


Evolutionary and Mutational Characterization of the First H5N8 Subtype Influenza A Virus in Humans


Abstract
:1. Introduction
2. Results
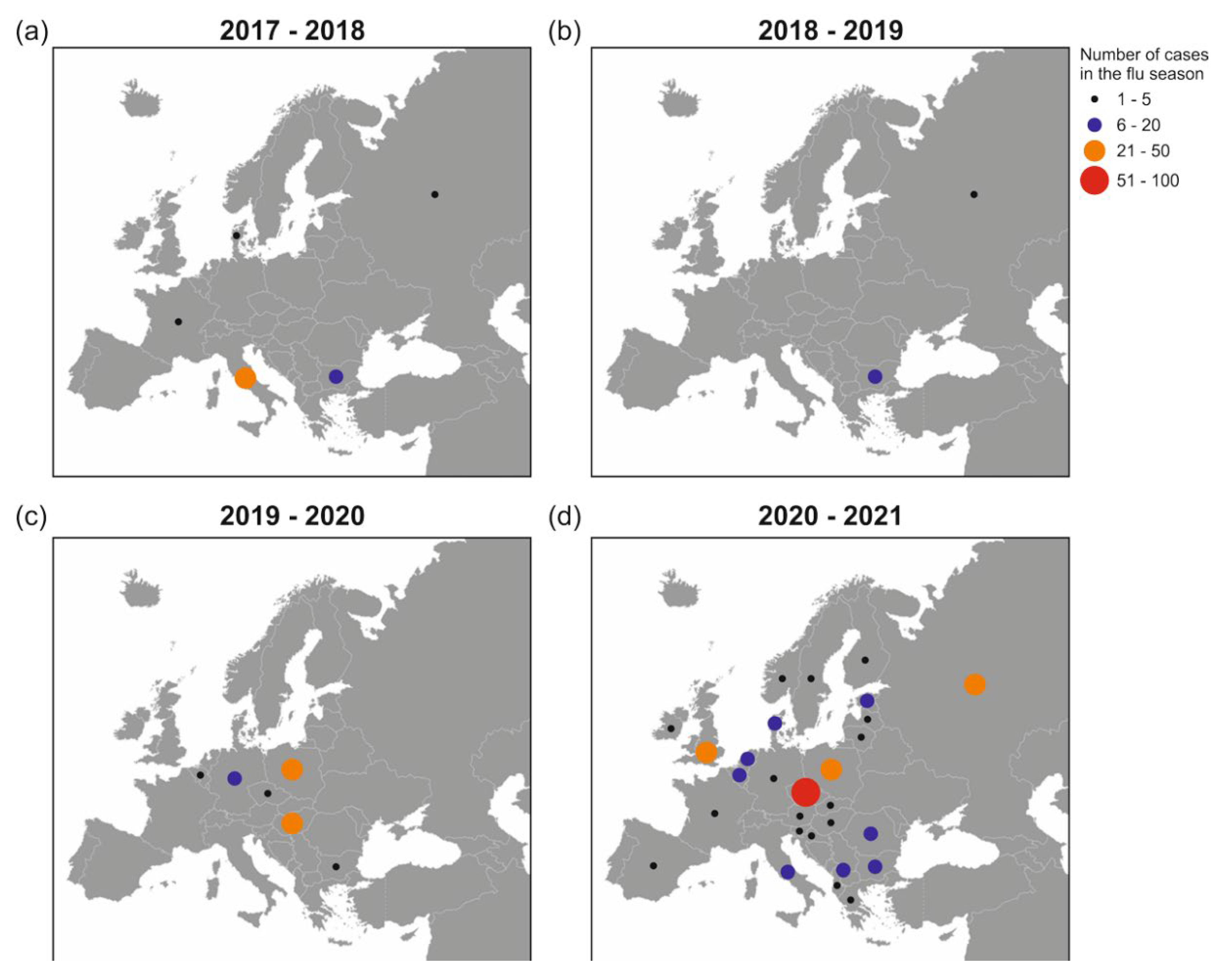
2.1. GISAID Database Recorded H5N8 Outbreaks in Europe during 2017–2021
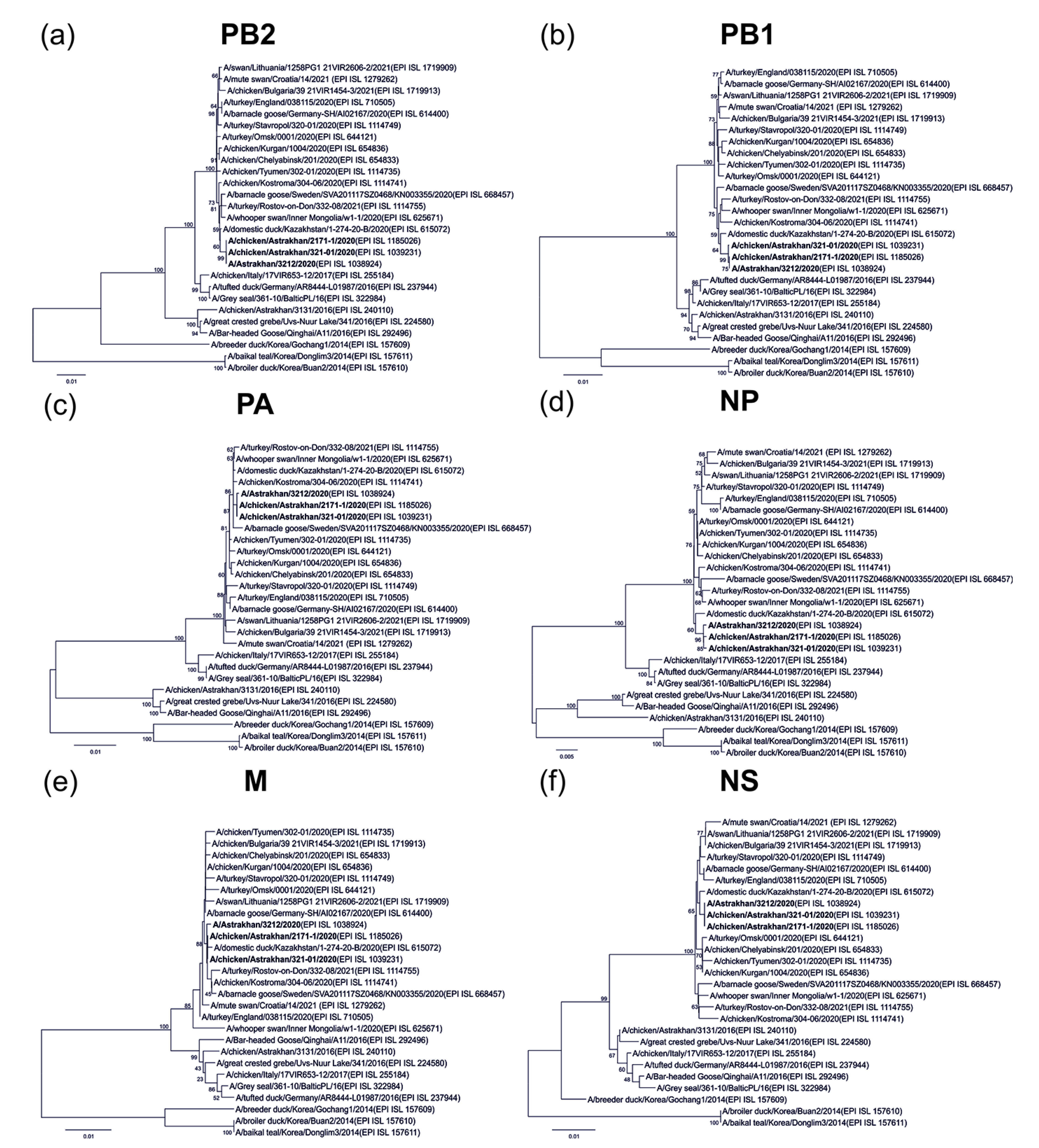
2.2. Human-Origin and Chicken Viruses Are Closely Related but Not Homologous to an Earlier Virus in Astrakhan
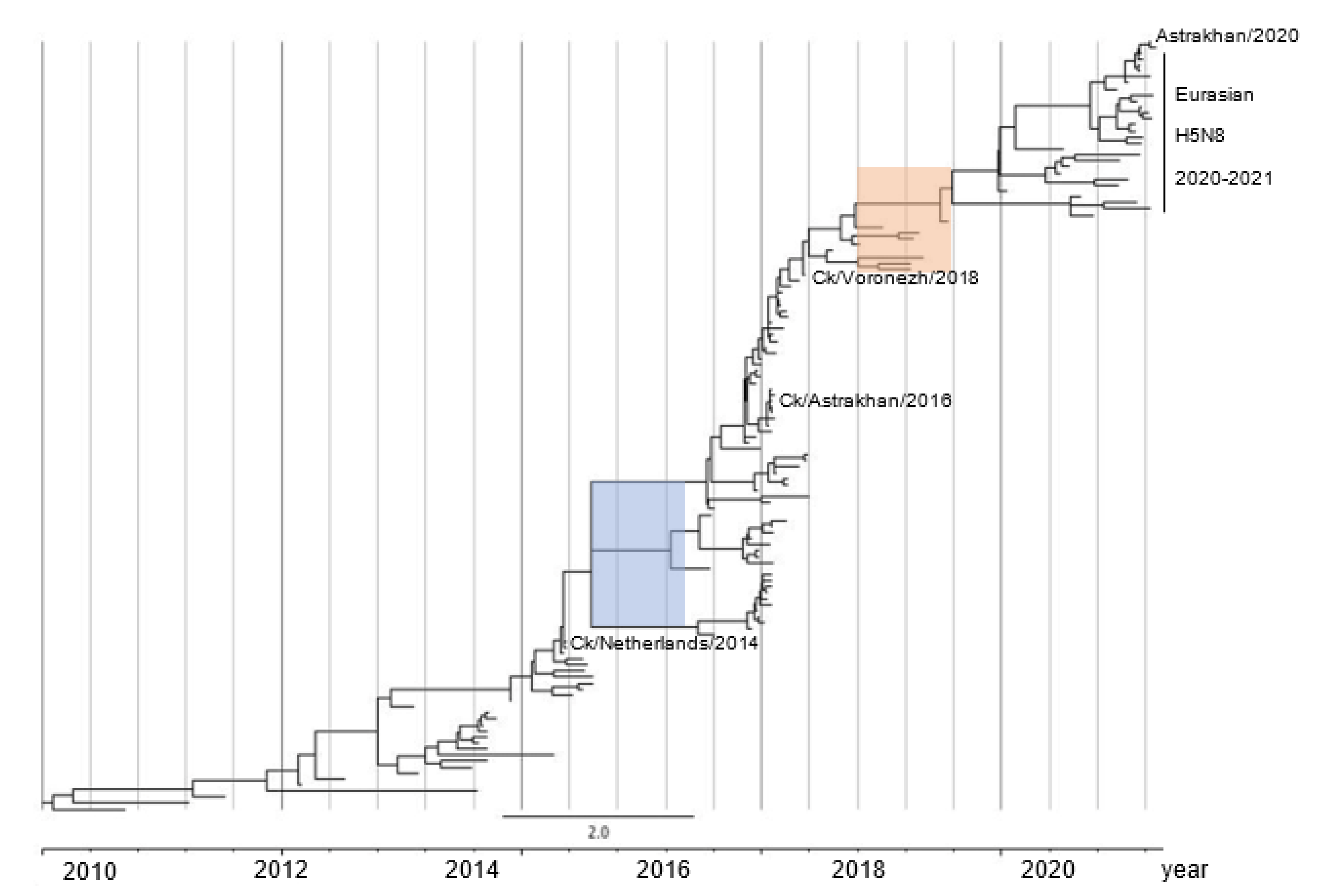
2.3. H5N8 Virus Took Two Evolutionary Steps during 2015–2016 and after 2018
2.4. Molecular Features of HA and NA Proteins of the Human H5N8 Virus
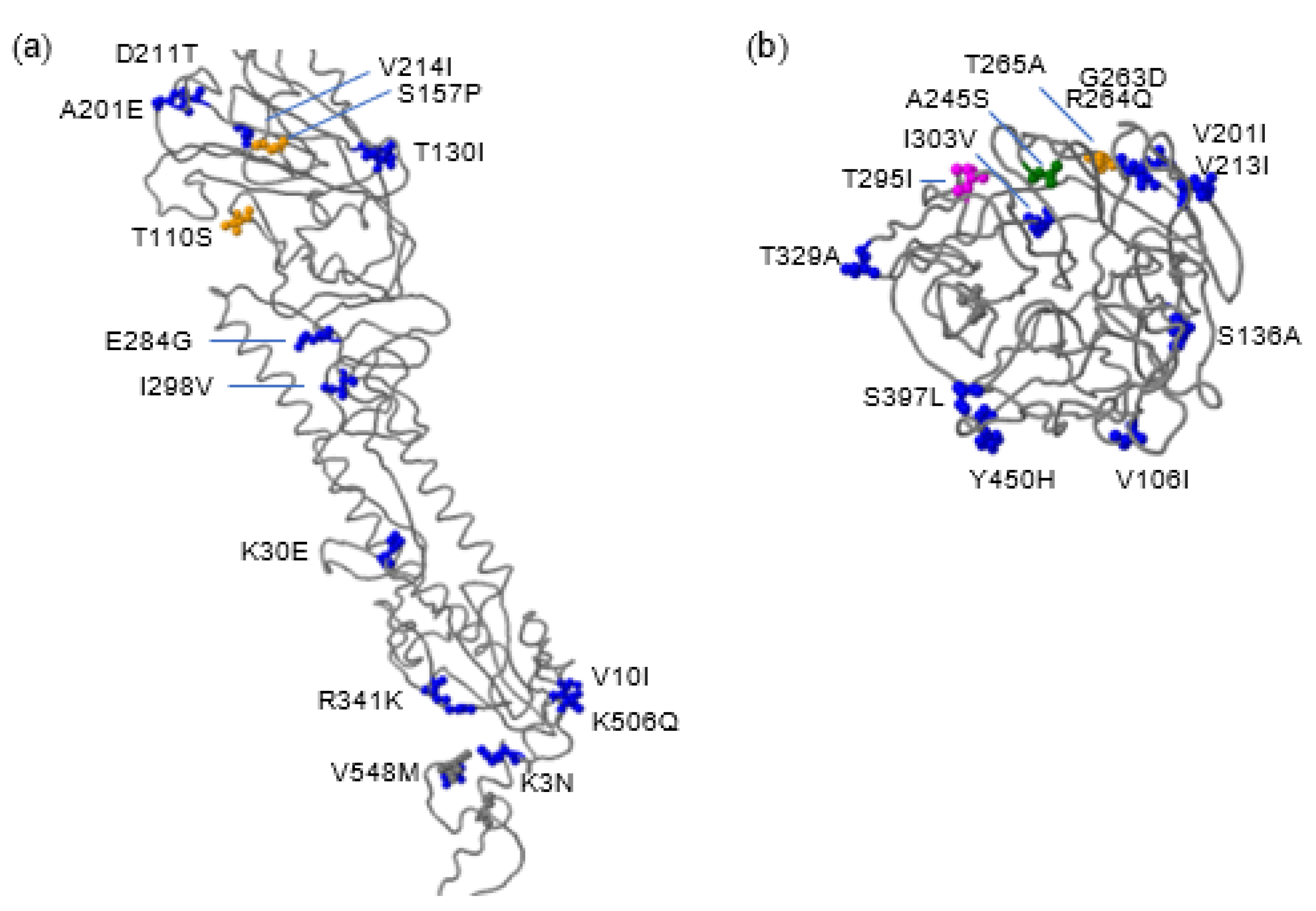
2.5. Probable Human Adaptive Mutations Are Identified in Astrakhan H5N8 Viruses in 2020
3. Discussion
4. Materials and Methods
4.1. Viral Strains
4.2. Phylogenetic Analysis
4.3. Time to Most Recent Common Ancestor (tMRCA) Analysis
4.4. Mutational Analysis with the FluSurver to an Ancestral H5N8 Strain
4.5. Molecular Comparison between Human and Avian H5N8 Viruses in Astrakhan
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sutton, T.C. The Pandemic Threat of Emerging H5 and H7 Avian Influenza Viruses. Viruses 2018, 10, 461. [Google Scholar] [CrossRef] [Green Version]
- Claas, E.C.; Osterhaus, A.D.; van Beek, R.; De Jong, J.C.; Rimmelzwaan, G.F.; Senne, D.A.; Krauss, S.; Shortridge, K.F.; Webster, R.G. Human influenza A H5N1 virus related to a highly pathogenic avian influenza virus. Lancet 1998, 351, 472–477. [Google Scholar] [CrossRef]
- Guan, Y.; Shortridge, K.F.; Krauss, S.; Webster, R.G. Molecular characterization of H9N2 influenza viruses: Were they the donors of the “internal” genes of H5N1 viruses in Hong Kong? Proc. Natl. Acad. Sci. USA 1999, 96, 9363–9367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, K.; Gu, M.; Zhong, L.; Duan, Z.; Zhang, Y.; Zhu, Y.; Zhao, G.; Zhao, M.; Chen, Z.; Hu, S.; et al. Characterization of three H5N5 and one H5N8 highly pathogenic avian influenza viruses in China. Vet. Microbiol. 2013, 163, 351–357. [Google Scholar] [CrossRef]
