
Pathogenicity and Its Implications in Taxonomy: The Brucella and Ochrobactrum Case

 , ,
, ,  and
and
Abstract
1. Introduction
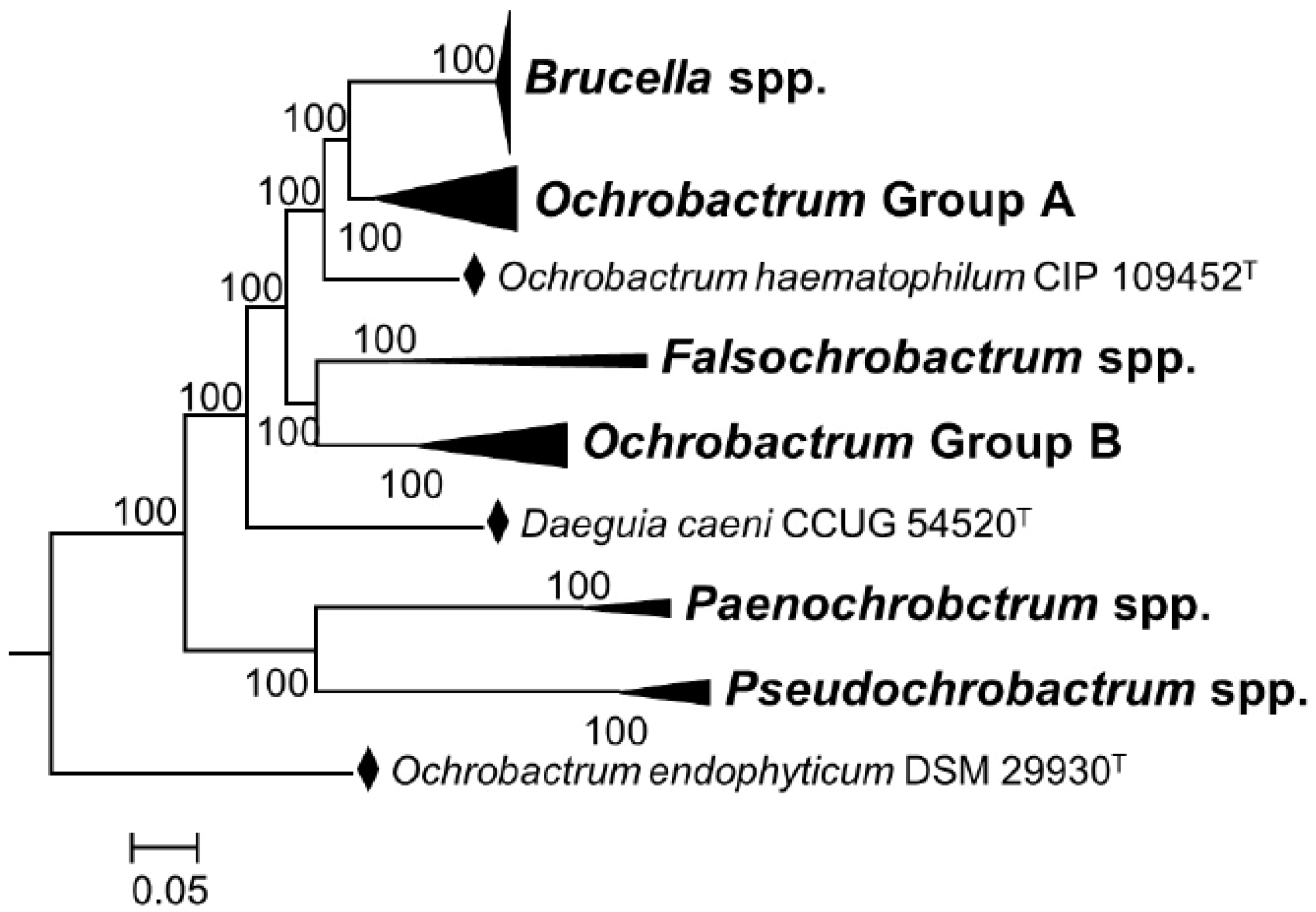
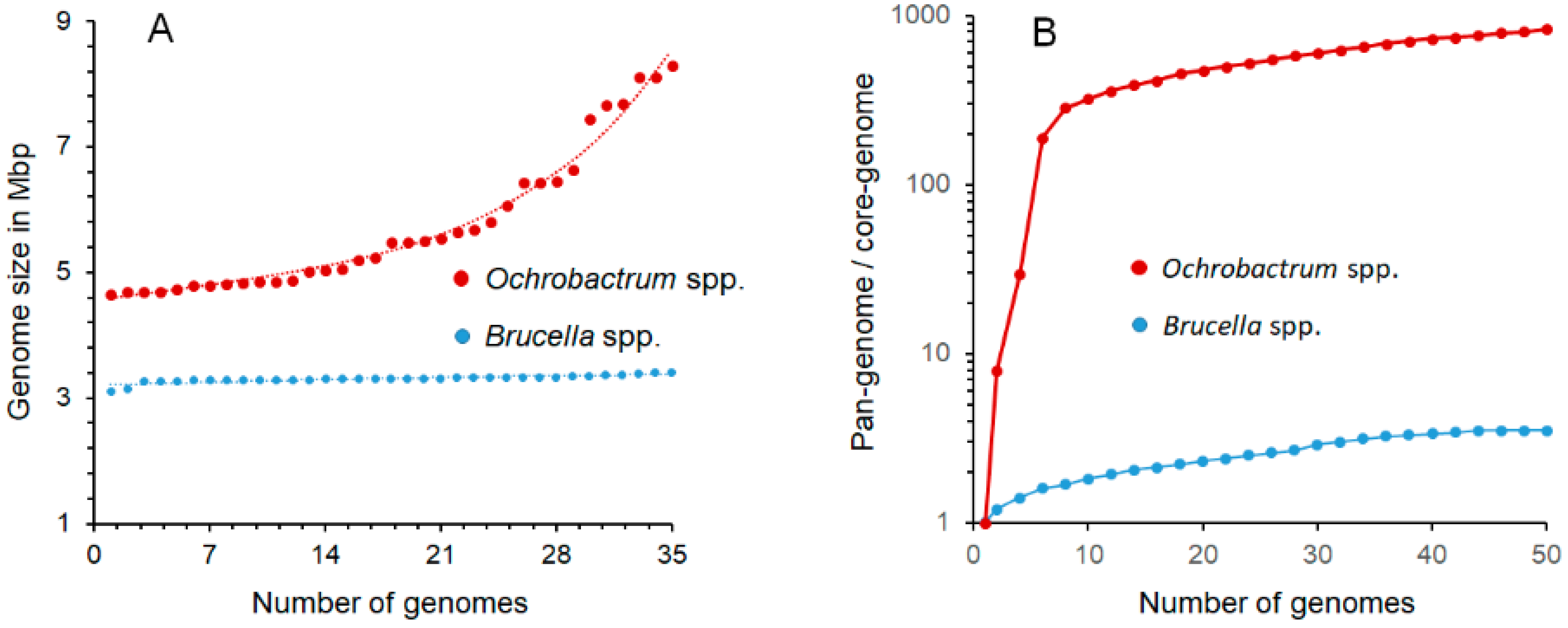
“The overall genomic divergence of the Brucella-Ochrobactrum clade was lower than in many clades harboring a single genus only. Brucella differs from Ochrobactrum regarding its pathogenic lifestyle, which may be reflected in the smaller genome size of Brucella. However, Ochrobactrum species are also known from clinical specimens, including its type species, and a more pronounced genome size reduction of pathogenic species nested within a partially non-pathogenic genus was observed elsewhere, as, e.g., in Mycobacterium leprae. Mycobacterium can also serve as an example for a genus harboring distinct risk groups, much like Burkholderia and Yersinia. Hence, the difference between Brucella and Ochrobactrum regarding their risk-group assignment could hardly be used as an argument against their inclusion in the same genus. Known phenotypic differences, if any, appeared to be restricted to autapomorphies of Brucella that may well be linked to its evolutionary adaptation to pathogenesis. Despite the differences in genome size, the gene-content analysis provided more support for the combined Brucella–Ochrobactrum clade than for the subclades”.[9] (Reproduced under Creative Commons CC-BY license)
2. The Concept of Genus
2.1. The Analytical Process
2.2. The Typological Process
2.3. The Analytical and Typological Processes Define the Genus
3. Pathogenicity and Its Taxonomical Implications: The Brucella and Ochrobactrum Case
3.1. Pathogenicity and False Equivalence Arguments
3.2. Cladistics, Genome Comparisons, and Pangenomes
3.3. The Significant Differences between Intracellular Pathogens and Opportunistic Free-Living Soil Bacteria in Evolutionary Paths and Population Structures
3.4. Pathogenicity, “Risk Groups”, and Taxonomy
4. The Practical Arguments Derived from Pathogenicity and Virulence
4.1. Animal Brucellosis and Ochrobactrum
4.2. Human Brucellosis versus Ochrobactrum Infections
5. Concluding Remarks
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Moreno, E. The one hundred year journey of the genus Brucella (Meyer and Shaw 1920). FEMS Microbiol. Rev. 2020, 45, fuaa045. [Google Scholar] [CrossRef] [PubMed]
- Corbel, M.J.; Brinley Morgan, W.J. Proposal for minimal standards for descriptions of new species and biotypes of the genus Brucella. Int. J. Syst. Evol. Microbiol. 1975, 25, 83–89. [Google Scholar]
- De Ley, J.; Mannheim, W.; Segers, P.; Lievens, A.; Deninj, M.; Vanhoucke, M.; Gillis, M. Ribosomal ribonucleic acid cistron similarities and taxonomic neighborhood of Brucella and CDC group Vd. Int. J. Syst. Bacteriol. 1987, 37, 35–42. [Google Scholar] [CrossRef]
- Dorsch, M.; Moreno, E.; Stackebrandt, E. Nucleotide sequence of the 16S rRNA from Brucella abortus. Nucleic Acids Res. 1989, 17, 1765. [Google Scholar] [CrossRef][Green Version]
- Moreno, E.; Stackebrandt, E.; Dorsch, M.; Wolters, J.; Busch, M.; Mayer, H. Brucella abortus 16S rRNA and lipid A reveal a phylogenetic relationship with members of the alpha-2 subdivision of the class Proteobacteria. J. Bacteriol. 1990, 172, 3569–3576. [Google Scholar] [CrossRef]
- Holmes, B.; Popoff, M.; Kiredjian, M.; Kersters, K. Ochrobactrum anthropi gen. nov., sp. nov. from human clinical specimens and previously known as Group Vd. Int. J. Syst. Bacteriol. 1988, 38, 406–416. [Google Scholar] [CrossRef]
- Velasco, J.; Romero, C.; López-Goñi, I.; Leiva, J.; Díaz, R.; Moriyón, I. Evaluation of the relatedness of Brucella spp. and Ochrobactrum anthropi and description of Ochrobactrum intermedium sp. nov., a new species with a closer relationship to Brucella spp. Int. J. Syst. Bacteriol. 1998, 48, 759–768. [Google Scholar] [CrossRef]
