
Diverse Cell Death Mechanisms Are Simultaneously Activated in Macrophages Infected by Virulent Mycobacterium tuberculosis

 , , ,
, , , 
Abstract
:1. Introduction
2. Results
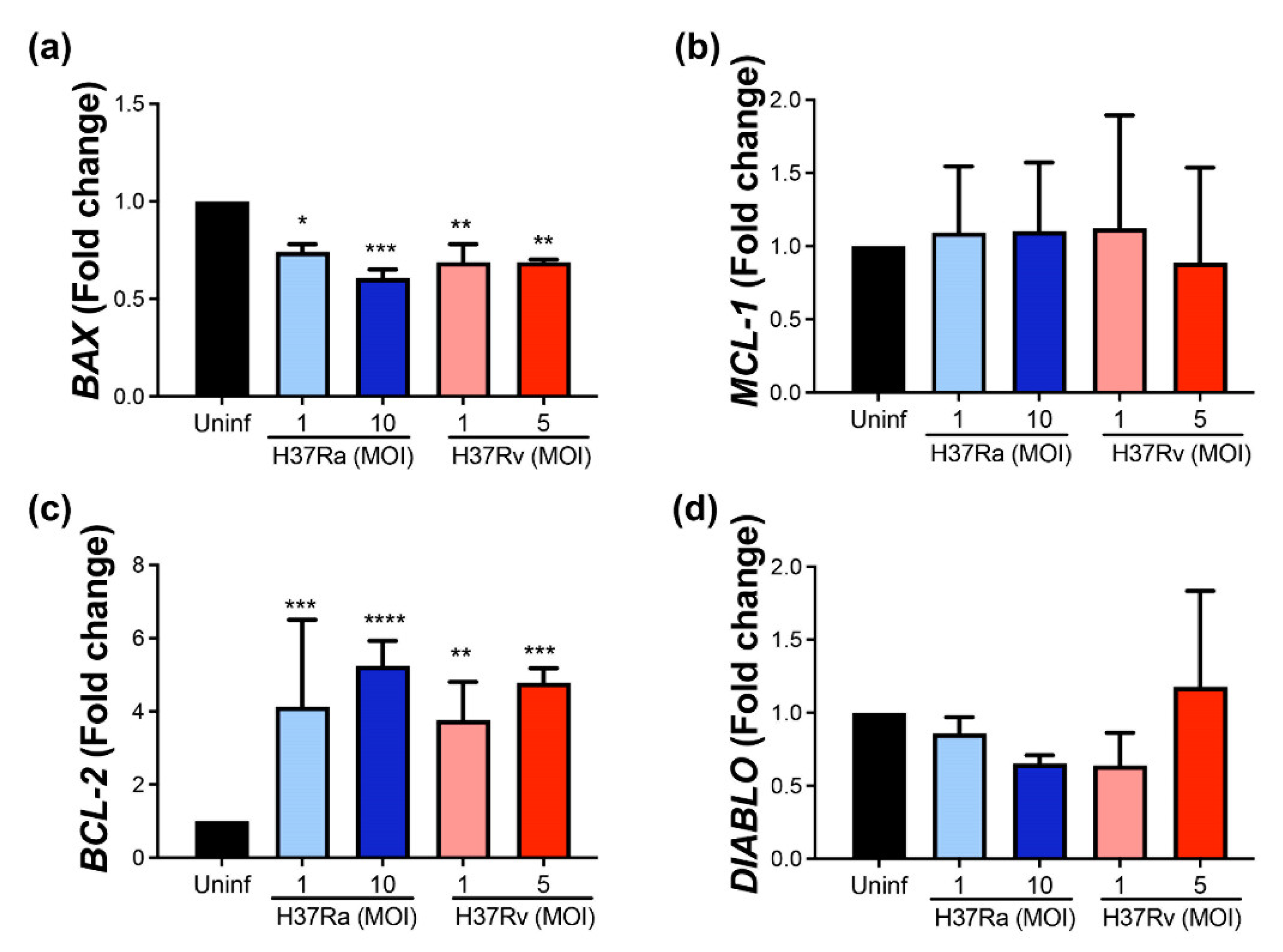
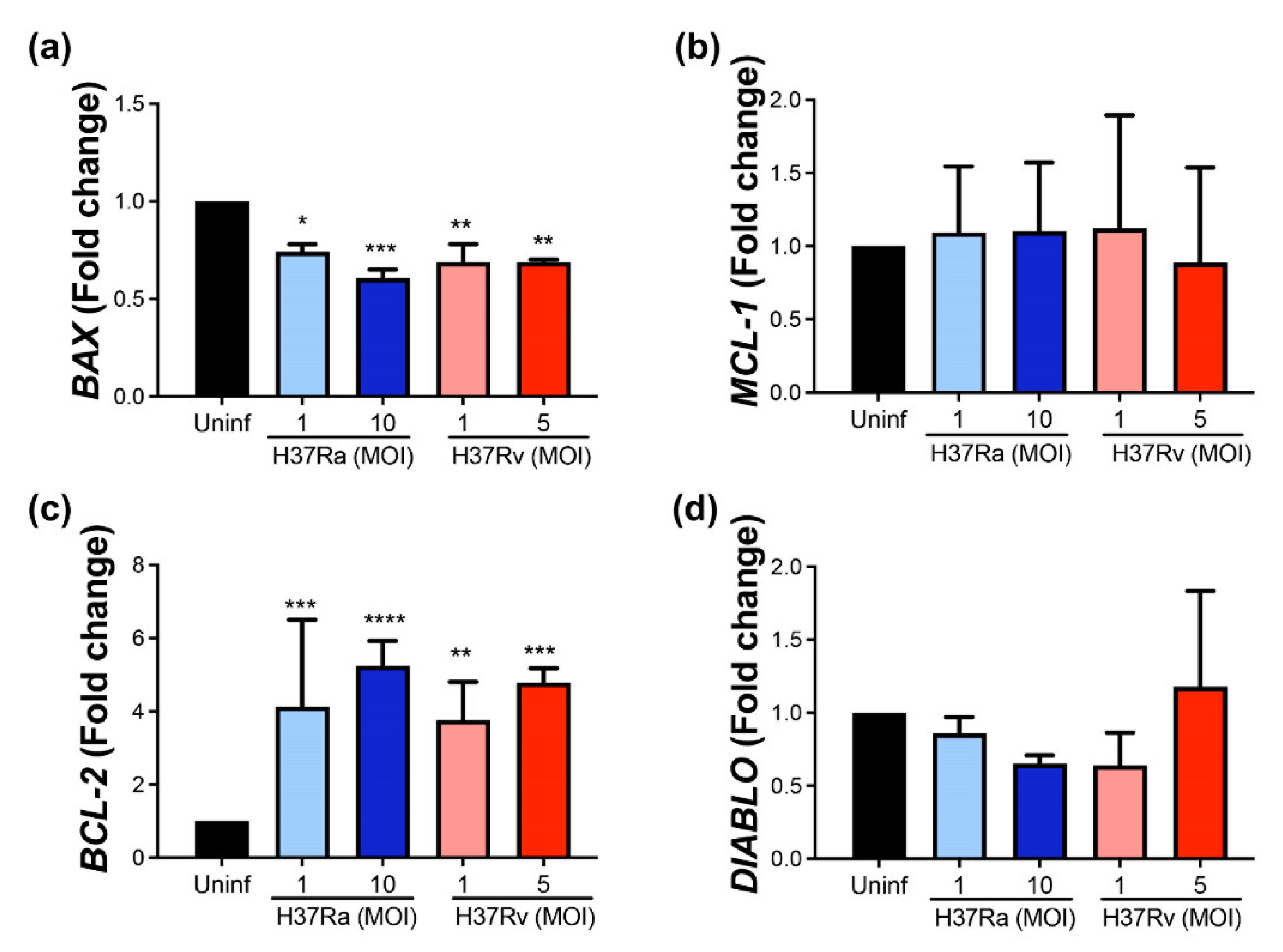
2.1. M. tb Increases BCL-2 and Decreases BAX at the Transcriptional Level, Independently of Its Virulence
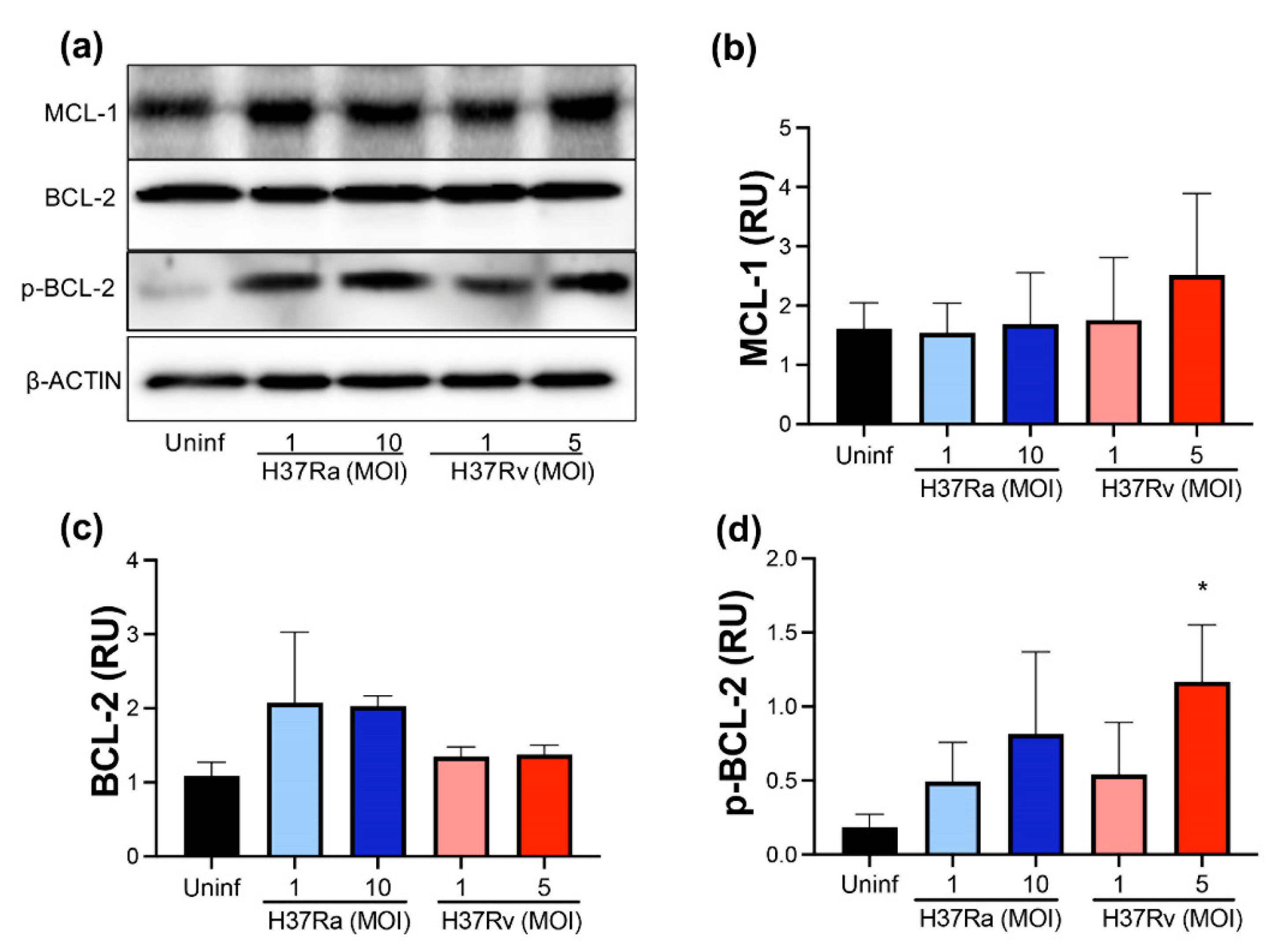
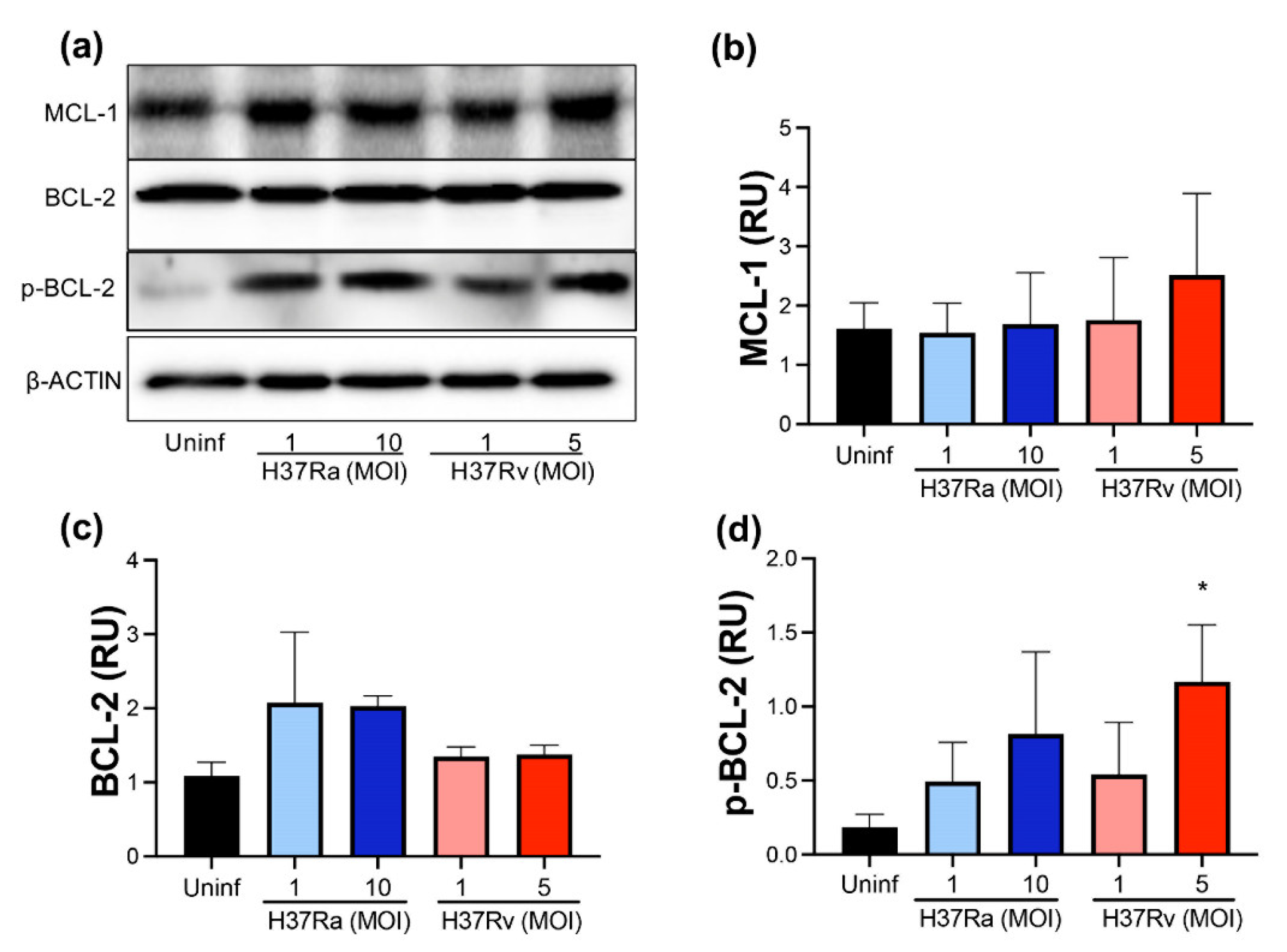
2.2. Virulent M. tb Increases p-BCL-2
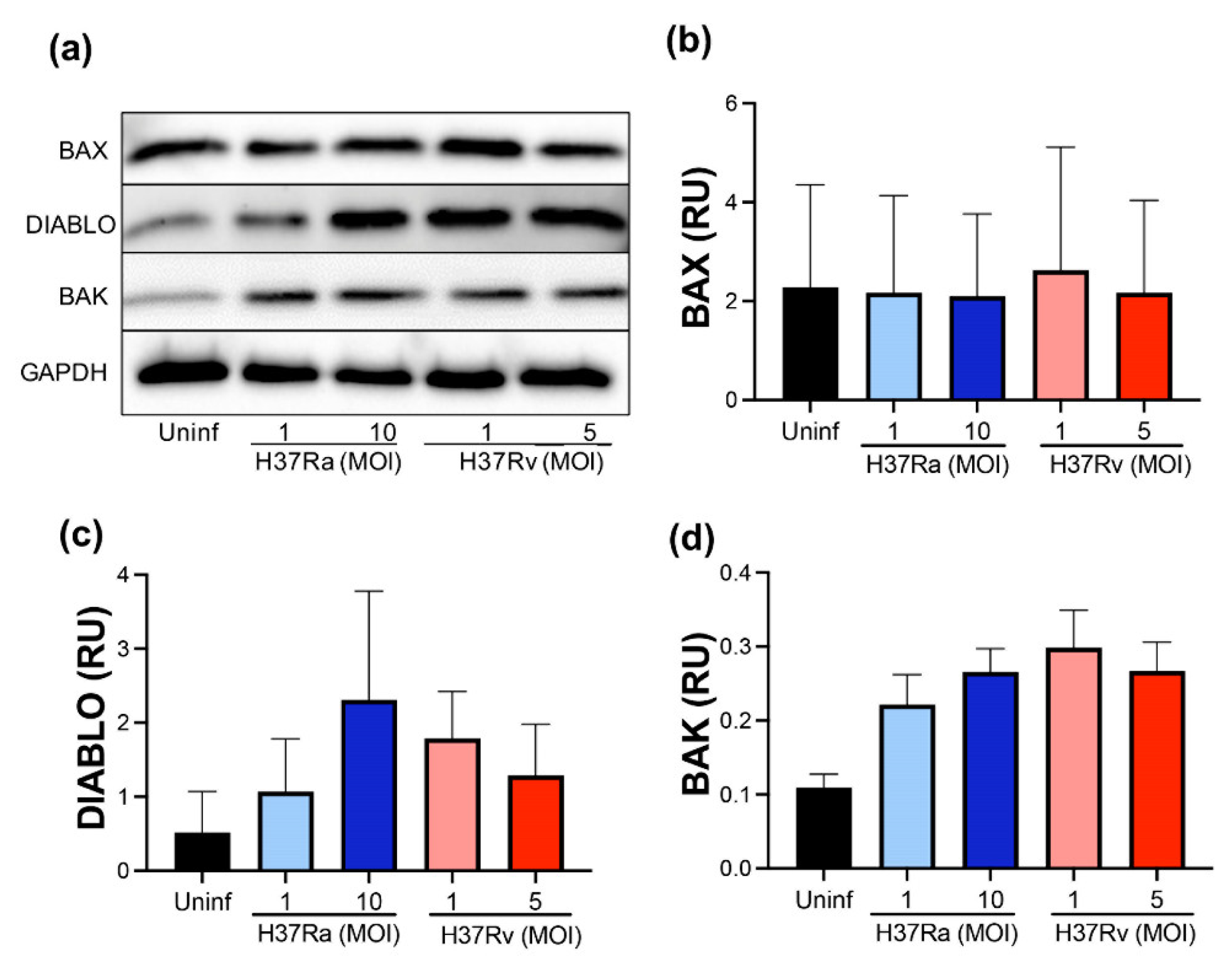
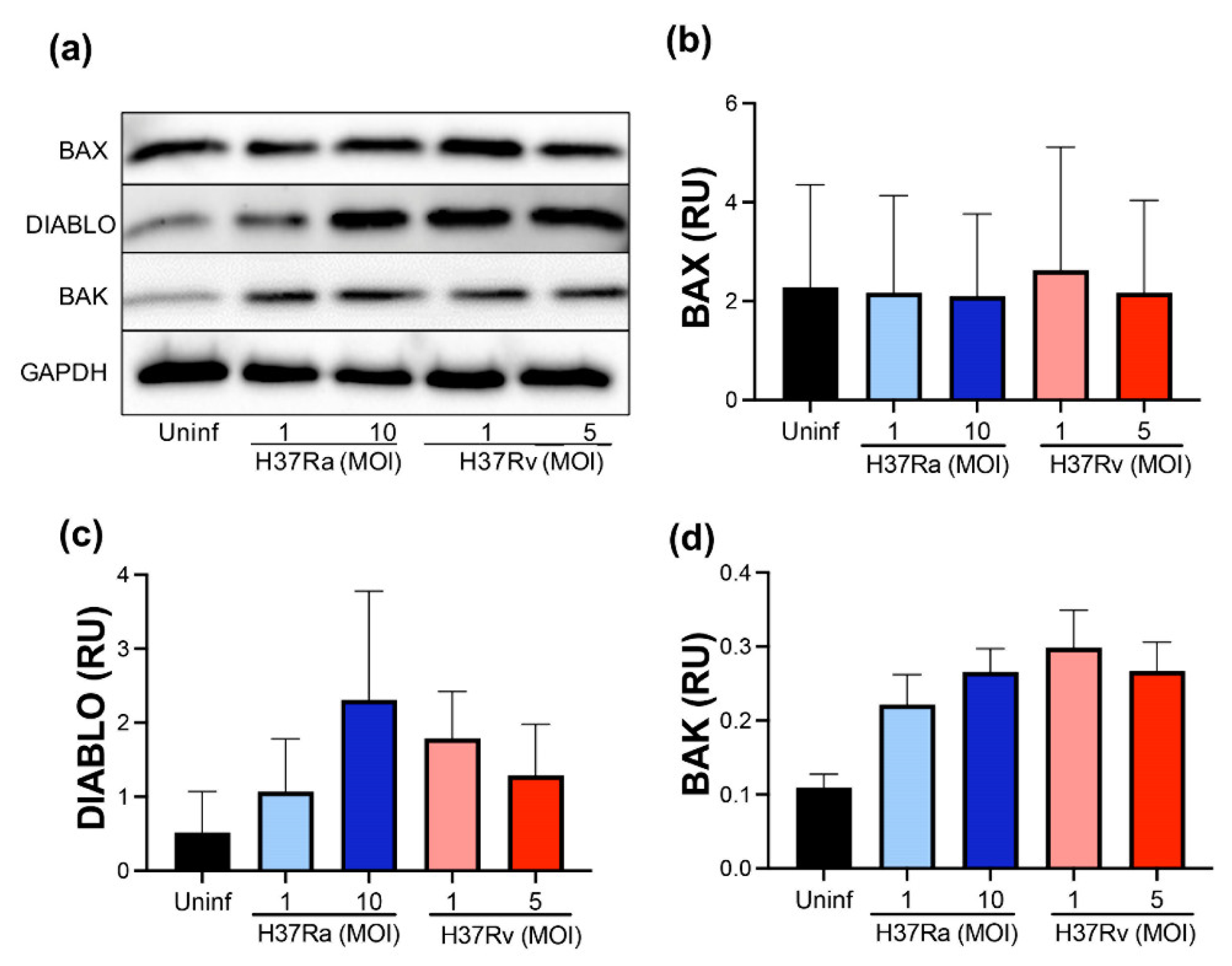
