Evaluation and utility of mitochondrial ribosomal genes for molecular systematics of parasitic nematodes. Parasit. Vec. 2020, 13, 1–13. [Google Scholar] [CrossRef]
- Lawton, S.P.; Lim, R.M.; Dukes, J.P.; Cook, R.T.; Walker, A.J.; Kirk, R.S. Identification of a major causative agent of human cercarial dermatitis, Trichobilharzia franki (Müller and Kimmig 1994), in southern England and its evolutionary relationships with other European populations. Parasit. Vec. 2014, 7, 1–10. [Google Scholar] [CrossRef]
- Brant, S.V. The occurrence of the avian schistosome Allobilharzia visceralis Kolarova, Rudolfova, Hampl et Skirnisson, 2006 (Schistosomatidae) in the tundra swan, Cygnus columbianus (Anatidae), from North America. Folia Parasitol. 2007, 54, 99–104. [Google Scholar] [CrossRef]
- Brant, S.V.; Jouet, D.; Ferté, H.; Loker, E.S. Anserobilharzia gen. n. (Digenea, Schistosomatidae) and redescription of A. brantae (Farr & Blankemeyer, 1956) comb. n. (syn. Trichobilharzia brantae), a parasite of geese (Anseriformes). Zootaxa 2013, 3670, 193–206. [Google Scholar] [PubMed]
- Sun, Z.; Pan, T.; Hu, C.; Sun, L.; Ding, H.; Wang, H.; Zhang, C.; Jin, H.; Chang, Q.; Kan, X.; et al. Rapid and recent diversification patterns in Anseriformes birds: Inferred from molecular phylogeny and diversification analyses. PLoS ONE 2017, 12, e0184529. [Google Scholar] [CrossRef] [PubMed]
- Sandom, C.; Faurby, S.; Sandel, B.; Svenning, J.C. Global late Quaternary megafauna extinctions linked to humans, not climate change. Proc. Roy. Soc. B Biol. Sci. 2014, 281, 20133254. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.F. Life history of Heterobilharzia americana Price 1929, a schistosome of the raccoon and other mammals in southeastern United States. J. Parasitol. 1962, 48, 728–739. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.F. Susceptibility of mammalian hosts to experimental infection with Heterobilharzia americana. J. Parasitol. 1962, 48, 740–745. [Google Scholar] [CrossRef]
- Loker, E.S.; Dolginow, S.Z.; Pape, S.; Topper, C.D.; Alda, P.; Pointier, J.P.; Ebbs, E.T.; Sanchez, M.C.; Verocai, G.G.; DeJong, R.J.; et al. An outbreak of canine schistosomiasis in Utah: Acquisition of a new snail host (Galba humilis) by Heterobilharzia americana, a pathogenic parasite on the move. One Health 2021, 13, 100280. [Google Scholar] [CrossRef]
- Correa, A.C.; Escobar, J.S.; Durand, P.; Renaud, F.; David, P.; Jarne, P.; Pointier, J.P.; Hurtrez-Boussès, S. Bridging gaps in the molecular phylogeny of the Lymnaeidae (Gastropoda: Pulmonata), vectors of Fascioliasis. BMC Evol. Biol. 2010, 10, 1–12. [Google Scholar] [CrossRef]
- Blair, D.; Islam, K.S. The life-cycle and morphology of Trichobilharzia australis n. sp. (Digenea: Schistosomatidae) from the nasal blood vessels of the black duck (Anas superciliosa) in Australia, with a review of the genus Trichobilharzia. Syst. Parasitol. 1983, 5, 89–117. [Google Scholar] [CrossRef]
- Rudge, J.W.; Lu, D.-B.; Fang, G.-R.; Wang, T.-P.; Basáñez, M.-G.; Webster, J.P. Parasite genetic differentiation by habitat type and host species: Molecular epidemiology of Schistosoma japonicum in hilly and marshland areas of Anhui Province, China. Mol. Ecol. 2009, 18, 2134–2147. [Google Scholar] [CrossRef]