- Leclercq, S.O.; Cloeckaert, A.; Zygmunt, M.S.; Foster, J.T. Taxonomic organization of the family Brucellaceae based on a phylogenomic approach. Front. Microbiol. 2020, 10, 3083. [Google Scholar] [CrossRef]
- Hördt, A.; López, M.G.; Meier-Kolthoff, J.P.; Schleuning, M.; Weinhold, L.-M.; Tindall, B.J.; Gronow, S.; Kyrpides, N.C.; Woyke, T.; Göker, M. Analysis of 1000+ type-strain genomes substantially improves taxonomic classification of Alphaproteobacteria. Front. Microbiol. 2020, 11, 468. [Google Scholar] [CrossRef]
- Malik, V. The genus: A natural or arbitrary entity. Plant. Arch. 2017, 17, 251–257. [Google Scholar]
- Bartlett, H.H. The concept of the genus: I. History of the generic concept in botany. Bull. Torrey Bot. Club 1940, 67, 349–362. [Google Scholar] [CrossRef]
- Rosselló-Móra, R. Towards a taxonomy of Bacteria and Archaea based on interactive and cumulative data repositories. Environ. Microbiol. 2012, 14, 318–334. [Google Scholar] [CrossRef] [PubMed]
- Puigbo, P.; Wolf, Y.I.; Koonin, E.V. The tree and net components of prokaryote evolution. Genome Biol. Evol. 2010, 2, 745–756. [Google Scholar] [CrossRef]
- Barco, R.A.; Garrity, G.M.; Scott, J.J.; Amend, J.P.; Nealson, K.H.; Emerson, D. A genus definition for bacteria and archaea based on a standard genome relatedness index. mBio 2020, 11, e02475-19. [Google Scholar] [CrossRef] [PubMed]
- Tindall, B.J.; Rosselló-Móra, R.; Busse, H.J.; Ludwig, W.; Kämpfer, P. Notes on the characterization of prokaryote strains for taxonomic purposes. Int. J. Syst. Evol. Microbiol. 2010, 60, 249–266. [Google Scholar] [CrossRef]
- Yarza, P.; Yilmaz, P.; Pruesse, E.; Glöckner, F.O.; Ludwig, W.; Schleifer, K.H.; Whitman, W.B.; Euzéby, J.; Amann, R.; Rosselló-Móra, R. Uniting the classification of cultured and uncultured bacteria and archaea using 16S rRNA gene sequences. Nat. Rev. Microbiol. 2014, 12, 635–645. [Google Scholar] [CrossRef]
- Puppe, F. Heuristic classification. In Systematic Introduction to Expert Systems; Springer: Berlin/Heidelberg, Germany, 1993; pp. 131–148. [Google Scholar]
- Mayr, E. Systematics and the Origin of Species; Columbia University Press: New York, NY, USA, 1942. [Google Scholar]
- Wood, B.; Collard, M. The human genus. Science 1999, 284, 65–71. [Google Scholar] [CrossRef]
- Teyssier, C.; Marchandin, H.; Masnou, A.; Jeannot, J.; de Buochberg, M.S.; Jumas-Bilak, E. Pulsed-field gel electrophoresis to study the diversity of whole-genome organization in the genus Ochrobactrum. Electrophoresis 2005, 26, 2898–2907. [Google Scholar] [CrossRef]
- Moreno, E. Genome evolution within the alpha Proteobacteria: Why do some bacteria not possess plasmids and others exhibit more than one different chromosome? FEMS Microbiol. Rev. 1998, 22, 255–275. [Google Scholar] [CrossRef]
- Gohil, K.; Rajput, V.; Dharne, M. Pan-genomics of Ochrobactrum species from clinical and environmental origins reveals distinct populations and possible links. Genomics 2020, 112, 3003–3012. [Google Scholar] [CrossRef]
- Wattam, A.R.; Foster, J.T.; Mane, S.P.; Beckstrom-Sternberg, S.M.; Beckstrom-Sternberg, J.M.; Dickerman, A.W.; Keim, P.; Pearson, T.; Shukla, M.; Ward, D.V.; et al. Comparative phylogenomics and evolution of the Brucellae reveal a path to virulence. J. Bacteriol. 2014, 196, 920–930. [Google Scholar] [CrossRef]
- Suárez-Esquivel, M.; Chaves-Olarte, E.; Moreno, E.; Guzmán-Verri, C. Brucella genomics: Macro and micro evolution. Int. J. Mol. Sci. 2020, 21, 7749. [Google Scholar] [CrossRef] [PubMed]