2.3. M. tb Infection Does Not Modify Pro-Apoptotic Proteins
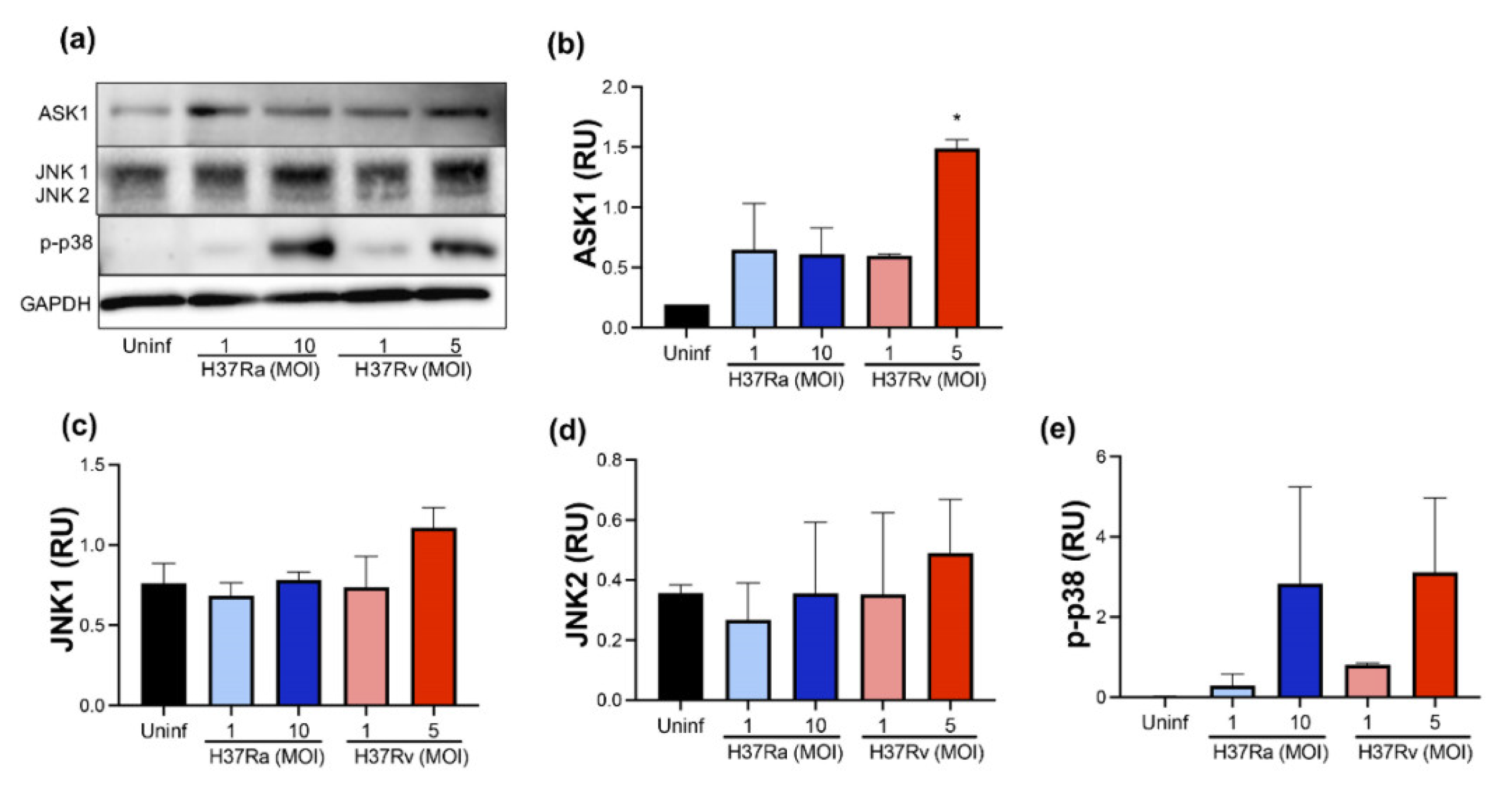
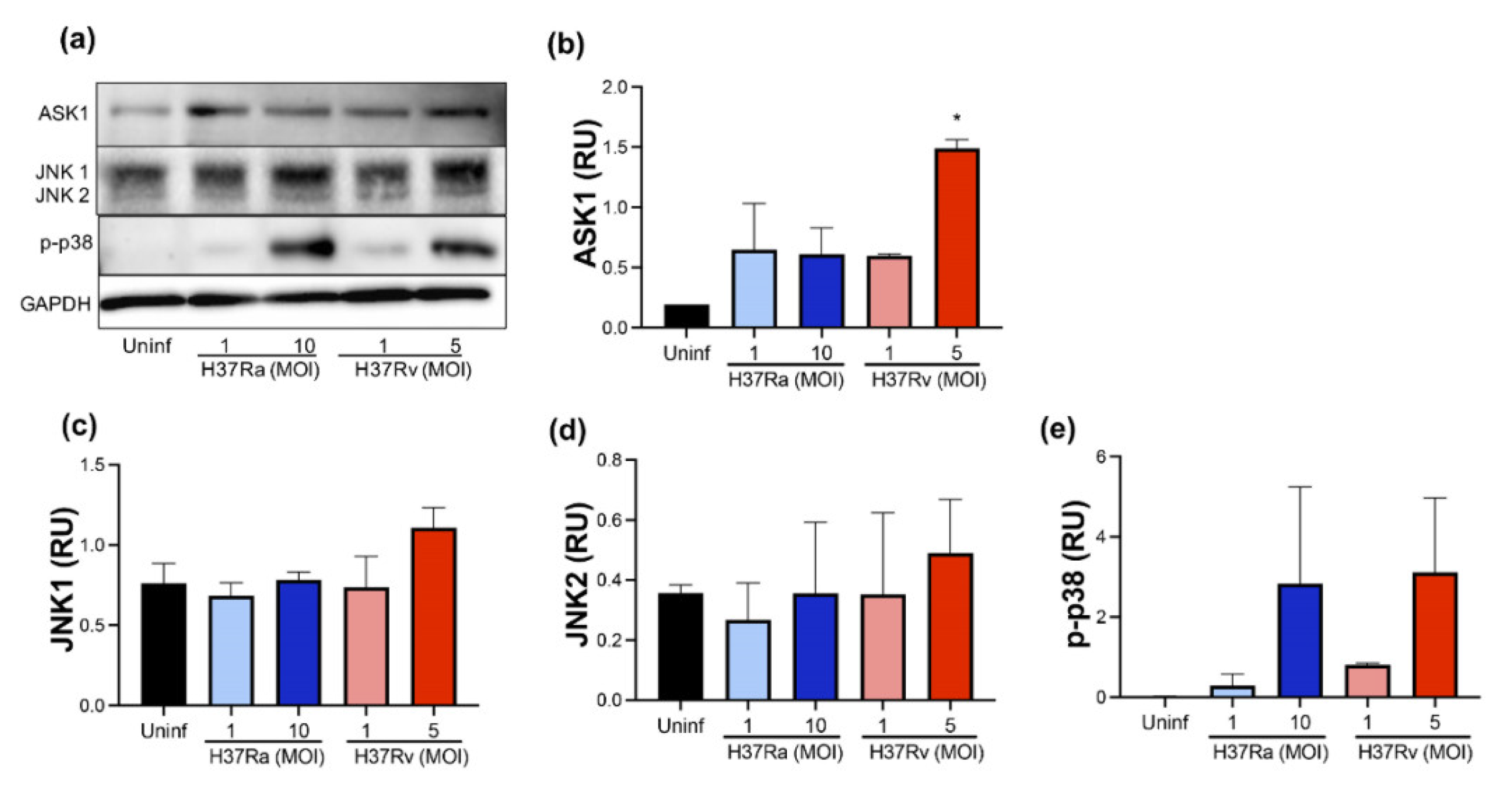
2.4. High MOI of Virulent M. tb Increases ASK1 and p-P38 Expression
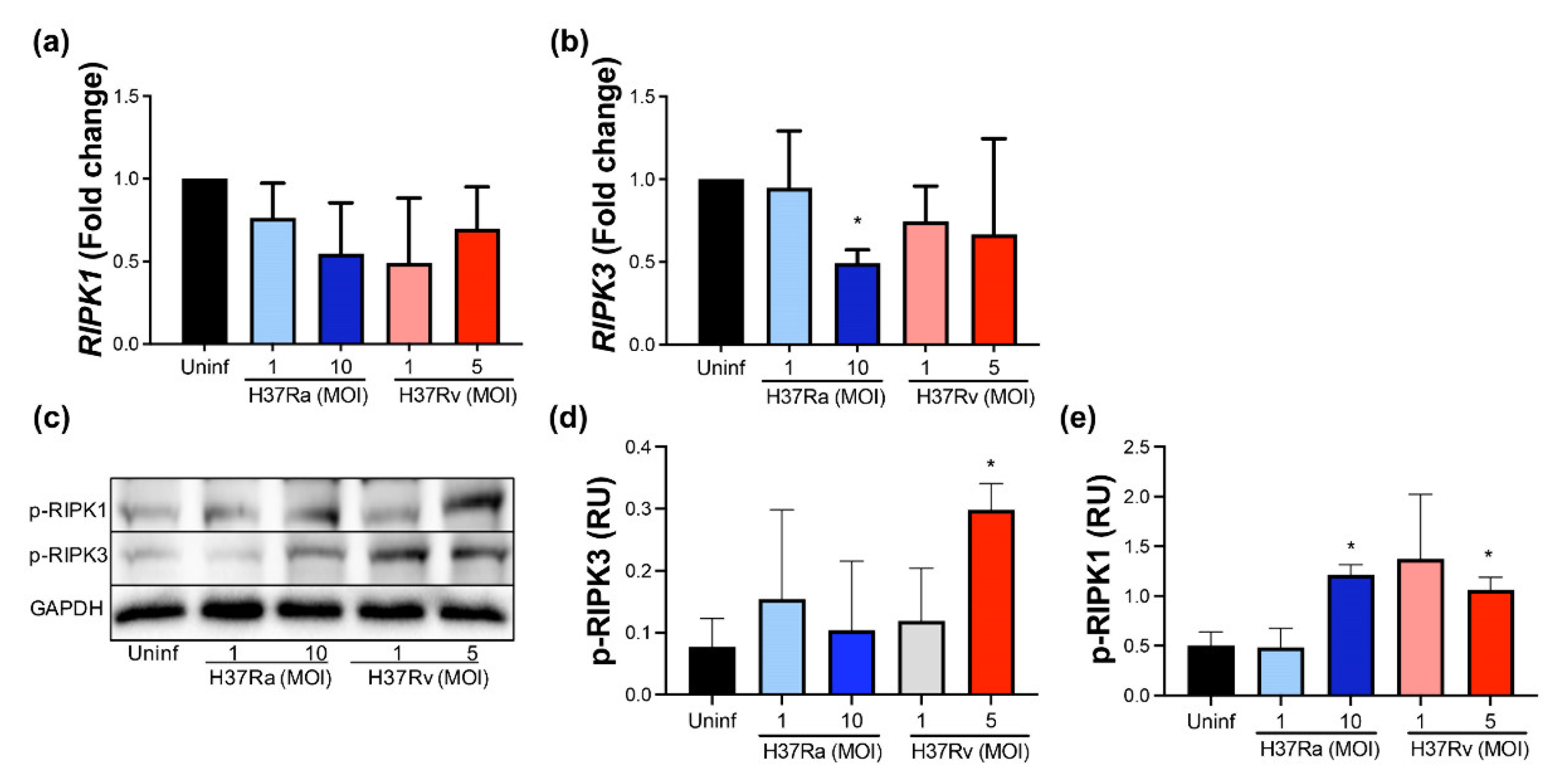
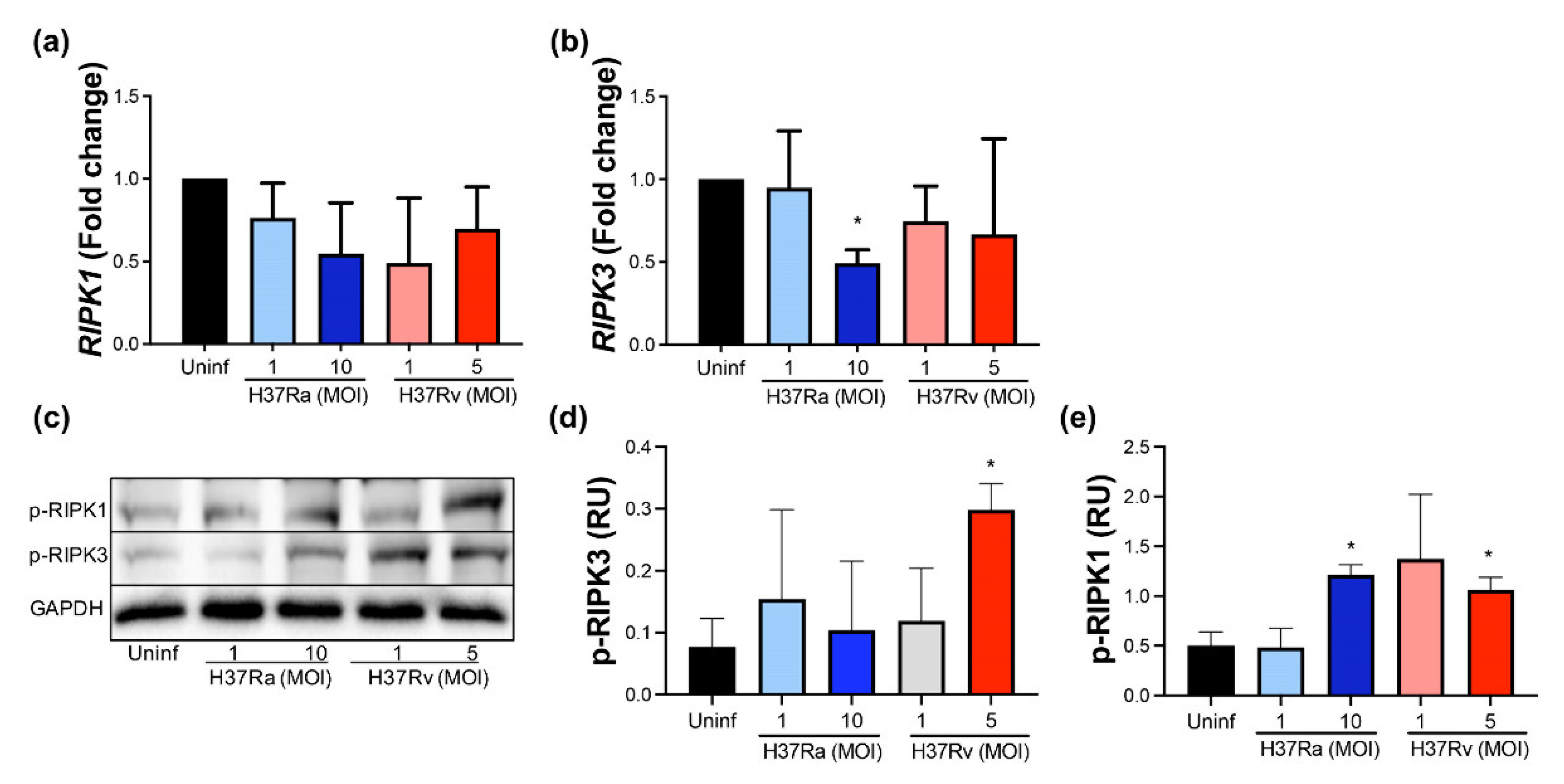
2.5. A High MOI of H37Ra Decreases RIPK3 Expression at the Transcriptional Level, Whereas a High MOI of Either M. tb Strain Increases the Phosphorylated RIPK1 Protein Level
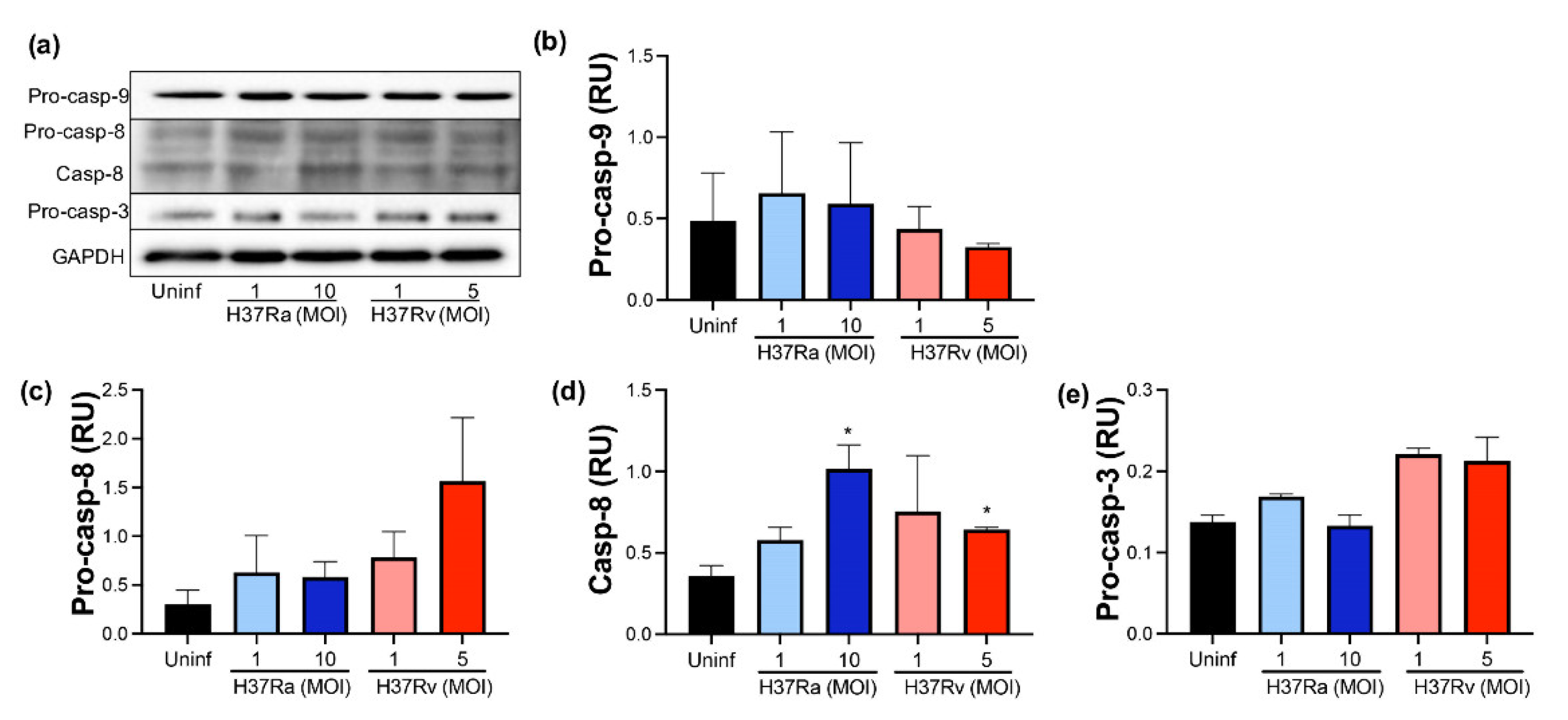
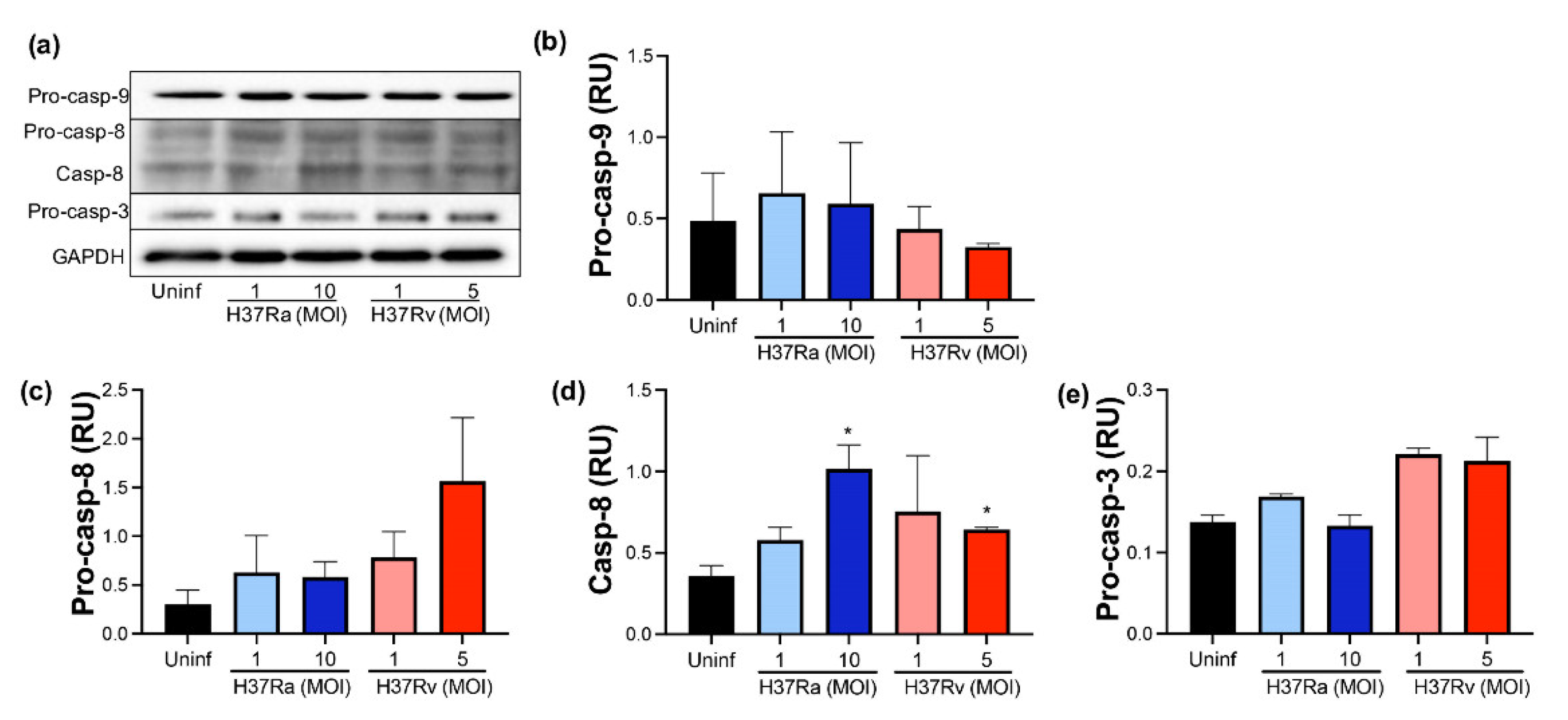