- Kim, S.H.; Hur, M.; Suh, J.H.; Woo, C.; Wang, S.J.; Park, E.R.; Hwang, J.; An, I.J.; Jo, S.D.; Shin, J.H.; et al. Molecular characterization of highly pathogenic avian influenza H5N8 viruses isolated from Baikal teals found dead during a 2014 outbreak in Korea. J. Vet. Sci. 2016, 17, 299–306. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.H.; Torchetti, M.K.; Winker, K.; Ip, H.S.; Song, C.S.; Swayne, D.E. Intercontinental Spread of Asian-Origin H5N8 to North America through Beringia by Migratory Birds. J. Virol. 2015, 89, 6521–6524. [Google Scholar] [CrossRef] [Green Version]
- Global Consortium for H5N8 and Related Influenza Viruses. Role for migratory wild birds in the global spread of avian influenza H5N8. Science 2016, 354, 213–217. [Google Scholar] [CrossRef] [Green Version]
- El-Shesheny, R.; Barman, S.; Feeroz, M.M.; Hasan, M.K.; Jones-Engel, L.; Franks, J.; Turner, J.; Seiler, P.; Walker, D.; Friedman, K.; et al. Genesis of Influenza A(H5N8) Viruses. Emerg. Infect. Dis. 2017, 23, 1368–1371. [Google Scholar] [CrossRef] [PubMed]
- Adlhoch, C.; Gossner, C.; Koch, G.; Brown, I.; Bouwstra, R.; Verdonck, F.; Penttinen, P.; Harder, T. Comparing introduction to Europe of highly pathogenic avian influenza viruses A(H5N8) in 2014 and A(H5N1) in 2005. Eurosurveillance 2014, 19, 20996. [Google Scholar] [CrossRef] [Green Version]
- Lycett, S.J.; Pohlmann, A.; Staubach, C.; Caliendo, V.; Woolhouse, M.; Beer, M.; Kuiken, T.; Global Consortium for H5N8 and Related Influenza Viruses. Genesis and spread of multiple reassortants during the 2016/2017 H5 avian influenza epidemic in Eurasia. Proc. Natl. Acad. Sci. USA 2020, 117, 20814–20825. [Google Scholar] [CrossRef]