- Huyse, T.; Audenaert, V.; Volckaert, F.A.M. Speciation and host-parasite relationships in the parasite genus Gyrodactylus (Monogenea, Platyhelminthes) infecting gobies of the genus Pomatoschistus (Gobiidae, Teleostei). Int. J. Parasitol. 2003, 33, 1679–1689. [Google Scholar] [CrossRef]
- Huyse, T.; Poulin, R.; Théron, A. Speciation in parasites: A population genetics approach. Trend. Parasitol. 2005, 21, 469–475. [Google Scholar] [CrossRef] [PubMed]
- Lotfy, W.M.; Brant, S.V.; DeJong, R.J.; Le, T.H.; Demiaszkiewicz, A.; Rajapakse, R.P.; Perera, V.B.; Laursen, J.R.; Loker, E.S. Evolutionary origins, diversification, and biogeography of liver flukes (Digenea, Fasciolidae). Am. J. Trop. Med. Hyg. 2008, 79, 248–255. [Google Scholar] [CrossRef] [PubMed]
- Laidemitt, M.R.; Zawadzki, E.T.; Brant, S.V.; Mutuku, M.W.; Mkoji, G.M.; Loker, E.S. Loads of trematodes: Discovering hidden diversity of paramphistomoids in Kenyan ruminants. Parasitology 2017, 144, 131–147. [Google Scholar] [CrossRef] [PubMed]
- De Buron, I.; Colon, B.L.; Siegel, S.V.; Oberstaller, J.; Rivero, A.; Kyle, D.E. First evidence of polychaete intermediate hosts for Neospirorchis spp. marine turtle blood flukes (Trematoda: Spirorchiidae). Int. J. Parasitol. 2018, 48, 1097–1106. [Google Scholar] [CrossRef] [PubMed]
- Cribb, T.H.; Chick, R.C.; O’Connor, W.; O’Connor, S.; Johnson, D.; Sewell, K.B.; Cutmore, S.C. Evidence that blood flukes (Trematoda: Aporocotylidae) of chondrichthyans infect bivalves as intermediate hosts: Indications of an ancient diversification of the Schistosomatoidea. Int. J. Parasitol. 2017, 47, 885–891. [Google Scholar] [CrossRef]
- Laidemitt, M.R.; Anderson, L.C.; Wearing, H.J.; Mutuku, M.W.; Mkoji, G.M.; Loker, E.S. Antagonism between parasites within snail hosts impacts the transmission of human schistosomiasis. Elife 2019, 8, e50095. [Google Scholar] [CrossRef]
- Lu, D.B.; Wang, T.P.; Rudge, J.W.; Donnelly, C.A.; Fang, G.R.; Webster, J.P. Evolution in a multi-host parasite: Chronobiological circadian rhythm and population genetics of Schistosoma japonicum cercariae indicates contrasting definitive host reservoirs by habitat. Int. J. Parasitol. 2009, 39, 1581–1588. [Google Scholar] [CrossRef]
- Walker, J.C. Austrobilharzia terrigalensis: A schistosome dominant in interspecific interactions in the molluscan host. Int. J. Parasitol. 1979, 9, 137–140. [Google Scholar] [CrossRef]
- Southgate, V.R.; Brown, D.S.; Warlow, A.; Knowles, R.J.; Jones, A. The influence of Calicophoron microbothrium on the susceptibility of Bulinus tropicus to Schistosoma bovis. Parasitol. Res. 1989, 75, 381–391. [Google Scholar] [CrossRef]
- Da-Bing, L.; Wang, T.-P.; Rudge, J.W.; Donnelly, C.A.; Fang, G.-R.; Webster, J.P. Genetic diversity of Schistosoma japonicum miracidia from individual rodent hosts. Int. J. Parasitol. 2011, 41, 1371–1376. [Google Scholar]
- Sire, C.; Durand, P.; Pointier, J.-P.; Théron, A. Genetic diversity and recruitment pattern of Schistosoma mansoni in a Biomphalaria glabrata snail population: A field study using random-amplified polymorphic DNA markers. J. Parasitol. 1999, 85, 436. [Google Scholar] [CrossRef]