- Teyssier, C.; Marchandin, H.; Jean-Pierre, H.; Diego, I.; Darbas, H.; Jeannot, J.L.; Gouby, A.; Jumas-Bilak, E. Molecular and phenotypic features for identification of the opportunistic pathogens Ochrobactrum spp. J. Med. Microbiol. 2005, 54, 945–953. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Li, Y.; Zang, J.; Li, Y.; Bie, P.; Lu, Y.; Wu, Q. Analysis of pan-genome to identify the core genes and essential genes of Brucella spp. Mol. Genet. Genom. 2016, 291, 905–912. [Google Scholar] [CrossRef]
- Sankarasubramanian, J.; Vishnu, U.S.; Gunasekaran, P.; Rajendhran, J. Identification of genetic variants of Brucella spp. through genome-wide association studies. Infect. Genet. Evol. 2017, 56, 92–98. [Google Scholar] [CrossRef] [PubMed]
- Meyer, M.E. Current concepts in the Taxonomy of the genus Brucella. In Animal Brucellosis; Nielsen, K.H., Duncan, J.R., Eds.; CRC Press: Boca Raton, FL, USA, 1990; pp. 1–17. [Google Scholar]
- Jäckel, C.; Hertwig, S.; Scholz, H.C.; Nockler, K.; Reetz, J.; Hammerl, J.A. Prevalence, host range, and comparative genomic analysis of temperate Ochrobactrum phages. Front. Microbiol. 2017, 8, 201. [Google Scholar] [CrossRef] [PubMed]
- Ashford, R.T.; Muchowski, J.; Koylass, M.; Scholz, H.C.; Whatmore, A.M. Application of whole genome sequencing and pan-family multi-locus sequence analysis to characterize relationships within the family Brucellaceae. Front. Microbiol. 2020, 11, 1329. [Google Scholar] [CrossRef]
- Hammerl, J.A.; Göllner, C.; Al Dahouk, S.; Nöckler, K.; Reetz, J.; Hertwig, S. Analysis of the first temperate broad host range brucellaphage (BiPBO1) isolated from B. inopinata. Front. Microbiol. 2016, 7, 24. [Google Scholar] [CrossRef]
- Rouli, L.; Merhej, V.; Fournier, P.-E.; Raoult, D. The bacterial pangenome as a new tool for analysing pathogenic bacteria. New Microbes New Infect. 2015, 7, 72–85. [Google Scholar] [CrossRef]
- Moreno, E.; Cloeckaert, A.; Moriyón, I. Brucella evolution and taxonomy. Vet. Microbiol. 2002, 90, 209–227. [Google Scholar] [CrossRef]
- Moreno, E. In search of a bacterial species definition. Rev. Biol. Trop. 1997, 45, 735–771. [Google Scholar]
- Barquero-Calvo, E.; Conde-Álvarez, R.; Chacón-Díaz, C.; Quesada-Lobo, L.; Martirosyan, A.; Guzmán-Verri, C.; Iriarte, M.; Manček-Keber, M.; Jerala, R.; Gorvel, J.-P.; et al. The differential interaction of Brucella and Ochrobactrum with innate immunity reveals traits related to the evolution of stealthy pathogens. PLoS ONE 2009, 4, e5893. [Google Scholar] [CrossRef] [PubMed]
- Velasco, J.; Bengoechea, J.A.; Brandenburg, K.; Lindner, B.; Seydel, U.; González, D.; Zähringer, U.; Moreno, E.; Moriyón, I. Brucella abortus and its closest phylogenetic relative, Ochrobactrum spp., differ in outer membrane permeability and cationic peptide resistance. Infect. Immun. 2000, 68, 3210–3218. [Google Scholar] [CrossRef] [PubMed]
- Diaz, M.; Wegmann, U.; Akinyemi, N.; Oguntoyinbo, F.A.; Sayavedra, L.; Mayer, M.J.; Narbad, A. Complete genome sequence of Ochrobactrum haematophilum FI11154, isolated from kunu-zaki, a nigerian millet-based fermented food. Genome Announc. 2018, 6, e00428-18. [Google Scholar] [CrossRef] [PubMed]
- Wattam, A.R.; Inzana, T.J.; Williams, K.P.; Mane, S.P.; Shukla, M.; Almeida, N.F.; Dickerman, A.W.; Mason, S.; Moriyón, I.; O’Callaghan, D.; et al. Comparative genomics of early-diverging Brucella strains reveals a novel lipopolysaccharide biosynthesis pathway. mBio 2012, 3, 27. [Google Scholar] [CrossRef] [PubMed]
- Barbier, T.; Nicolas, C.; Letesson, J.-J. Brucella adaptation and survival at the crossroad of metabolism and virulence. FEBS Lett. 2011, 585, 2929–2934. [Google Scholar] [CrossRef]