- Pyankova, O.G.; Susloparov, I.M.; Moiseeva, A.A.; Kolosova, N.P.; Onkhonova, G.S.; Danilenko, A.V.; Vakalova, E.V.; Shendo, G.L.; Nekeshina, N.N.; Noskova, L.N.; et al. Isolation of clade 2.3.4.4b A(H5N8), a highly pathogenic avian influenza virus, from a worker during an outbreak on a poultry farm, Russia, December 2020. Eurosurveillance 2021, 26, 2100439. [Google Scholar] [CrossRef]
- Shin, D.L.; Siebert, U.; Lakemeyer, J.; Grilo, M.; Pawliczka, I.; Wu, N.H.; Valentin-Weigand, P.; Haas, L.; Herrler, G. Highly Pathogenic Avian Influenza A(H5N8) Virus in Gray Seals, Baltic Sea. Emerg. Infect. Dis. 2019, 25, 2295–2298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herve, S.; Schmitz, A.; Briand, F.X.; Gorin, S.; Queguiner, S.; Niqueux, E.; Paboeuf, F.; Scoizec, A.; Le Bouquin-Leneveu, S.; Eterradossi, N.; et al. Serological Evidence of Backyard Pig Exposure to Highly Pathogenic Avian Influenza H5N8 Virus during 2016–2017 Epizootic in France. Pathogens 2021, 10, 621. [Google Scholar] [CrossRef] [PubMed]
- Schulein, A.; Ritzmann, M.; Christian, J.; Schneider, K.; Neubauer-Juric, A. Exposure of wild boar to Influenza A viruses in Bavaria: Analysis of seroprevalences and antibody subtype specificity before and after the panzootic of highly pathogenic avian influenza viruses A (H5N8). Zoonoses Public Health 2021, 68, 503–515. [Google Scholar] [CrossRef]
- Floyd, T.; Banyard, A.C.; Lean, F.Z.X.; Byrne, A.M.P.; Fullick, E.; Whittard, E.; Mollett, B.C.; Bexton, S.; Swinson, V.; Macrelli, M.; et al. Encephalitis and Death in Wild Mammals at a Rehabilitation Center after Infection with Highly Pathogenic Avian Influenza A(H5N8) Virus, United Kingdom. Emerg. Infect. Dis. 2021, 27, 2856–2863. [Google Scholar] [CrossRef]
- Bauer, K.; Richter, M.; Wutzler, P.; Schmidtke, M. Different neuraminidase inhibitor susceptibilities of human H1N1, H1N2, and H3N2 influenza A viruses isolated in Germany from 2001 to 2005/2006. Antivir. Res. 2009, 82, 34–41. [Google Scholar] [CrossRef]