2.6. Virulent M. tb Favors an Increase of Processed Caspase-8
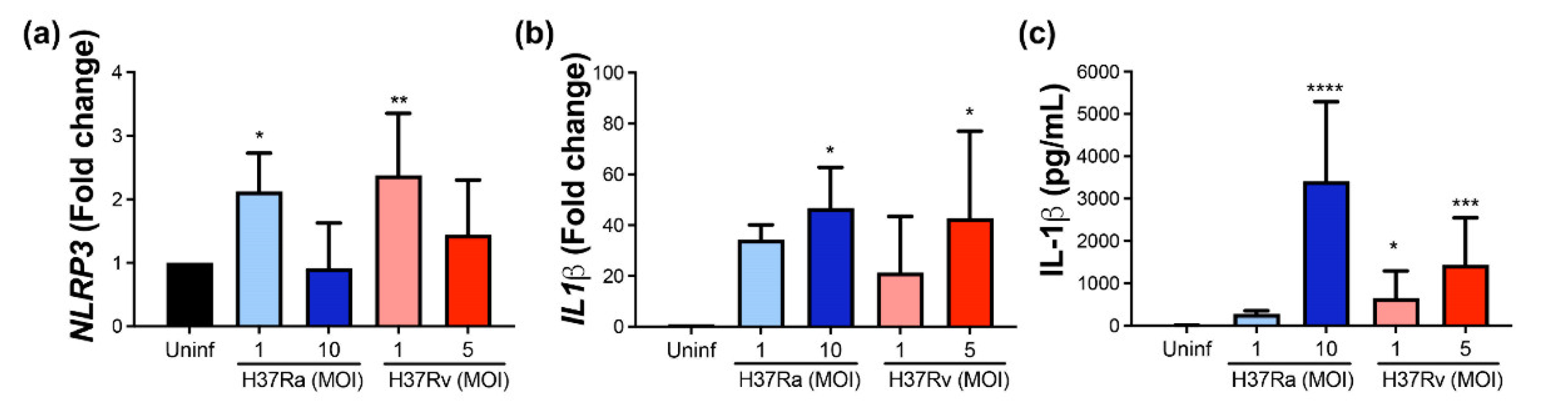
2.7. Virulent M. tb Increases the Expression of NLRP3 and IL-1β, Molecules Involved in Pyroptosis
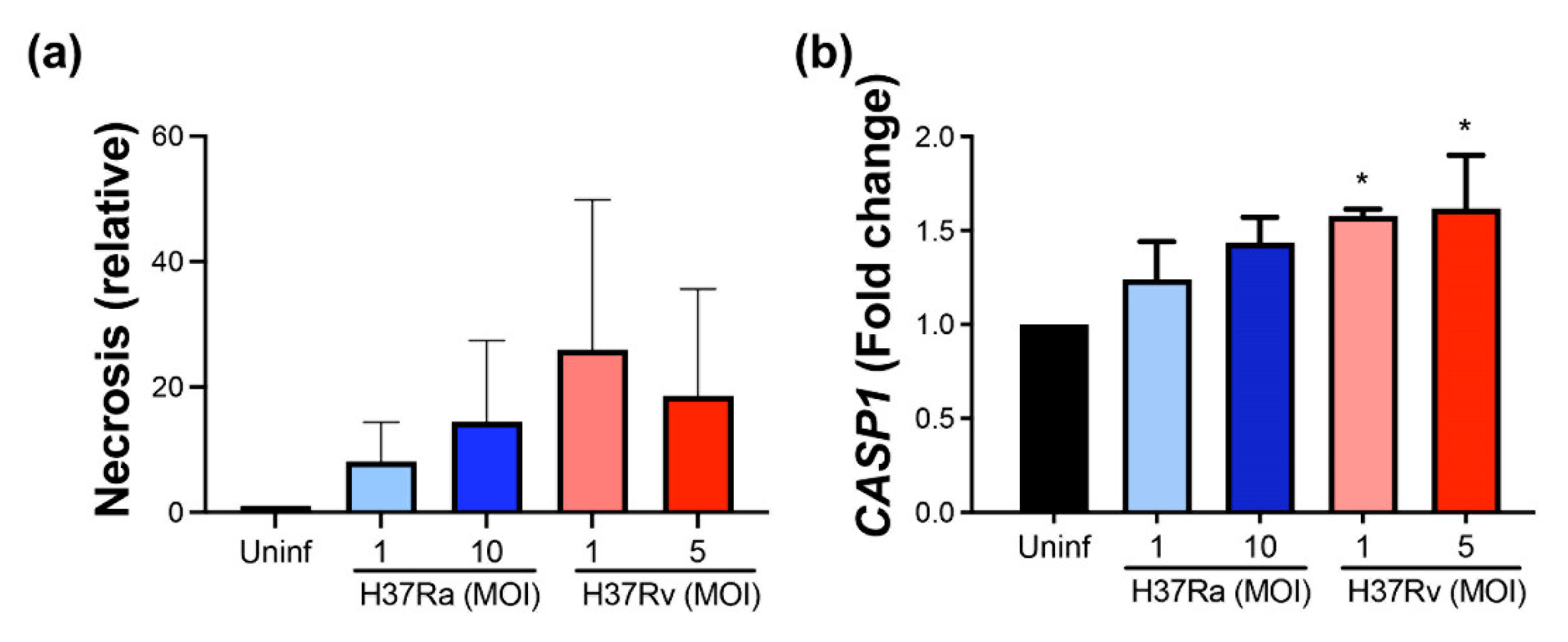
2.8. Virulent M. tb Favors Further Necrosis and the CASP1 Expression at the Transcriptional Level
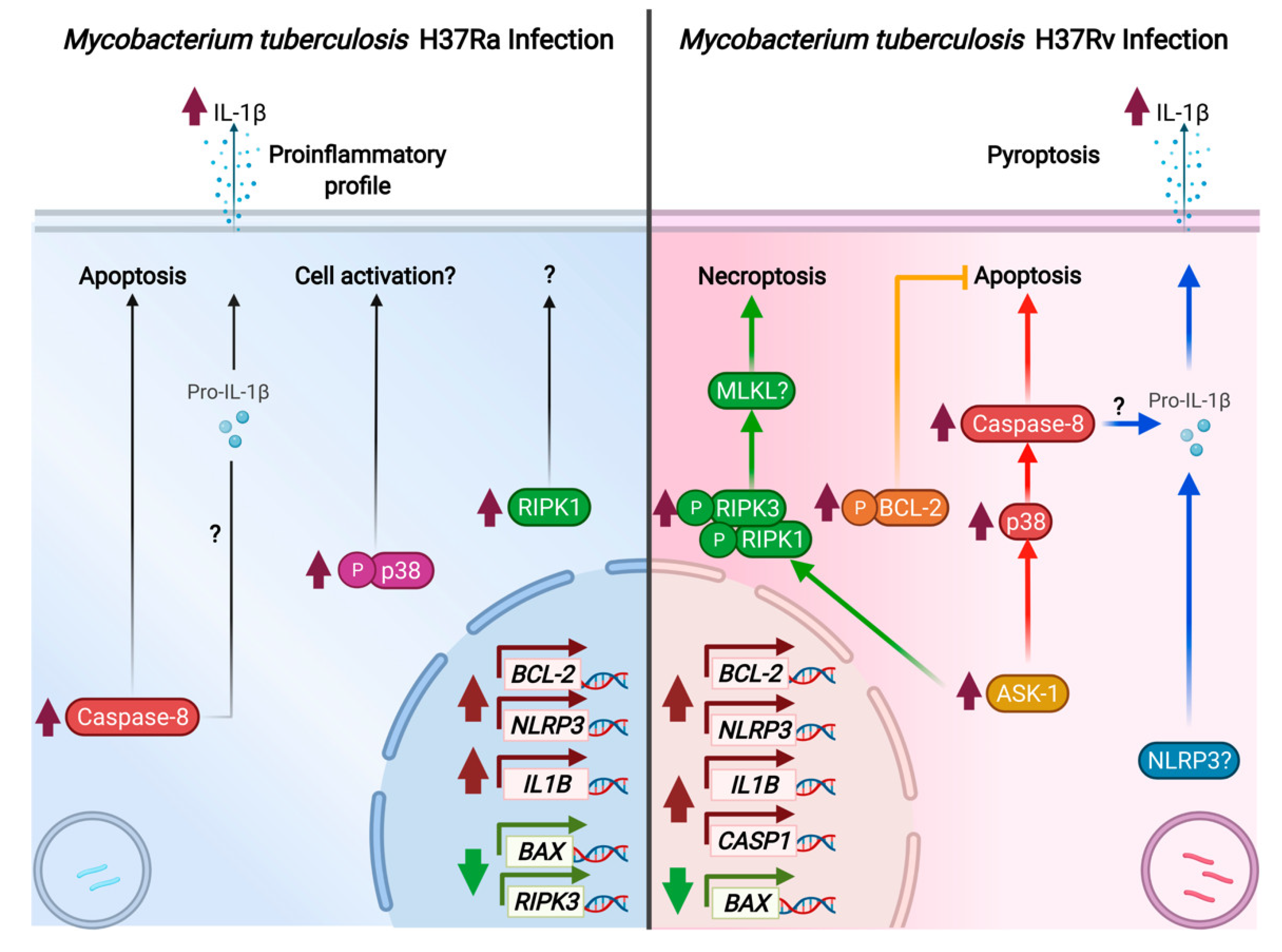
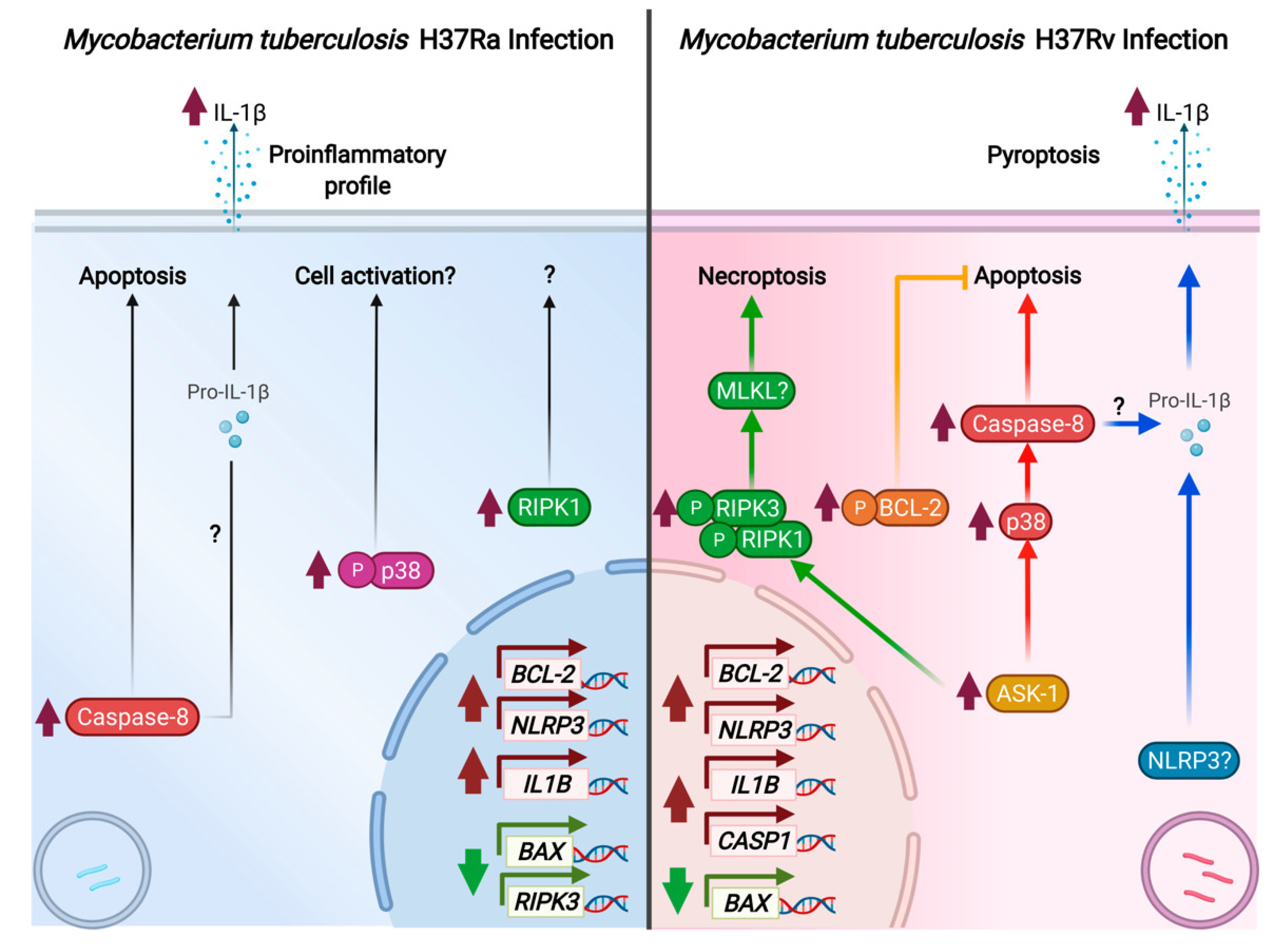
3. Discussion
4. Materials and Methods
4.1. Ethical Approval
4.2. Enrichment of CD14+ Cells and Generation of Monocyte-Derived Macrophages (MDM)