- Barbier, T.; Zúñiga-Ripa, A.; Moussa, S.; Plovier, H.; Sternon, J.F.; Lázaro-Antón, L.; Conde-Álvarez, R.; De Bolle, X.; Iriarte, M.; Moriyón, I.; et al. Brucella central carbon metabolism: An update. Crit. Rev. Microbiol. 2017, 44, 182–211. [Google Scholar] [CrossRef]
- Ronneau, S.; Moussa, S.; Barbier, T.; Conde-Álvarez, R.; Zúñiga-Ripa, A.; Moriyón, I.; Letesson, J.-J. Brucella, nitrogen and virulence. Crit Rev. Microbiol. 2014, 7828, 507–525. [Google Scholar] [CrossRef]
- Ozdemir, G.; Ozturk, T.; Ceyhan, N.; Isler, R.; Cosar, T. Heavy metal biosorption by biomass of Ochrobactrum anthropi producing exopolysaccharide in activated sludge. Bioresour. Technol. 2003, 90, 71–74. [Google Scholar] [CrossRef]
- Morais, P.V.; Branco, R.; Francisco, R. Chromium resistance strategies and toxicity: What makes Ochrobactrum tritici 5bvl1 a strain highly resistant. Biometals 2011, 24, 401–410. [Google Scholar] [CrossRef]
- Bathe, S.; Achouak, W.; Hartmann, A.; Heulin, T.; Schloter, M.; Lebuhn, M. Genetic and phenotypic microdiversity of Ochrobactrum spp. FEMS Microbiol. Ecol. 2006, 56, 272–280. [Google Scholar] [CrossRef] [PubMed]
- Trujillo, M.E.; Willems, A.; Abril, A.; Planchuelo, A.M.; Rivas, R.; Ludena, D.; Mateos, P.F.; Martinez-Molina, E.; Velazquez, E. Nodulation of Lupinus albus by strains of Ochrobactrum lupini sp. nov. Appl. Environ. Microbiol. 2005, 71, 1318–1327. [Google Scholar] [CrossRef] [PubMed]
- Roop, R.M.; Barton, I.S.; Hopersberger, D.; Martin, D.W. Uncovering the hidden credentials of Brucella virulence. Microbiol. Mol. Biol. Rev. 2021, 85, e00021-19. [Google Scholar] [CrossRef] [PubMed]
- Ryan, M.P.; Tony Pembroke, J. The genus Ochrobactrum as major opportunistic pathogens. Microorganisms 2020, 8, 1797. [Google Scholar] [CrossRef]
- González-Espinoza, G.; Arce-Gorvel, V.; Mémet, S.; Gorvel, J.P. Brucella: Reservoirs and niches in animals and humans. Pathogens 2021, 10, 186. [Google Scholar] [CrossRef]
- Conde-Álvarez, R.; Arce-Gorvel, V.; Iriarte, M.; Manček-Keber, M.; Barquero-Calvo, E.; Palacios-Chaves, L.; Chacón-Díaz, C.; Chaves-Olarte, E.; Martirosyan, A.; von Bargen, K.; et al. The lipopolysaccharide core of Brucella abortus acts as a shield against innate immunity recognition. PLoS Pathog. 2012, 8, e1002675. [Google Scholar] [CrossRef]
- Celli, J.; De Chastellier, C.; Franchini, D.M.; Pizarro-Cerda, J.; Moreno, E.; Gorvel, J.-P. Brucella evades macrophage killing via VirB-dependent sustained interactions with the endoplasmic reticulum. J. Exp. Med. 2003, 198, 545–556. [Google Scholar] [CrossRef]
- Zhu, M.; Zhao, X.; Zhu, Q.; Zhang, Z.; Dai, Y.; Chen, L.; Liang, Z. Clinical characteristics of patients with Ochrobactrum anthropi bloodstream infection in a Chinese tertiary-care hospital: A 7-year study. J. Infect. Public Health 2018, 11, 873–877. [Google Scholar] [CrossRef]
- Moreno, E.; Moriyón, I. The genus Brucella. In The Prokaryotes. Volume 5. Proteobacteria; Falkow, S., Rosenberg, E., Schleifer, K.H., Stackebrandt, E., Dworkin, M., Eds.; Springer: Berlin/Heidelberg, Germany, 2007; Volume 3, pp. 315–455. [Google Scholar]
- Corner, L.A.A. Three aspects of bovine brucellosis: Epidemiology, the role of bulls and vaccines. N. S. W. Vet. Proc. 1983, 19, 47–48. [Google Scholar]
- Kampfer, P.; Huber, B.; Busse, H.J.; Scholz, H.C.; Tomaso, H.; Hotzel, H.; Melzer, F. Ochrobactrum pecoris sp. nov., isolated from farm animals. Int. J. Syst. Evol. Microbiol. 2011, 61, 2278–2283. [Google Scholar] [CrossRef]
- Yagupsky, P.; Morata, P.; Colmenero, J.D. Laboratory diagnosis of human brucellosis. Clin. Microbiol. Rev. 2019, 33, e00073-19. [Google Scholar] [CrossRef]