- Boltz, D.A.; Douangngeun, B.; Phommachanh, P.; Sinthasak, S.; Mondry, R.; Obert, C.; Seiler, P.; Keating, R.; Suzuki, Y.; Hiramatsu, H.; et al. Emergence of H5N1 avian influenza viruses with reduced sensitivity to neuraminidase inhibitors and novel reassortants in Lao People’s Democratic Republic. J. Gen. Virol. 2010, 91, 949–959. [Google Scholar] [CrossRef] [PubMed]
- Sui, J.; Hwang, W.C.; Perez, S.; Wei, G.; Aird, D.; Chen, L.M.; Santelli, E.; Stec, B.; Cadwell, G.; Ali, M.; et al. Structural and functional bases for broad-spectrum neutralization of avian and human influenza A viruses. Nat. Struct. Mol. Biol. 2009, 16, 265–273. [Google Scholar] [CrossRef]
- Rudrawar, S.; Kerry, P.S.; Rameix-Welti, M.A.; Maggioni, A.; Dyason, J.C.; Rose, F.J.; van der Werf, S.; Thomson, R.J.; Naffakh, N.; Russell, R.J.; et al. Synthesis and evaluation of novel 3-C-alkylated-Neu5Ac2en derivatives as probes of influenza virus sialidase 150-loop flexibility. Org. Biomol. Chem. 2012, 10, 8628–8639. [Google Scholar] [CrossRef]
- Brown, E.G.; Bailly, J.E. Genetic analysis of mouse-adapted influenza A virus identifies roles for the NA, PB1, and PB2 genes in virulence. Virus Res. 1999, 61, 63–76. [Google Scholar] [CrossRef]
- Matrosovich, M.N.; Gambaryan, A.S.; Teneberg, S.; Piskarev, V.E.; Yamnikova, S.S.; Lvov, D.K.; Robertson, J.S.; Karlsson, K.A. Avian influenza A viruses differ from human viruses by recognition of sialyloligosaccharides and gangliosides and by a higher conservation of the HA receptor-binding site. Virology 1997, 233, 224–234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, M.W.; Liao, H.Y.; Huang, Y.; Jan, J.T.; Huang, C.C.; Ren, C.T.; Wu, C.Y.; Cheng, T.J.; Ho, D.D.; Wong, C.H. Broadly neutralizing DNA vaccine with specific mutation alters the antigenicity and sugar-binding activities of influenza hemagglutinin. Proc. Natl. Acad. Sci. USA 2011, 108, 3510–3515. [Google Scholar] [CrossRef] [Green Version]