4.3. In Vitro Infection Assays
4.4. Analysis of Gene Expression by Quantitative Real-Time PCR
4.5. Western Blot Assay
4.6. ELISA Sandwich Assays
4.7. Cell Death Detection ELISA
4.8. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antibody | Clone | Fluorochrome/ Catalog Number | Company |
|---|---|---|---|
| CD14 | Monoclonal HCD14 | FITC | BioLegend |
| CD2 | Monoclonal TS1/8 | BV421 | BioLegend |
| CD19 | Monoclonal HIB19 | PE Cy7 | BioLegend |
| BCL-2 | Polyclonal | 2872 | Cell Signaling |
| Phospho-BCL-2 (Ser70) | Monoclonal, 5-H2 | 2827S | Cell Signaling |
| MCL-1 | Polyclonal | 4572s | Cell Signaling |
| BAK | Monoclonal, D2D3 | 6947s | Cell Signaling |
| Caspase-9 | Monoclonal, 13D9C60 | 679101 | BioLegend |
| Caspase-8 | Monoclonal, 84131 | MAB704 | R&D |
| Caspase-3 | Monoclonal, Poly6341 | 634101 | BioLegend |
| ASK1 | Polyclonal | AF3575 | R&D |
| JNK1/2 | Monoclonal, 252323 | MAB2076 | R&D |
| Phospho-RIP1 (Ser166) | Polyclonal | PA5-104645 | Thermo Scientific |
| Phospho-RIP3 (Ser232) | Polyclonal | PA5-105701 | Thermo Scientific |
| Phospho-p38 MAP Kinase (T180/Y182) | Polyclonal | AF869 | R&D |
| BAX | Monoclonal, D2E11 | 5023S | Cell Signaling |
| DIABLO | Monoclonal, D5S3R | 15108S | Cell Signaling |
| Β-ACTIN | Monoclonal, D6A8 | 8457 | Cell Signaling |
| GAPDH | Monoclonal, D16H11 | 5174 | Cell Signaling |
| Sheep | Polyclonal | HAF016 | R&D |
| rabbit | Polyclonal | HAF008 | R&D |
| mouse | Polyclonal | HAF007 | R&D |
References
- World Health Organization (WHO). Global Tuberculosis Report. 2021. Available online: https://www.who.int/teams/global-tuberculosis-programme/tb-reports/global-tuberculosis-report-2021 (accessed on 28 November 2021).