- Theron, A.; Sire, C.; Rognon, A.; Prugnolle, F.; Durand, P. Molecular ecology of Schistosoma mansoni transmission inferred from the genetic composition of larval and adult infrapopulations within intermediate and definitive hosts. Parasitology 2004, 129, 571–585. [Google Scholar] [CrossRef] [PubMed]
- Crellen, T.; Allan, F.; David, S.; Durrant, C.; Huckvale, T.; Holroyd, N.; Emery, A.M.; Rollinson, D.; Aanensen, D.M.; Berriman, M.; et al. Whole genome resequencing of the human parasite Schistosoma mansoni reveals population history and effects of selection. Sci. Rep. 2016, 6, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Červená, B.; Brant, S.V.; Fairet, E.; Shirley, M.H.; Petrželková, K.J.; Modrý, D. Schistosoma mansoni in Gabon: Emerging or Ignored? Am. J. Trop. Med. Hyg. 2006, 95, 849–851. [Google Scholar] [CrossRef] [PubMed]
- Leaché, A.D.; Chavez, A.S.; Jones, L.N.; Grummer, J.A.; Gottscho, A.D.; Linkem, C.W. Phylogenomics of phrynosomatid lizards: Conflicting signals from sequence capture versus restriction site associated DNA sequencing. Genom. Biol. Evol. 2015, 7, 706–719. [Google Scholar] [CrossRef]
- Blouin, M.S.; Yowell, C.A.; Courtney, C.H.; Dame, J.B. Host movement and the genetic structure of populations of parasitic nematodes. Genet. 1995, 141, 1007–1014. [Google Scholar] [CrossRef]
- Gorton, M.J.; Kasl, E.L.; Detwiler, J.T.; Criscione, C.D. Testing local-scale panmixia provides insights into the cryptic ecology, evolution, and epidemiology of metazoan animal parasites. Parasitology 2012, 139, 981–997. [Google Scholar] [CrossRef]
- Helmer, N.; Blatterer, H.; Hörweg, C.; Reier, S.; Sattmann, H.; Schindelar, J.; Szucsich, N.U.; Haring, E. First record of Trichobilharzia physellae (Talbot, 1936) in Europe, a possible causative agent of cercarial dermatitis. Pathogens 2021, 10, 1473. [Google Scholar] [CrossRef]
- Maddison, W.P. Missing data versus missing characters in phylogenetic analysis. Syst. Biol. 1993, 42, 576–581. [Google Scholar] [CrossRef]
- Streicher, J.W.; Schulte, J.A.; Wiens, J.J. How should genes and taxa be sampled for phylogenomic analyses with missing data? An empirical study in iguanian lizards. Syst. Biol. 2016, 65, 128–145. [Google Scholar] [CrossRef]
- Heled, J.; Drummond, A.J. Bayesian inference of species trees from multilocus data. Mol. Biol. Evol. 2009, 27, 570–580. [Google Scholar] [CrossRef] [PubMed]
- Bayzid, M.S.; Warnow, T. Estimating optimal species trees from incomplete gene trees under deep coalescence. J. Comput. Biol. 2012, 19, 591–605. [Google Scholar] [CrossRef] [PubMed]
- Corl, A.; Ellegren, H. Sampling strategies for species trees: The effects on phylogenetic inference of the number of genes, number of individuals, and whether loci are mitochondrial, sex-linked, or autosomal. Mol. Phylo. Evol. 2013, 67, 358–366. [Google Scholar] [CrossRef] [PubMed]
- Yazdi, H.P.; Ellegren, H. A genetic map of ostrich Z chromosome and the role of inversions in avian sex chromosome evolution. Genom. Biol. Evol. 2018, 10, 2049–2060. [Google Scholar] [CrossRef] [PubMed]
- Olson, P.D.; Cribb, T.H.; Tkach, V.V.; Bray, R.A.; Littlewood, D.T.J. Phylogeny and classification of the Digenea (Platyhelminthes: Trematoda). Int. J. Parasitol. 2003, 33, 733–755. [Google Scholar] [CrossRef]