- Kaverin, N.V.; Rudneva, I.A.; Govorkova, E.A.; Timofeeva, T.A.; Shilov, A.A.; Kochergin-Nikitsky, K.S.; Krylov, P.S.; Webster, R.G. Epitope mapping of the hemagglutinin molecule of a highly pathogenic H5N1 influenza virus by using monoclonal antibodies. J. Virol. 2007, 81, 12911–12917. [Google Scholar] [CrossRef] [Green Version]
- Gu, W.; Shi, J.; Cui, P.; Yan, C.; Zhang, Y.; Wang, C.; Zhang, Y.; Xing, X.; Zeng, X.; Liu, L.; et al. Novel H5N6 reassortants bearing the clade 2.3.4.4b HA gene of H5N8 virus have been detected in poultry and caused multiple human infections in China. Emerg. Microbes Infect. 2022, 11, 1174–1185. [Google Scholar] [CrossRef] [PubMed]
- Imai, H.; Shinya, K.; Takano, R.; Kiso, M.; Muramoto, Y.; Sakabe, S.; Murakami, S.; Ito, M.; Yamada, S.; Le, M.T.; et al. The HA and NS genes of human H5N1 influenza A virus contribute to high virulence in ferrets. PLoS Pathog. 2010, 6, e1001106. [Google Scholar] [CrossRef] [Green Version]
- Blaurock, C.; Blohm, U.; Luttermann, C.; Holzerland, J.; Scheibner, D.; Schafer, A.; Groseth, A.; Mettenleiter, T.C.; Abdelwhab, E.M. The C-terminus of non-structural protein 1 (NS1) in H5N8 clade 2.3.4.4 avian influenza virus affects virus fitness in human cells and virulence in mice. Emerg. Microbes Infect. 2021, 10, 1760–1776. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.; Li, L.; Yan, Z.; Gan, T.; Li, L.; Chen, R.; Chen, R.; Zheng, Z.; Hong, W.; Wang, J.; et al. Dual E627K and D701N mutations in the PB2 protein of A(H7N9) influenza virus increased its virulence in mammalian models. Sci. Rep. 2015, 5, 14170. [Google Scholar] [CrossRef] [Green Version]