- Mertens, C.; Marques, O.; Horvat, N.K.; Simonetti, M.; Muckenthaler, M.U.; Jung, M. The Macrophage Iron Signature in Health and Disease. Int. J. Mol. Sci. 2021, 22, 8457. [Google Scholar] [CrossRef] [PubMed]
- Shapouri-Moghaddam, A.; Mohammadian, S.; Vazini, H.; Taghadosi, M.; Esmaeili, S.A.; Mardani, F.; Seifi, B.; Mohammadi, A.; Afshari, J.T.; Sahebkar, A. Macrophage Plasticity, Polarization, and Function in Health and Disease. J. Cell. Physiol. 2018, 233, 6425–6440. [Google Scholar] [CrossRef] [PubMed]
- Orecchioni, M.; Ghosheh, Y.; Pramod, A.B.; Ley, K. Macrophage Polarization: Different Gene Signatures in M1(Lps+) vs. Classically and M2(LPS-) vs. Alternatively Activated Macrophages. Front. Immunol. 2019, 10, 1084. [Google Scholar] [CrossRef]
- Beham, A.W.; Puellmann, K.; Laird, R.; Fuchs, T.; Streich, R.; Breysach, C.; Raddatz, D.; Oniga, S.; Peccerella, T.; Findeisen, P.; et al. A TNF-Regulated Recombinatorial Macrophage Immune Receptor Implicated in Granuloma Formation in Tuberculosis. PLoS Pathog. 2011, 7, e1002375. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Cruz, A.; Vesin, D.; Ramon-Luing, L.; Zuñiga, J.; Quesniaux, V.F.J.; Ryffel, B.; Lascurain, R.; Garcia, I.; Chávez-Galán, L. CD3+ Macrophages Deliver Proinflammatory Cytokines by a CD3- and Transmembrane TNF-Dependent Pathway and Are Increased at the BCG-Infection Site. Front. Immunol. 2019, 10, 2550. [Google Scholar] [CrossRef] [PubMed]
- Chavez-Galan, L.; Vesin, D.; Blaser, G.; Uysal, H.; Benmerzoug, S.; Rose, S.; Ryffel, B.; Quesniaux, V.F.J.; Garcia, I. Myeloid Cell TNFR1 Signaling Dependent Liver Injury and Inflammation upon BCG Infection. Sci. Rep. 2019, 9, 5297. [Google Scholar] [CrossRef]
- Ramon-Luing, L.A.; Carranza, C.; Téllez-Navarrete, N.A.; Medina-Quero, K.; Gonzalez, Y.; Torres, M.; Chavez-Galan, L. Mycobacterium Tuberculosis H37Rv Strain Increases the Frequency of CD3+TCR+ Macrophages and Affects Their Phenotype, but Not Their Migration Ability. Int. J. Mol. Sci. 2021, 23, 329. [Google Scholar] [CrossRef]
- Moule, M.G.; Cirillo, J.D. Mycobacterium Tuberculosis Dissemination Plays a Critical Role in Pathogenesis. Front. Cell. Infect. Microbiol. 2020, 10, 65. [Google Scholar] [CrossRef]
- Mohareer, K.; Asalla, S.; Banerjee, S. Cell Death at the Cross Roads of Host-Pathogen Interaction in Mycobacterium Tuberculosis Infection. Tuberculosis 2018, 113, 99–121. [Google Scholar] [CrossRef]
- Xu, X.; Lai, Y.; Hua, Z.C. Apoptosis and Apoptotic Body: Disease Message and Therapeutic Target Potentials. Biosci. Rep. 2019, 39, 20180992. [Google Scholar] [CrossRef] [Green Version]
- Warren, C.F.A.; Wong-Brown, M.W.; Bowden, N.A. BCL-2 Family Isoforms in Apoptosis and Cancer. Cell Death Dis. 2019, 10, 177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peña-Blanco, A.; García-Sáez, A.J. Bax, Bak and beyond—Mitochondrial Performance in Apoptosis. FEBS J. 2018, 285, 416–431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Theobald, S.J.; Gräb, J.; Fritsch, M.; Suárez, I.; Eisfeld, H.S.; Winter, S.; Koch, M.; Hölscher, C.; Pasparakis, M.; Kashkar, H.; et al. Gasdermin D Mediates Host Cell Death but Not Interleukin-1β Secretion in Mycobacterium Tuberculosis-Infected Macrophages. Cell Death Discov. 2021, 7, 327. [Google Scholar] [CrossRef] [PubMed]
- Beckwith, K.S.; Beckwith, M.S.; Ullmann, S.; Sætra, R.S.; Kim, H.; Marstad, A.; Åsberg, S.E.; Strand, T.A.; Haug, M.; Niederweis, M.; et al. Plasma Membrane Damage Causes NLRP3 Activation and Pyroptosis during Mycobacterium Tuberculosis Infection. Nat. Commun. 2020, 11, 2270. [Google Scholar] [CrossRef]
- Chávez-Galán, L.; Ramon-Luing, L.A.; Torre-Bouscoulet, L.; Pérez-Padilla, R.; Sada-Ovalle, I. Pre-Exposure of Mycobacterium Tuberculosis-Infected Macrophages to Crystalline Silica Impairs Control of Bacterial Growth by Deregulating the Balance between Apoptosis and Necrosis. PLoS ONE 2013, 8, e80971. [Google Scholar] [CrossRef]
- Afriyie-Asante, A.; Dabla, A.; Dagenais, A.; Berton, S.; Smyth, R.; Sun, J. Mycobacterium Tuberculosis Exploits Focal Adhesion Kinase to Induce Necrotic Cell Death and Inhibit Reactive Oxygen Species Production. Front. Immunol. 2021, 12, 4401. [Google Scholar] [CrossRef]
- Zhao, X.; Khan, N.; Gan, H.; Tzelepis, F.; Nishimura, T.; Park, S.Y.; Divangahi, M.; Remold, H.G. Bcl-XL Mediates RIPK3-Dependent Necrosis in M. Tuberculosis-Infected Macrophages. Mucosal Immunol. 2017, 10, 1553. [Google Scholar] [CrossRef] [Green Version]
- Tenev, T.; Bianchi, K.; Darding, M.; Broemer, M.; Langlais, C.; Wallberg, F.; Zachariou, A.; Lopez, J.; MacFarlane, M.; Cain, K.; et al. The Ripoptosome, a Signaling Platform That Assembles in Response to Genotoxic Stress and Loss of IAPs. Mol. Cell 2011, 43, 432–448. [Google Scholar] [CrossRef]
- Pajuelo, D.; Gonzalez-Juarbe, N.; Tak, U.; Sun, J.; Orihuela, C.J.; Niederweis, M. NAD+ Depletion Triggers Macrophage Necroptosis, a Cell Death Pathway Exploited by Mycobacterium Tuberculosis. Cell Rep. 2018, 24, 429. [Google Scholar] [CrossRef] [Green Version]
- Davis, R.J. Signal Transduction by the JNK Group of MAP Kinases. Cell 2000, 103, 239–252. [Google Scholar] [CrossRef] [Green Version]
- Yue, J.; López, J.M. Understanding MAPK Signaling Pathways in Apoptosis. Int. J. Mol. Sci. 2020, 21, 2346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bergsbaken, T.; Fink, S.L.; Cookson, B.T. Pyroptosis: Host Cell Death and Inflammation. Nat. Rev. Microbiol. 2009, 7, 99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carranza, C.; Chavez-Galan, L. Several Routes to the Same Destination: Inhibition of Phagosome-Lysosome Fusion by Mycobacterium Tuberculosis. Am. J. Med. Sci. 2019, 357, 184–194. [Google Scholar] [CrossRef] [PubMed]
- Chai, Q.; Wang, L.; Liu, C.H.; Ge, B. New Insights into the Evasion of Host Innate Immunity by Mycobacterium Tuberculosis. Cell. Mol. Immunol. 2020, 17, 901–913. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.K.; Silwal, P.; Jo, E.K. Host-Pathogen Dialogues in Autophagy, Apoptosis, and Necrosis during Mycobacterial Infection. Immune Netw. 2020, 20, e37. [Google Scholar] [CrossRef]
- Chávez-Galán, L.; Sada-Ovalle, I.; Baez-Saldaña, R.; Chávez, R.; Lascurain, R. Monocytes from Tuberculosis Patients That Exhibit Cleaved Caspase 9 and Denaturalized Cytochrome c Are More Susceptible to Death Mediated by Toll-like Receptor 2. Immunology 2012, 135, 299–311. [Google Scholar] [CrossRef] [Green Version]
- Khan, N.; Downey, J.; Sanz, J.; Kaufmann, E.; Blankenhaus, B.; Pacis, A.; Pernet, E.; Ahmed, E.; Cardoso, S.; Nijnik, A.; et al. M. tuberculosis Reprograms Hematopoietic Stem Cells to Limit Myelopoiesis and Impair Trained Immunity. Cell 2020, 183, 752–770.e22. [Google Scholar] [CrossRef]
- de Zuani, M.; Frič, J. Train the Trainer: Hematopoietic Stem Cell Control of Trained Immunity. Front. Immunol. 2022, 13, 827250. [Google Scholar] [CrossRef]
- Aguilo, J.I.; Alonso, H.; Uranga, S.; Marinova, D.; Arbués, A.; de Martino, A.; Anel, A.; Monzon, M.; Badiola, J.; Pardo, J.; et al. ESX-1-Induced Apoptosis Is Involved in Cell-to-Cell Spread of Mycobacterium Tuberculosis. Cell. Microbiol. 2013, 15, 1994–2005. [Google Scholar] [CrossRef]
- Arnett, E.; Schlesinger, L.S. Live and Let Die: TB Control by Enhancing Apoptosis. Immunity 2021, 54, 1625–1627. [Google Scholar] [CrossRef]