- De León, G.P.P.; Hernández-Mena, D. Testing the higher-level phylogenetic classification of Digenea (Platyhelminthes, Trematoda) based on nuclear rDNA sequences before entering the age of the ‘next-generation’ Tree of Life. J. Helm. 2019, 93, 260–276. [Google Scholar] [CrossRef]
- Short, R.B.; Holliman, R.B. Austrobilharzia penneri, a new schistosome from marine snails. J. Parasitol. 1961, 47, 447–450. [Google Scholar] [CrossRef]
- Brant, S.V.; Cohen, A.N.; James, D.; Hui, L.; Hom, A.; Loker, E.S. Cercarial dermatitis transmitted by exotic marine snail. Emerg. Infect. Dis. 2010, 16, 1357–1365. [Google Scholar] [CrossRef]
- Al-Kandari, W.Y.; Al-Bustan, S.A.; Isaac, A.M.; George, B.A.; Chandy, B.S. Molecular identification of Austrobilharzia species parasitizing Cerithidea cingulata (Gastropoda: Potamididae) from Kuwait Bay. J. Helm. 2012, 29, 470–478. [Google Scholar] [CrossRef]
- Hutson, K.S.; Vaughan, D.B.; Blair, D. First record of a ‘fish’ blood fluke (Digenea: Aporocotylidae) from a marine mammal: Cardicola dhangali n. sp. Int. J. Parasitol Parasit. Wildlf. 2019, 10, 23–28. [Google Scholar] [CrossRef]
- Appleton, C.C. The eggs of some blood-flukes (Trematoda: Schistosomatidae) from South African birds. Afr. Zool. 1982, 17, 147–150. [Google Scholar] [CrossRef][Green Version]
- Derkarabetian, S.; Benavides, L.R.; Giribet, G. Sequence capture phylogenomics of historical ethanol-preserved museum specimens: Unlocking the rest of the vault. Mol. Ecol. Res. 2019, 19, 1531–1544. [Google Scholar] [CrossRef] [PubMed]
- Raxworthy, C.J.; Smith, B.T. Mining museums for historical DNA: Advances and challenges in museomics. Trend. Ecol. Evol. 2021, 36, 1049–1060. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Faircloth, B.C. PHYLUCE is a software package for the analysis of conserved genomic loci. Bioinformatics 2016, 32, 786–788. [Google Scholar] [CrossRef] [PubMed]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [PubMed]
- Harris, R.S. Improved Pairwise Alignment of Genomic DNA; The Pennsylvania State University: State College, PA, USA, 2007. [Google Scholar]
- Li, D.; Liu, C.-M.; Luo, R.; Sadakane, K.; Lam, T.-W. MEGAHIT: An ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 2015, 31, 1674–1676. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; Linder, C.R.; Warnow, T. RAxML and FastTree: Comparing two methods for large-scale maximum likelihood phylogeny estimation. PLoS ONE 2011, 6, e27731. [Google Scholar] [CrossRef]
- Wiens, J.J. Missing data and the design of phylogenetic analyses. J. Biomed. Inform. 2006, 39, 34–42. [Google Scholar] [CrossRef] [PubMed]
- Bouckaert, R.; Heled, J.; Kühnert, D.; Vaughan, T.; Wu, C.H.; Xie, D.; Suchard, M.A.; Rambaut, A.; Drummond, A.J. BEAST 2: A software platform for Bayesian evolutionary analysis. PLoS Comp. Biol. 2014, 10, e1003537. [Google Scholar] [CrossRef] [PubMed]
- Rambaut, A.; Drummond, A.J.; Xie, D.; Baele, G.; Suchard, M.A. Posterior summarization in Bayesian phylogenetics using Tracer 1.7. Syst. Biol. 2018, 67, 901–904. [Google Scholar] [CrossRef] [PubMed]
- Maddison, W. Reconstructing character evolution on polytomous cladograms. Cladistics 1989, 5, 365–377. [Google Scholar] [CrossRef] [PubMed]