- Abed, Y.; Baz, M.; Boivin, G. A novel neuraminidase deletion mutation conferring resistance to oseltamivir in clinical influenza A/H3N2 virus. J. Infect. Dis. 2009, 199, 180–183. [Google Scholar] [CrossRef] [PubMed]
- Monto, A.S.; McKimm-Breschkin, J.L.; Macken, C.; Hampson, A.W.; Hay, A.; Klimov, A.; Tashiro, M.; Webster, R.G.; Aymard, M.; Hayden, F.G.; et al. Detection of influenza viruses resistant to neuraminidase inhibitors in global surveillance during the first 3 years of their use. Antimicrob. Agents Chemother. 2006, 50, 2395–2402. [Google Scholar] [CrossRef] [Green Version]
- Yeo, J.Y.; Gan, S.K. Peering into Avian Influenza A(H5N8) for a Framework towards Pandemic Preparedness. Viruses 2021, 13, 2276. [Google Scholar] [CrossRef]
- Shu, Y.; McCauley, J. GISAID: Global initiative on sharing all influenza data—From vision to reality. Eurosurveillance 2017, 22, 30494. [Google Scholar] [CrossRef] [Green Version]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: Molecular Evolutionary Genetics Analysis version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef] [Green Version]
- Bouckaert, R.; Vaughan, T.G.; Barido-Sottani, J.; Duchene, S.; Fourment, M.; Gavryushkina, A.; Heled, J.; Jones, G.; Kuhnert, D.; De Maio, N.; et al. BEAST 2.5: An advanced software platform for Bayesian evolutionary analysis. PLoS Comput. Biol. 2019, 15, e1006650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qu, B.; Li, X.; Cardona, C.J.; Xing, Z. Reassortment and adaptive mutations of an emerging avian influenza virus H7N4 subtype in China. PLoS ONE 2020, 15, e0227597. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Virus (Year) | Ak/3212/ 2020 | Ck/Ak/321-01/ 2020 | Ck/Ak/2171-1/ 2020 | Ck/RoD/308-02/ 2020 | Ck/V/1488/ 2018 | Ck/RoD/44/ 2017 | Ck/V/18/ 2017 | Ck/Ak/3131/ 2016 | Ck/NL/14015526/ 2014 | |
|---|---|---|---|---|---|---|---|---|---|---|
| \Host Residue | human | chicken | chicken | chicken | chicken | chicken | chicken | chicken | chicken | |