- Yagel, Y.; Sestito, S.; Motro, Y.; Shnaiderman-Torban, A.; Khalfin, B.; Sagi, O.; Navon-Venezia, S.; Steinman, A.; Moran-Gilad, J. Genomic characterization of antimicrobial resistance, virulence, and phylogeny of the genus Ochrobactrum. Antibiotics 2020, 9, 177. [Google Scholar] [CrossRef]
- Corbel, M.J.; Alton, G.G.; Banai, M.; Díaz, R.; Dranovskaia, B.A.; Elberg, S.S.; Garin-Bastuji, B.; Kolar, J.; Mantovani, A.; Mousa, A.M.; et al. Brucellosis in Humans and Animals; WHO Press: Geneva, Switzerland, 2006; Volume 52, ISBN 9241547138. [Google Scholar]
- Ariza, J. Brucellosis: An update. The perspective from the Mediterranean basin. Rev. Med. Microbiol. 1999, 10, 125–135. [Google Scholar]
- García-Lobo, J.M.; Agüero, J.; Solera, J. La duradera y notoria sensibilidad de Brucella a las tetraciclinas. Enferm. Infecc. Microbiol. Clin. 1999, 17, 366–367. [Google Scholar]
- Rouzic, N.; Desmier, L.; Cariou, M.-E.; Gay, E.; Foster, J.T.; Williamson, C.H.D.; Schmitt, F.; Le Henaff, M.; Le Coz, A.; Lorléac’h, A. First case of brucellosis caused by an amphibian-type Brucella. Clin. Infect. Dis. 2021, 72, e404–e407. [Google Scholar] [CrossRef]
- Thoma, B.; Straube, E.; Scholz, H.C.; Al-Dahouk, S.; Zoller, L.; Pfeffer, M.; Neubauer, H.; Tomaso, H. Identification and antimicrobial susceptibilities of Ochrobactrum spp. Int. J. Med. Microbiol. 2009, 299, 209–220. [Google Scholar] [CrossRef]
- Blasco, J.M.; Moreno, E.; Moriyón, I. Brucellosis vaccines and vaccine candidates. In Veterinary Vaccines. Principles and Applications, 1st ed.; Metwally, S., Viljoen, G.J., El Idrissi, A., Eds.; FAO: Rome, Italy; Wiley & Sons: Hobeken, NJ, USA, 2021; pp. 295–316. [Google Scholar]
- Nicoletti, P. Relationship between animal and human disease. In Brucellosis: Clinical and Laboratory Aspects; Young, E.D., Corbel, M.J., Eds.; CRC Press: Boca Raton, FL, USA, 1989; pp. 41–52. ISBN 0-8493-6661-5. [Google Scholar]
- Mayr, E. Two empires or three? Proc. Natl. Acad. Sci. USA 1998, 95, 9720–9723. [Google Scholar] [CrossRef]
- Seferbekova, Z.; Zabelkin, A.; Yakovleva, Y.; Afasizhev, R.; Dranenko, N.O.; Alexeev, N.; Gelfand, M.S.; Bochkareva, O.O. High rates of genome rearrangements and pathogenicity of Shigella spp. Front. Microbiol. 2021, 12, 12. [Google Scholar] [CrossRef]
- Ryu, Y.J.; Koh, W.-J.; Daley, C.L. Diagnosis and treatment of nontuberculous mycobacterial lung disease: Clinicians’ perspectives. Tuberc. Respir. Dis. 2016, 79, 74–84. [Google Scholar] [CrossRef]
- Johnson, M.M.; Odell, J.A. Nontuberculous mycobacterial pulmonary infections. J. Thorac. Dis. 2014, 6, 210. [Google Scholar]
- de los Ángeles Giusti, M.; Pistorio, M.; Lozano, M.J.; Torres Tejerizo, G.A.; Salas, M.E.; Martini, M.C.; López, J.L.; Draghi, W.O.; Del Papa, M.F.; Pérez-Mendoza, D.; et al. Genetic and functional characterization of a yet-unclassified rhizobial Dtr (DNA-transfer-and-replication) region from a ubiquitous plasmid conjugal system present in Sinorhizobium meliloti, in Sinorhizobium medicae, and in other nonrhizobial Gram-negatives. Plasmid 2012, 67, 199–210. [Google Scholar] [CrossRef]
- Doolittle, W.F.; Zhaxybayeva, O. On the origin of prokaryotic species. Genome Res. 2009, 19, 744–756. [Google Scholar] [CrossRef]
- Dicenzo, G.C.; Finan, T.M. The divided bacterial genome: Structure, function, and evolution. Microbiol. Mol. Biol. Rev. 2017, 81, e00019-17. [Google Scholar] [CrossRef]
- Eberhard, W.G. Evolution in bacterial plasmids and levels of selection. Q. Rev. Biol. 1990, 65, 3–22. [Google Scholar] [CrossRef]
- El-Sayed, W.S.; Ibrahim, M.K.; Abu-Shady, M.; El-Beih, F.; Ohmura, N.; Saiki, H.; Ando, A. Isolation and identification of a novel strain of the genus Ochrobactrum with phenol-degrading activity. J. Biosci. Bioeng. 2003, 96, 310–312. [Google Scholar] [CrossRef]