- Whitfield, J.B.; Lockhart, P.J. Deciphering ancient rapid radiations. Trend. Ecol. Evol. 2007, 22, 258–265. [Google Scholar] [CrossRef] [PubMed]
- Eckert, A.J.; Carstens, B.C. Does gene flow destroy phylogenetic signal? The performance of three methods for estimating species phylogenies in the presence of gene flow. Mol. Phylogenetics Evol. 2008, 49, 832–842. [Google Scholar] [CrossRef] [PubMed]
- Kutschera, V.E.; Bidon, T.; Hailer, F.; Rodi, J.L.; Fain, S.R.; Janke, A. Bears in a forest of gene trees: Phylogenetic inference is complicated by incomplete lineage sorting and gene flow. Mol. Biol. Evol. 2014, 31, 2004–2017. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
| Source | AO | SB | SB + M | DAS + SB | DAS | DAS + HS | HS | Loci |
|---|---|---|---|---|---|---|---|---|
| [25] | N | ns | ns | ns | Y | partial | Y | 28S |
| [26] | N | ns | ns | N | Y | N | Y | 18S, 28S + cox1 |
| [27] | Y | Y | N | partial | partial | partial | Y | 18S, 28S + cox1 |
| [28] | Y | N | N | N | Y | partial | N | 18S, 28S, ITS + cox1 |
| [37] | ns | Y | Ns | ns | ns | Ns | ns | 18S, 28S + cox1 |
| [21] | Y | ns | Ns | Y | Y | N | Y | 18S, 28S, ITS + cox1 |
| [38] | N | partial | N | N | Y | N | Y | 18S, 28S + cox1 |
| [34] | ns | Y | Y | Y | N | N | Y | 28S |
| [35] | Y | N | N | N | Y | N | Y | 28S, ITS + cox1 |
| [39] | Y | partial | N | Y | Y | partial | Y | 28S + cox1 |
| [40] | Y | partial | N | partial | Y | partial | Y | 28S + cox1 |
| Taxa | Sample ID | Host | Locality | NCBI | MSB:Para:# |
|---|---|---|---|---|---|
| Anserobilharzia brantae | W335 | Gyraulus parvus | CAN | SRR19593566 | 14745 |
| W351 | Branta canadensis | CAN | SRR19593565 | 7984 | |
| W333 | Gyraulus parvus | CAN | SRR19593554 | 14744 | |
| Austrobilharzia variglandis | SAL63.81 | Larus delawarensis | CAN | SRR19593543 | 32451 |
| SAL63.80 | Larus delawarensis | CAN | SRR19593533 | 29053 | |
| Bilharziella polonica | W930 | Anas platyrhynchos | UA | SRR19593532 | 32667 |
| Bivitellobilharzia nairi | W465.2 | Rhinoceros unicornis | NP | SRR19593531 | 29075 |
| Dendritobilharzia pulverulenta | W926 | Anas crecca | USA | SRR19593530 | 29034 |
| W836 | Anas discors | USA | SRR19593529 | 20795 | |
| Gigantobilharzia huronensis | W414/513 | Physa sp. | USA | SRR19593528 | 18687/25488 |
| W678 | Physa sp. | USA | SRR19593564 | 29074 | |
| Heterobilharzia americana | W805 | Procyon lotor | USA | SRR19593563 | 19286 |
| Macrobilharzia macrobilharzia | W931 | Anhinga anhinga | USA | SRR19593562 | 32668 |
| Schistosomatium douthitti | SAL95_60 | Stagnicola sp. | CAN | SRR19593561 | 2861 |
| Schistosoma bovis | PM1 | Bulinus sp. | KE | SRR19593560 | 32666 |
| Schistosoma nasalae | W546 | Indoplanorbis exustus | NP | SRR19593559 | — |
| Schistosoma spindale | W545 | Indoplanorbis exustus | NP | SRR19593559 | — |
| Schistosoma edwardiense | W957 | Biomphalaria sp. | KE | SRR19593557 | 32609 |
| Schistosoma sp. | W555 | Indoplanorbis sp. | NP | SRR19593556 | — |
| Ornithobilharzia canaliculata | W393 | Larus occidentalis | USA | SRR19593555 | 18542 |
| Trichobilharzia querquedulae | W664 | Spatula smithii | ZA | SRR19593553 | 19000 |