| PB2 | 354 | V | V | V | V | I | I | I | I | I |
| 356 | I | I | I | I | V | V | V | V | V | |
| 522 | H | H | H | H | Q | Q | Q | Q | L | |
| 556 | S | S | S | N | N | N | N | N | N | |
| 677 | G | G | G | E | E | E | E | E | E | |
| PB1 | 142 | S | S | S | S | A | A | A | A | A |
| 181 | V | V | V | V | I | I | I | I | I | |
| 384 | P | P | P | P | S | S | S | S | S | |
| 614 | D | D | D | E | E | E | E | E | E | |
| PB1-F2 | del. | 1-38 aa | 1-38 aa | 1-38 aa | 1-38 aa | 1-38 aa | 1-38 aa | 1-38 aa | 1-38 aa | No |
| PA | 115 | D | D | D | N | N | N | N | N | N |
| 224 | A | A | A | A | S | S | S | S | S | |
| 343 | S | S | S | S | A | A | A | A | V | |
| 545 | V | V | V | V | I | I | I | I | I | |
| 598 | T | A | A | A | A | A | A | A | A | |
| HA | 236 | D | D | D | D | N | N | N | N | N |
| 522 | A | A | A | A | V | V | V | V | V | |
| CS | REKRRKR↓GLF | REKRRKR↓GLF | REKRRKR↓GLF | REKRRKR↓GLF | REKRRKR↓GLF | REKRRKR↓GLF | REKRRKR↓GLF | REKRRKR↓GLF | RERRRKR↓GLF | |
| RBS | QRG | QRG | QRG | QRG | QRG | QRG | QRG | QRG | QRG | |
| NP | 16 | S | S | S | G | G | G | G | G | G |
| 129 | S | S | S | A | A | A | A | T | A | |
| 201 | V | V | V | V | I | I | I | I | I | |
| 219 | F | F | F | F | Y | Y | Y | Y | Y | |
| 371 | I | I | I | I | M | M | M | M | M | |
| NA | stalk del. | No | No | No | No | No | No | No | No | No |
| 28 | L | L | L | L | V | V | V | V | V | |
| 29 | N | S | S | S | S | S | S | S | S | |
| 51 | I | I | I | V | V | V | V | V | V | |
| 107 | I | I | I | I | V | V | V | V | V | |
| 202 | I | I | I | I | V | V | V | V | V | |
| 214 | I | I | I | I | V | V | V | V | V | |
| 246 | S | S | S | S | A | A | A | A | A | |
| 266 | A | A | A | A | T | T | T | T | T | |
| 296 | I | I | I | M | T | T | T | T | T | |
| 360 | M | M | M | M | V | V | V | V | V | |