- Veeranagouda, Y.; Paul, P.V.E.; Gorla, P.; Siddavattam, D.; Karegoudar, T.B. Complete mineralisation of dimethylformamide by Ochrobactrum sp. DGVK1 isolated from the soil samples collected from the coalmine leftovers. Appl. Microbiol. Biotechnol. 2006, 71, 369–375. [Google Scholar] [CrossRef]
- Zhang, R.; Cui, Z.; Jiang, J.; He, J.; Gu, X.; Li, S. Diversity of organophosphorus pesticide-degrading bacteria in a polluted soil and conservation of their organophosphorus hydrolase genes. Can. J. Microbiol. 2005, 51, 337–343. [Google Scholar] [CrossRef]
- Kim, Y.-M.; Park, K.; Joo, G.-J.; Jeong, E.-M.; Kim, J.-E.; Rhee, I.-K. Glutathione-dependent biotransformation of the fungicide chlorothalonil. J. Agric. Food Chem. 2004, 52, 4192–4196. [Google Scholar] [CrossRef]
- Ermakova, I.T.; Shushkova, T.V.; Sviridov, A.V.; Zelenkova, N.F.; Vinokurova, N.G.; Baskunov, B.P.; Leontievsky, A.A. Organophosphonates utilization by soil strains of Ochrobactrum anthropi and Achromobacter sp. Arch. Microbiol. 2017, 199, 665–675. [Google Scholar] [CrossRef]
- Margulis, L. Symbiosis in Cell Evolution: Life and Its Environment on the Early Earth; Freeman, W.H., Ed.; W H Freeman & Co.: San Francisco, CA, USA, 1981; ISBN 0716712555. [Google Scholar]
- Woese, C.R. Bacterial evolution. Microbiol. Rev. 1987, 51, 221–271. [Google Scholar] [CrossRef]
- Gao, Y.; Wu, M. Microbial trait evolution is dominated by frequent and rare pulsed evolution. bioRxiv 2021. [Google Scholar] [CrossRef]
- de Bagües, M.P.J.; Iturralde, M.; Arias, M.A.; Pardo, J.; Cloeckaert, A.; Zygmunt, M.S. The new strains Brucella inopinata BO1 and Brucella species 83-210 behave biologically like classic infectious Brucella species and cause death in murine models of infection. J. Infect. Dis. 2014, 210, 467–472. [Google Scholar] [CrossRef] [PubMed]
- Barquero-Calvo, E.; Chaves-Olarte, E.; Weiss, D.S.; Guzmán-Verri, C.; Chacón-Díaz, C.; Rucavado, A.; Moriyón, I.; Moreno, E. Brucella abortus uses a stealthy strategy to avoid activation of the innate immune system during the onset of infection. PLoS ONE 2007, 2, e631. [Google Scholar] [CrossRef] [PubMed]
- Martirosyan, A.; Moreno, E.; Gorvel, J.-P.P. An evolutionary strategy for a stealthy intracellular Brucella pathogen. Immunol. Rev. 2011, 240, 211–234. [Google Scholar] [CrossRef] [PubMed]
- Fontana, C.; Conde-Álvarez, R.; Ståhle, J.; Holst, O.; Iriarte, M.; Zhao, Y.; Arce-Gorvel, V.; Hanniffy, S.S.; Gorvel, J.-P.P.; Moriyón, I.; et al. Structural studies of lipopolysaccharide-defective mutants from Brucella melitensis identify a core oligosaccharide critical in virulence. J. Biol. Chem. 2016, 291, 7727–7741. [Google Scholar] [CrossRef]
- Martirosyan, A.; Gorvel, J.-P. Brucella evasion of adaptive immunity. Future Microbiol. 2013, 8, 147–154. [Google Scholar] [CrossRef]
- Berg, G.; Eberl, L.; Hartmann, A. The rhizosphere as a reservoir for opportunistic human pathogenic bacteria. Environ. Microbiol. 2005, 7, 1673–1685. [Google Scholar] [CrossRef]
- Cieslak, T.J.; Drabick, C.J.; Robb, M.L. Pyogenic infections due to Ochrobactrum anthropi. Clin. Infect. Dis. 1996, 22, 845–847. [Google Scholar] [CrossRef]
- Kettaneh, A.; Weill, F.X.; Poilane, I.; Fain, O.; Thomas, M.; Herrmann, J.L.; Hocqueloux, L. Septic shock caused by Ochrobactrum anthropi in an otherwise healthy host. J. Clin. Microbiol. 2003, 41, 1339–1341. [Google Scholar] [CrossRef]
- Ozdemir, D.; Soypacaci, Z.; Sahin, I.; Bicik, Z.; Sencan, I. Ochrobactrum anthropi endocarditis and septic shock in a patient with no prosthetic valve or rheumatic heart disease: Case report and review of the literature. Jpn. J. Infect. Dis. 2006, 59, 264–265. [Google Scholar]