| Trichobilharzia stagnicolae | W233 | Stagnicola emarginata | USA | SRR19593552 | 19509 |
| Trichobilharzia cf. szidati | W620A | Lymnaea stagnalis | USA | SRR19593551 | 259 |
| Avian Schistosomatidae sp. A | W613 | Melanitta deglandi | USA | SRR19593550 | 25264 |
| Avian Schistosomatidae lineage 2 | W634 | Chilina perrieri | AR | SRR19593549 | 7969 |
| Avian Schistosomatidae sp. B | W399 | Physa acuta | USA | SRR19593548 | 18677 |
| Avian Schistosomatidae sp. C2 | W402 | Gyraulus parvus | USA | SRR19593547 | 18680 |
| Avian Schistosomatidae sp. C4 | W847 | Biomphalaria glabrata | BR | SRR19593546 | 25514 |
| Avian Schistosomatidae sp. C11 | W607 | Anas americana | USA | SRR19593545 | 25258 |
| Avian Schistosomatidae sp. C11 | W616A | Gyraulus sp. | USA | SRR19593544 | 19650 |
| Avian Schistosomatidae sp. D | W342 | Gyraulus parvus | CAN | SRR19593542 | 18619 |
| Avian Schistosomatidae sp. G | W877 | Ceratophallus sp. | KE | SRR19593541 | 32612 |
| Marinabilharzia patagonense | W637A | Siphonaria lessoni | AR | SRR19593540 | 18935 |
| Avian Schistosomatidae lineage 3 | C1 | Chilina neuquenensi | AR | SRR19593539 | 7970 |
| Avian Schistosomatidae sp. M1 | W216 | Haminoea japonica | USA | SRR19593538 | 18660 |
| Schistosomatidae sp. W688 | W688 | Indoplanorbis exustus | NP | SRR19593537 | 18710 |
| Outgroups | |||||
| Vasotrema cf. robustum | W411 | Physa sp. | USA | SRR19593536 | 18690 |
| Vasotrema cf. robustum | PS | Physa sp. | USA | SRR19593535 | 18715 |
| Spirorchiidae | W702 | Biomphalaria straminea | BR | SRR19593534 | 20804 |
| Published Genomes | |||||
| Allobilharzia visceralis | SAMEA2201407 | ||||
| Schistosoma hematobium | |||||
| Schistosoma mansoni | |||||
| Schistosoma japonicum | |||||
| Schistosomatium douthitti | SAMEA1920831 | ||||
| Trichobilharzia szidati | PRJEB461 | ||||
| Trichobilharzia regenti | SAMEA2422295 | ||||
| Echinostoma caproni | PRJEB127 | ||||
| Taxa | Sample ID | Life Stage | DNA [µg] | No. of Contigs | No. of bp | UCEs |
|---|---|---|---|---|---|---|
| Schistosomatidae | ||||||
| Anserobilharzia brantae | W352/W340 | C | 0.562 | 1158 | 330,978 | 48 |
| W335 | C | nq | 16,743 | 4,870,006 | 1334 | |
| W351 | C | >0.005 | 5907 | 1,836,149 | 551 | |
| Austrobilharzia variglandis | W396/359 | A | 0.0352 | 145 | 35,487 | 6 |
| W697 | A | 0.0193 | 8344 | 2,495,016 | 1440 | |
| Bilharziella polonica | W930 | A | 0.0155 | 17,081 | 5,897,771 | 1677 |
| Bivitellobilharzia nairi | W465 | M | 0.078 | 18,455 | 5,052,402 | 1336 |
| BN2011 | M | nq | 3696 | 1,162,332 | 47 | |
| Dendritobilharzia pulverulenta | W836 | A | nq | 26,978 | 8,405,837 | 1737 |
| W926 | A | 0.0944 | 47,498 | 14,794,680 | 1631 | |
| Gigantobilharzia huronensis | W414/513 | C | 0.0375 | 242,326 | 72,500,699 | 1650 |
| W678 | C | 0.0325 | 48,623 | 16,046,122 | 1582 | |
| Heterobilharzia americana | W805 | A | 0.133 | 442,043 | 139,396,852 | 1784 |
| Macrobilharzia macrobilharzia | W931 | A | 0.0472 | 3508 | 1,580,643 | 1590 |
| Marinabilharzia patagonense | W637A | C | 0.163 | 34,871 | 13,736,793 | 1749 |
| Ornithobilharzia canaliculata | W393 | A | 0.15 | 31,569 | 8,873,305 | 1441 |
| Schistosomatium douthitti | SAL95.60 | C | nq | 15,755 | 4,482,586 | 597 |
| Schistosoma bovis | PM1 | C | nq | 6316 | 4,181,809 | 1873 |