| 470 | G | G | G | G | K | K | K | K | K | |
| M1 | 139 | A | A | A | A | T | T | T | T | T |
| 248 | L | L | L | L | M | M | M | M | M | |
| M2 | 12 | K | K | K | K | R | R | R | R | R |
| 18 | N | N | N | N | K | K | K | K | R | |
| NS1 | del. | 218-237 aa | 218-237 aa | 218-237 aa | 218-237 aa | 218-237 aa | 218-237 aa | 218-237 aa | 218-237 aa | No |
| 3 | P | P | P | P | S | S | S | S | S | |
| 48 | T | T | T | T | N | N | N | N | S | |
| 53 | S | S | S | S | G | G | G | G | D | |
| 56 | A | A | A | A | T | T | T | T | T | |
| 60 | E | E | E | E | A | A | A | A | A | |
| 66 | K | K | K | K | E | E | E | E | E | |
| 70 | K | K | K | K | E | E | E | E | G | |
| 84 | G | G | G | G | S | S | S | S | V | |
| 109 | R | R | R | R | Q | Q | Q | Q | Q | |
| 114 | P | P | P | P | S | S | S | S | S | |
| 189 | N | N | N | N | D | D | D | D | D | |
| 207 | D | D | D | D | N | N | N | N | N | |
| NS2 | 3 | P | P | P | P | S | S | S | S | S |
| 31 | I | I | I | I | M | M | M | M | M |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ding, L.; Li, J.; Li, X.; Qu, B. Evolutionary and Mutational Characterization of the First H5N8 Subtype Influenza A Virus in Humans. Pathogens 2022, 11, 666. https://doi.org/10.3390/pathogens11060666
Ding L, Li J, Li X, Qu B. Evolutionary and Mutational Characterization of the First H5N8 Subtype Influenza A Virus in Humans. Pathogens. 2022; 11(6):666. https://doi.org/10.3390/pathogens11060666
Chicago/Turabian StyleDing, Lin, Jie Li, Xue Li, and Bingqian Qu. 2022. "Evolutionary and Mutational Characterization of the First H5N8 Subtype Influenza A Virus in Humans" Pathogens 11, no. 6: 666. https://doi.org/10.3390/pathogens11060666
APA StyleDing, L., Li, J., Li, X., & Qu, B. (2022). Evolutionary and Mutational Characterization of the First H5N8 Subtype Influenza A Virus in Humans. Pathogens, 11(6), 666. https://doi.org/10.3390/pathogens11060666