- Spink, W.W. The natural course of brucellosis. In The Nature of Brucellosis; The University of Minnesota Press: Minneapolis, MN, USA, 1956; pp. 145–170. [Google Scholar]
- Scholz, H.C.; Banai, M.; Cloeckaert, A.; Kämpfer, P.; Whatmore, A.M. Brucella. In Bergey’s Manual of Systematics of Archaea and Bacteria; Wiley: Hoboken, NJ, USA, 2018; pp. 1–38. ISBN 9781118960608. [Google Scholar]
- Moreno, E. Retrospective and prospective perspectives on zoonotic brucellosis. Front. Microbiol. 2014, 5, 213. [Google Scholar] [CrossRef]
- Alonso, C.A.; Kwabugge, Y.A.; Anyanwu, M.U.; Torres, C.; Chah, K.F. Diversity of Ochrobactrum species in food animals, antibiotic resistance phenotypes and polymorphisms in the blaOCH gene. FEMS Microbiol. Lett. 2017, 364, 364. [Google Scholar] [CrossRef]
- Alvarado-Capó, Y.; González, N.P.; García-Aguila, L.; Freire-Seijo, M.; Martínez, Y.; Kosky, R.G. Ochrobactrum anthropi, contaminants of in vitro culture of sugarcane cells and tissues. Biotecnol. Veg. 2007, 7, 211–214. [Google Scholar]
- Bielanski, A.; Bergeron, H.; Lau, P.C.K.; Devenish, J. Microbial contamination of embryos and semen during long term banking in liquid nitrogen. Cryobiology 2003, 46, 146–152. [Google Scholar] [CrossRef]
- Choi, G.-M.; Kim, K.M.; Yun, C.-S.; Lee, S.Y.; Kim, S.Y.; Wee, J.-H.; Im, W.-T. Ochrobactrum soli sp. nov., Isolated from a Korean cattle farm. Curr. Microbiol. 2020, 77, 1104–1110. [Google Scholar] [CrossRef] [PubMed]
- Cobo, F.; Concha, A. Environmental microbial contamination in a stem cell bank. Lett. Appl. Microbiol. 2007, 44, 379–386. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, K.; Tamura, T.; Okamura, Y.; Kabamoto, A.; Moriwaki, S.; Eto, M. Isolation and characterization of Ochrobactrum intermedium from imported cattle serologically diagnosed with bovine brucellosis. J. Jpn. Vet. Med. Assoc. 2010, 63, 615–619. [Google Scholar] [CrossRef]
- Ducrotoy, M.J.; Bertu, W.J.; Matope, G.; Cadmus, S.; Conde-Álvarez, R.; Gusi, A.M.; Welburn, S.; Ocholi, R.; Blasco, J.M.; Moriyón, I. Brucellosis in Sub-Saharan Africa: Current challenges for management, diagnosis and control. Acta Trop. 2017, 165, 179–193. [Google Scholar] [CrossRef]
- Deshmukh, A.; Hagen, F.; Al Sharabasi, O.; Abraham, M.; Wilson, G.; Doiphode, S.; Al Maslamani, M.; Meis, J.F. In vitro antimicrobial susceptibility testing of human Brucella melitensis isolates from Qatar between 2014–2015. BMC Microbiol. 2015, 15, 121. [Google Scholar] [CrossRef]
- Rubinstein, E.; Lang, R.; Shasha, B.; Hagar, B.; Diamanstein, L.; Joseph, G.; Anderson, M.; Harrison, K. In vitro susceptibility of Brucella melitensis to antibiotics. Antimicrob. Agents Chemother. 1991, 35, 1925–1927. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ariza, J.; Bosilkovski, M.; Cascio, A.; Colmenero, J.D.; Corbel, M.J.; Falagas, M.E.; Memish, Z.A.; Roushan, M.R.H.; Rubinstein, E.; Sipsas, N.V.; et al. Perspectives for the treatment of brucellosis in the 21st century: The Ioannina recommendations. PLoS Med. 2007, 4, e317. [Google Scholar] [CrossRef] [PubMed]
- Judicial Commission Proposal to emend the International Code of Nomenclature of Bacteria. Int. J Syst. Bacteriol. 1985, 35, 123. [CrossRef]
- Bouza, E.; Sánchez-Carrillo, C.; Hernan-Gómez, S.; González, M.J. Laboratory-acquired brucellosis: A Spanish national survey. J. Hosp. Infect. 2005, 61, 80–83. [Google Scholar] [CrossRef] [PubMed]
- Baron, E.J.; Miller, J.M. Bacterial and fungal infections among diagnostic laboratory workers: Evaluating the risks. Diagn. Microbiol. Infect. Dis. 2008, 60, 241–246. [Google Scholar] [CrossRef]
- Singh, K. Laboratory-acquired infections. Clin. Infect. Dis. 2009, 49, 142–147. [Google Scholar] [CrossRef]