| Schistosoma nasale | W546 | C | 0.0107 | 41,764 | 14,799,490 | 1830 |
| Schistosoma spindale | W545 | C | 0.0062 | 15,206 | 5,394,821 | 1885 |
| Schistosoma edwardiense | W957 | C | nq | 21210 | 8,383,839 | 1800 |
| Schistosoma sp. | W555 | C | 0.1455 | 15,865 | 7,245,729 | 1862 |
| Trichobilharzia querquedulae | W929 | A | 0.0224 | 70,532 | 21,183,008 | 1576 |
| Trichobilharzia stagnicolae | W233 | C | 0.0531 | 18,675 | 13,662 | 658 |
| Trichobilharzia cf. szidati | W620A | C | nq | 48,011 | 15,967,522 | 1123 |
| Avian Schistosomatidae sp. A | W613 | A | 0.091 | 10,458 | 3,571,057 | 1511 |
| Avian Schistosomatidae sp. B | W399 | C | 0.0905 | 9349 | 2,995,586 | 1115 |
| Avian Schistosomatidae sp. C2 | W402 | C | 0.0845 | 15,168 | 4,939,724 | 849 |
| Avian Schistosomatidae sp. C4 | W847 | C | nq | 35,118 | 10,814,130 | 1732 |
| Avian Schistosomatidae sp. C11 | W607 | C | 0.265 | 34,656 | 53,303 | 1599 |
| Avian Schistosomatidae sp. C11 | W616A | C | 0.084 | 32,003 | 10,790,620 | 1409 |
| Avian Schistosomatidae sp. D | W342 | C | 0.43 | 13,126 | 3,804,186 | 171 |
| Avian Schistosomatidae sp. G | W877 | C | nq | 1227 | 356,239 | 330 |
| Avian Schistosomatidae lineage 2 | W634 | C | nq | 6368 | 1,553,874 | 419 |
| Avian Schistosomatidae lineage 3 | C1 | C | nq | 2576 | 764,111 | 458 |
| Schistosomatidae sp. W688 | W688 | C | 0.0337 | 8006 | 3,890,904 | 1428 |
| Non-Schistosomatidae | ||||||
| Vasotrema cf. robustum | W411 | C | nq | 17,681 | 4,449,659 | 372 |
| Spirorchiidae | W702 | C | nq | 47,339 | 15,477,172 | 885 |
| Vasotrema cf. robustum | PS | C | nq | 41,958 | 12,949,767 | 473 |
| Spirorchiidae | Aporocotylidae | |
|---|---|---|
| Trichobilharzia regenti | 84.90 | 80.24 |
| Heterobilharzia americana | 84.30 | 79.50 |
| Schistosoma mansoni | 83.60 | 80.84 |
| Macrobilharzia macrobilharzia | 85.13 | 80.48 |
| AO Clade | 83.40 | 80.56 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ebbs, E.T.; Loker, E.S.; Bu, L.; Locke, S.A.; Tkach, V.V.; Devkota, R.; Flores, V.R.; Pinto, H.A.; Brant, S.V. Phylogenomics and Diversification of the Schistosomatidae Based on Targeted Sequence Capture of Ultra-Conserved Elements. Pathogens 2022, 11, 769. https://doi.org/10.3390/pathogens11070769
Ebbs ET, Loker ES, Bu L, Locke SA, Tkach VV, Devkota R, Flores VR, Pinto HA, Brant SV. Phylogenomics and Diversification of the Schistosomatidae Based on Targeted Sequence Capture of Ultra-Conserved Elements. Pathogens. 2022; 11(7):769. https://doi.org/10.3390/pathogens11070769
Chicago/Turabian StyleEbbs, Erika T., Eric S. Loker, Lijing Bu, Sean A. Locke, Vasyl V. Tkach, Ramesh Devkota, Veronica R. Flores, Hudson A. Pinto, and Sara V. Brant. 2022. "Phylogenomics and Diversification of the Schistosomatidae Based on Targeted Sequence Capture of Ultra-Conserved Elements" Pathogens 11, no. 7: 769. https://doi.org/10.3390/pathogens11070769
APA StyleEbbs, E. T., Loker, E. S., Bu, L., Locke, S. A., Tkach, V. V., Devkota, R., Flores, V. R., Pinto, H. A., & Brant, S. V. (2022). Phylogenomics and Diversification of the Schistosomatidae Based on Targeted Sequence Capture of Ultra-Conserved Elements. Pathogens, 11(7), 769. https://doi.org/10.3390/pathogens11070769