- Dallenga, T.; Repnik, U.; Corleis, B.; Eich, J.; Reimer, R.; Griffiths, G.W.; Schaible, U.E. M. tuberculosis-Induced Necrosis of Infected Neutrophils Promotes Bacterial Growth Following Phagocytosis by Macrophages. Cell Host Microbe 2017, 22, 519–530.e3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lerner, T.R.; Borel, S.; Greenwood, D.J.; Repnik, U.; Russell, M.R.G.; Herbst, S.; Jones, M.L.; Collinson, L.M.; Griffiths, G.; Gutierrez, M.G. Mycobacterium Tuberculosis Replicates within Necrotic Human Macrophages. J. Cell Biol. 2017, 216, 583–594. [Google Scholar] [CrossRef] [PubMed]
- Rodel, H.E.; Ferreira, I.A.T.M.; Ziegler, C.G.K.; Ganga, Y.; Bernstein, M.; Hwa, S.H.; Nargan, K.; Lustig, G.; Kaplan, G.; Noursadeghi, M.; et al. Aggregated Mycobacterium Tuberculosis Enhances the Inflammatory Response. Front. Microbiol. 2021, 12, 3436. [Google Scholar] [CrossRef]
- Dadsena, S.; Zollo, C.; García-Sáez, A.J. Mechanisms of Mitochondrial Cell Death. Biochem. Soc. Trans. 2021, 49, 663–674. [Google Scholar] [CrossRef] [PubMed]
- Divangahi, M.; Chen, M.; Gan, H.; Desjardins, D.; Hickman, T.T.; Lee, D.M.; Fortune, S.; Behar, S.M.; Remold, H.G. Mycobacterium Tuberculosis Evades Macrophage Defenses by Inhibiting Plasma Membrane Repair. Nat. Immunol. 2009, 10, 899. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, M.; Divangahi, M.; Gan, H.; Shin, D.S.J.; Hong, S.; Lee, D.M.; Serhan, C.N.; Behar, S.M.; Remold, H.G. Lipid Mediators in Innate Immunity against Tuberculosis: Opposing Roles of PGE2 and LXA4 in the Induction of Macrophage Death. J. Exp. Med. 2008, 205, 2791. [Google Scholar] [CrossRef] [Green Version]
- Chen, M.; Gan, H.; Remold, H.G. A Mechanism of Virulence: Virulent Mycobacterium Tuberculosis Strain H37Rv, but Not Attenuated H37Ra, Causes Significant Mitochondrial Inner Membrane Disruption in Macrophages Leading to Necrosis. J Immunol 2006, 176, 3707–3716. [Google Scholar] [CrossRef] [Green Version]
- Sly, L.M.; Hingley-Wilson, S.M.; Reiner, N.E.; McMaster, W.R. Survival of Mycobacterium Tuberculosis in Host Macrophages Involves Resistance to Apoptosis Dependent upon Induction of Antiapoptotic Bcl-2 Family Member Mcl-1. J. Immunol. 2003, 170, 430–437. [Google Scholar] [CrossRef] [Green Version]
- Han, L.; Lu, Y.; Wang, X.; Zhang, S.; Wang, Y.; Wu, F.; Zhang, W.; Wang, X.; Zhang, L. Regulatory Role and Mechanism of the Inhibition of the Mcl-1 Pathway during Apoptosis and Polarization of H37Rv-Infected Macrophages. Medicine 2020, 99, e22438. [Google Scholar] [CrossRef]
- Adrain, C.; Creagh, E.M.; Martin, S.J. Apoptosis-Associated Release of Smac/DIABLO from Mitochondria Requires Active Caspases and Is Blocked by Bcl-2. EMBO J. 2001, 20, 6627–6636. [Google Scholar] [CrossRef] [Green Version]
- Pyrillou, K.; Burzynski, L.C.; Clarke, M.C.H. Alternative Pathways of IL-1 Activation, and Its Role in Health and Disease. Front. Immunol. 2020, 11, 3288. [Google Scholar] [CrossRef] [PubMed]
- Man, S.M.; Tourlomousis, P.; Hopkins, L.; Monie, T.P.; Fitzgerald, K.A.; Bryant, C.E. Salmonella Infection Induces Recruitment of Caspase-8 to the Inflammasome to Modulate IL-1β Production. J. Immunol. 2013, 191, 5239–5246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weng, D.; Marty-Roix, R.; Ganesan, S.; Proulx, M.K.; Vladimer, G.I.; Kaiser, W.J.; Mocarski, E.S.; Pouliot, K.; Chan, F.K.M.; Kelliher, M.A.; et al. Caspase-8 and RIP Kinases Regulate Bacteria-Induced Innate Immune Responses and Cell Death. Proc. Natl. Acad. Sci. USA 2014, 111, 7391–7396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.; Remold, H.G.; Ieong, M.H.; Kornfeld, H. Macrophage Apoptosis in Response to High Intracellular Burden of Mycobacterium Tuberculosis Is Mediated by a Novel Caspase-Independent Pathway. J. Immunol. 2006, 176, 4267–4274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Obsilova, V.; Honzejkova, K.; Obsil, T. Structural Insights Support Targeting ASK1 Kinase for Therapeutic Interventions. Int. J. Mol. Sci. 2021, 22, 13395. [Google Scholar] [CrossRef] [PubMed]
- Kishino, A.; Hayashi, K.; Maeda, M.; Jike, T.; Hidai, C.; Nomura, Y.; Oshima, T. Caspase-8 Regulates Endoplasmic Reticulum Stress-Induced Necroptosis Independent of the Apoptosis Pathway in Auditory Cells. Int. J. Mol. Sci. 2019, 20, 5896. [Google Scholar] [CrossRef] [Green Version]
- Bertheloot, D.; Latz, E.; Franklin, B.S. Necroptosis, Pyroptosis and Apoptosis: An Intricate Game of Cell Death. Cell. Mol. Immunol. 2021, 18, 1106–1121. [Google Scholar] [CrossRef]
- Amaral, E.P.; Namasivayam, S. Emerging Role for Ferroptosis in Infectious Diseases. Adv. Exp. Med. Biol. 2021, 1301, 59–79. [Google Scholar] [CrossRef]
- Halliwell, B. Reflections of an Aging Free Radical. Free Radic. Biol. Med. 2020, 161, 234–245. [Google Scholar] [CrossRef]
- Conrad, M.; Kagan, V.E.; Bayir, H.; Pagnussat, G.C.; Head, B.; Traber, M.G.; Stockwell, B.R. Regulation of Lipid Peroxidation and Ferroptosis in Diverse Species. Genes Dev. 2018, 32, 602–619. [Google Scholar] [CrossRef] [Green Version]
- Dodson, M.; Castro-Portuguez, R.; Zhang, D.D. NRF2 Plays a Critical Role in Mitigating Lipid Peroxidation and Ferroptosis. Redox Biol. 2019, 23, 101107. [Google Scholar] [CrossRef] [PubMed]
- Haschka, D.; Hoffmann, A.; Weiss, G. Iron in Immune Cell Function and Host Defense. Semin. Cell Dev. Biol. 2021, 115, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Amaral, E.P.; Costa, D.L.; Namasivayam, S.; Riteau, N.; Kamenyeva, O.; Mittereder, L.; Mayer-Barber, K.D.; Andrade, B.B.; Sher, A. A Major Role for Ferroptosis in Mycobacterium Tuberculosis–Induced Cell Death and Tissue Necrosis. J. Exp. Med. 2019, 216, 556–570. [Google Scholar] [CrossRef] [PubMed]
- Luo, K.; Stocker, R.; Britton, W.J.; Kikuchi, K.; Oehlers, S.H. Haem Oxygenase Limits Mycobacterium Marinum Infection-Induced Detrimental Ferrostatin-Sensitive Cell Death in Zebrafish. FEBS J. 2022, 289, 671–681. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Chen, X.; Gueydan, C.; Han, J. Plasma Membrane Changes during Programmed Cell Deaths. Cell Res. 2018, 28, 9–21. [Google Scholar] [CrossRef]
- Carranza, C.; Juárez, E.; Torres, M.; Ellner, J.J.; Sada, E.; Schwander, S.K. Mycobacterium Tuberculosis Growth Control by Lung Macrophages and CD8 Cells from Patient Contacts. Am. J. Respir. Crit. Care Med. 2006, 173, 238–245. [Google Scholar] [CrossRef] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ramon-Luing, L.A.; Olvera, Y.; Flores-Gonzalez, J.; Palacios, Y.; Carranza, C.; Aguilar-Duran, Y.; Vargas, M.A.; Gutierrez, N.; Medina-Quero, K.; Chavez-Galan, L. Diverse Cell Death Mechanisms Are Simultaneously Activated in Macrophages Infected by Virulent Mycobacterium tuberculosis. Pathogens 2022, 11, 492. https://doi.org/10.3390/pathogens11050492
Ramon-Luing LA, Olvera Y, Flores-Gonzalez J, Palacios Y, Carranza C, Aguilar-Duran Y, Vargas MA, Gutierrez N, Medina-Quero K, Chavez-Galan L. Diverse Cell Death Mechanisms Are Simultaneously Activated in Macrophages Infected by Virulent Mycobacterium tuberculosis. Pathogens. 2022; 11(5):492. https://doi.org/10.3390/pathogens11050492
Chicago/Turabian StyleRamon-Luing, Lucero A., Yessica Olvera, Julio Flores-Gonzalez, Yadira Palacios, Claudia Carranza, Yerany Aguilar-Duran, Marco Antonio Vargas, Neptali Gutierrez, Karen Medina-Quero, and Leslie Chavez-Galan. 2022. "Diverse Cell Death Mechanisms Are Simultaneously Activated in Macrophages Infected by Virulent Mycobacterium tuberculosis" Pathogens 11, no. 5: 492. https://doi.org/10.3390/pathogens11050492
APA StyleRamon-Luing, L. A., Olvera, Y., Flores-Gonzalez, J., Palacios, Y., Carranza, C., Aguilar-Duran, Y., Vargas, M. A., Gutierrez, N., Medina-Quero, K., & Chavez-Galan, L. (2022). Diverse Cell Death Mechanisms Are Simultaneously Activated in Macrophages Infected by Virulent Mycobacterium tuberculosis. Pathogens, 11(5), 492. https://doi.org/10.3390/pathogens11050492