- Lloyd, K.G.; Tahon, G. Science depends on nomenclature, but nomenclature is not science. Nat. Rev. Microbiol. 2022, 20, 123–124. [Google Scholar] [CrossRef]
- Li, S.Y.; Huang, Y.E.; Chen, J.Y.; Lai, C.H.; Mao, Y.C.; Huang, Y.T.; Liu, P.Y. Genomics of Ochrobactrum pseudogrignonense (newly named Brucella pseudogrignonensis) reveals a new blaoxa subgroup. Microb. Genom. 2021, 7, 000626. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Divergent Properties | Brucella | Ochrobactrum | References |
|---|---|---|---|
| Genome Size | 3.1–3.4 Mb | 4.7–8.3 Mb | [20,21] |
| Pangenome | Closed | Open | [20,22,23,24,25,26,27] |
| Plasmid | No | Variable (up to 6) | [20,21,23,25,28,29] |
| Phylogeny | Monophyletic | Polyphyletic | [8,30] |
| Active Phages | No | >4 | [29,31] |
| Lateral gene transfer | Absent | Present | [29] |
| Speciation type | Allopatric | Sympatric | [32,33,34] |
| Cell envelope permeability | Permeable to hydrophobic probes and resistant to destabilization by polycationic peptides | Impermeable to hydrophobic probes and sensitive to polycationic peptides | [35,36] |
| Metabolic redundancy | Low | High | [22,37] |
| Degradation of complex molecules | No | A large variety of such molecules | [38,39,40,41] |
| Removing toxic metals | No | Yes (some species/strains) | [42,43] |
| Capable to root nodulation | No | Yes (some species/strains) | [44,45] |
| Life style | Pathogen (class 3) | Saprophyte | [6,44,46] |
| Natural habitat | Intracellular | Soil and root plant surfaces | [24,35,44] |
| Transmission | Host-host interaction/animal products | Mostly iatrogenic | [47,48] |
| Virulence | Finely tuned | Fortuitus/opportunistic | [6,46,47] |
| Virulence mechanisms | Escape from the immune response/deviation of the intracellular trafficking | No true ones and virulence depending on host immune status | [35,46,49,50] |
| Infection dynamics | Long-lasting infection and low proinflammatory response | Acute proinflammatory/pyogenic; self-limiting in immunocompetent hosts | [46,47,51] |
| Animal disease | Very important | Seldom | [46,48,52,53,54] |
| Human health | Very important | Negligible | [22,46,47,48,52] |
| Diagnosis | Well-standardized serological methods | No serological tests are available or necessary | [52,55] |
| Treatment | WHO recommended long bi-therapy in uncomplicated cases | Based on antibiotic resistance/short monotherapy | [47,56,57] |
| Antibiotic resistance | Seldom and well-defined | High | [25,47,56,57,58,59,60,61] |
| Vaccine | Available (domestic ruminants) and critically important to control disease | Unnecessary | [62,63] |
| WHO/OIE/FAO regulations | Very important | Null | [57] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moreno, E.; Blasco, J.M.; Letesson, J.J.; Gorvel, J.P.; Moriyón, I. Pathogenicity and Its Implications in Taxonomy: The Brucella and Ochrobactrum Case. Pathogens 2022, 11, 377. https://doi.org/10.3390/pathogens11030377
Moreno E, Blasco JM, Letesson JJ, Gorvel JP, Moriyón I. Pathogenicity and Its Implications in Taxonomy: The Brucella and Ochrobactrum Case. Pathogens. 2022; 11(3):377. https://doi.org/10.3390/pathogens11030377
Chicago/Turabian StyleMoreno, Edgardo, José María Blasco, Jean Jacques Letesson, Jean Pierre Gorvel, and Ignacio Moriyón. 2022. "Pathogenicity and Its Implications in Taxonomy: The Brucella and Ochrobactrum Case" Pathogens 11, no. 3: 377. https://doi.org/10.3390/pathogens11030377
APA StyleMoreno, E., Blasco, J. M., Letesson, J. J., Gorvel, J. P., & Moriyón, I. (2022). Pathogenicity and Its Implications in Taxonomy: The Brucella and Ochrobactrum Case. Pathogens, 11(3), 377. https://doi.org/10.3390/pathogens11030377