Abstract
Schistosomiasis is a chronic helminthic disease of both humans and animals and the second most prevalent parasitic disease after malaria. Through a complex migration process, schistosome eggs trapped in the liver can lead to the formation of granulomas and subsequent schistosome-induced liver damage, which results in high mortality and morbidity. Although praziquantel can eliminate mature worms and prevent egg deposition, effective drugs to reverse schistosome-induced liver damage are scarce. High mobility group box 1 (HMGB1) is a multifunctional cytokine contributing to liver injury, inflammation, and immune responses in schistosomiasis by binding to cell-surface Toll-like receptors and receptors for advanced glycation end products. HMGB1 is increased in the serum of patients with schistosomiasis and enables hepatic stellate cells to adopt a proliferative myofibroblast-like phenotype, which is crucial to schistosome-induced granuloma formation. Inhibition of HMGB1 was found to generate protective responses against fibrotic diseases in animal models. Clinically, HMGB1 presents a potential target for treatment of the chronic sequelae of schistosomiasis. Here, the pivotal role of HMGB1 in granuloma formation and schistosome-induced liver damage, as well the potential of HMGB1 as a therapeutic target, are discussed.
1. Introduction
Schistosomiasis, or bilharzia, is an infectious disease endemic in tropical areas of Asia, Africa, and South America affecting over 230 million individuals worldwide [1]. Three main species of schistosomes (i.e., Schistosoma japonicum, S. haematobium, and S. mansoni) account for most human infections [2]. S. haematobium is endemic in Africa, some areas of the Arabian Peninsula and was recently identified on the island of Corsica [3]. S. japonicum is prevalent in the Philippines, Indonesia, and China, while S. mansoni is endemic in Africa, the Middle East, and several countries in South America, including Brazil and Venezuela, and the Caribbean [4]. The young worms first pierce the skin and enter the blood vessels, then move through the heart and lungs to the liver vasculature, where mating and maturation of the male and female worms occur [5]. From there, the parasite couples migrate to the favored egg laying sites. S. haematobium migrates to the vesicle venous plexus, which induces urogenital schistosomiasis, while mating pairs of S. japonicum and S. mansoni migrate to the mesenteric vessels and induce intestinal/hepatic schistosomiasis [5,6].
Schistosome eggs are responsible for the occurrence and spread of schistosomiasis. Each mature female S. mansoni lays about 800 eggs per day, while female S. japonicum can produce up to 3000 eggs per day [7]. The enormous amounts of eggs deposited in the host’s liver by S. japonicum and S. mansoni trigger the primary pathology of schistosomiasis, which includes egg-induced granuloma formation, subsequent chronic inflammation, and eventual fibrosis due to the host immune response [8,9]. The extensive deposition of extracellular matrix (ECM) in the periportal areas is a typical symptom of hepatic fibrosis, giving rise to blockage of the portal veins, portal hypertension, splenomegaly, and gastrointestinal varices [2]. The pertinacious fibrosis of chronic schistosomiasis brings about hepatic cirrhosis and is responsible for the high mortality rate of liver cancer in Asian countries [10,11]. In the advanced phase, the course of schistosome-induced liver diseases is often irreversible [12]. Although the chemotherapy agent praziquantel (PZQ) can effectively eliminate mature worms and prevent the accumulation of eggs, there is a lack of effective drugs directly targeting and reversing schistosome-induced hepatic fibrosis [13]. Without prompt treatment, liver fibrosis can further transform into portal hypertension and ascites, which are usually the main causes of death in patients with chronic schistosomiasis [14]. Therefore, the development of new treatments targeting factors that regulate hepatic fibrosis in schistosomiasis patients remains a priority.
High mobility group box 1 (HMGB1) is a highly conserved DNA-shepherding protein that is plentiful in the cell nucleus [15]. HMGB1 is actively secreted by multiple cell types, including macrophages, monocytes, dendritic cells, natural killer cells, endothelial cells, and platelets [16], and passively by necrotic and damaged cells [17,18]. Either mode can release substantial amounts of extracellular HMGB1, which participates in multiple biological functions. Serum and liver levels of HMGB1 are significantly increased in some schistosomiasis patients with inflammatory responses, suggesting a close association with disease progression [19]. Moreover, recent studies suggest that HMGB1 is significantly upregulated in patients with schistosome-induced hepatic fibrosis as well as animal models [20]. Meanwhile, inhibition of the translation and release of HMGB1 or blocking related signaling pathways via the HMGB1 receptor can protect against hepatic inflammation and fibrosis, suggesting that HMGB1 may be a vital factor in schistosome-induced liver disease as well as a potential therapeutic target [20,21,22]. Accordingly, targeting of HMGB1 could potentially reduce the occurrence and pathogenesis of schistosome-induced liver disease, although the underlying mechanism remains unclear.
Therefore, based on the extensive body of literature, the present review briefly summarizes the structure and functions of HMGB1, the development and migration of schistosomes in the host, factors that contribute to the regulation of schistosome-induced liver disease, the crucial role of HMGB1 in schistosome-induced liver disease, and HMGB1 as a potential target for treatment of schistosomiasis-associated inflammation and fibrosis.
2. HMGB1 Structure and Receptors
2.1. HMGB1 Structure
HMGB1, first isolated and identified in 1973 by Goodwin et al. [23], is a non-histone chromosome-binding protein composed of 215 amino acid (aa) residues and is named to describe the high electrophoretic mobility in polyacrylamide gels. The structure of human HMGB1 is composed of two functional DNA-binding domains, the A box (aa 9–79) and B box (aa 95–163), an N-terminus, and a C-terminus, known as the “acidic tail”, which consists of 30 glutamic and aspartic acid residues [24] (Figure 1). The sequence of HMGB1 is 98.5% identical in all mammals. The B box is the functional region of HMGB1 that promotes inflammation, while the A box is the antagonistic site that can competitively inhibit the pro-inflammatory effect of the B box [18]. Besides, HMGB1 carries two nuclear localization signals (NLSs): NLS 1, which is located in the A box (aa 28–44), and NLS 2, located in the B box (aa 179–185) [25]. Moreover, HMGB1 contains a Toll-like receptor (TLR) binding site (aa 89–108) and a receptor for advanced glycation end-products (RAGE) binding site (aa 150–183) [24]. Interestingly, the HMGB1 sequences of S. japonicum (SjHMGB1) and S. mansoni (SmHMGB1) are identical and share 60% similarity with mammalian HMGB1. Unlike human HMGB1, which contains an unbroken run of 30 glutamic or aspartic acid residues, SmHMGB1 and SjHMGB1 possess unusually short acidic C-terminal tails, composed of five acidic residues interrupted by two serine residues [26].
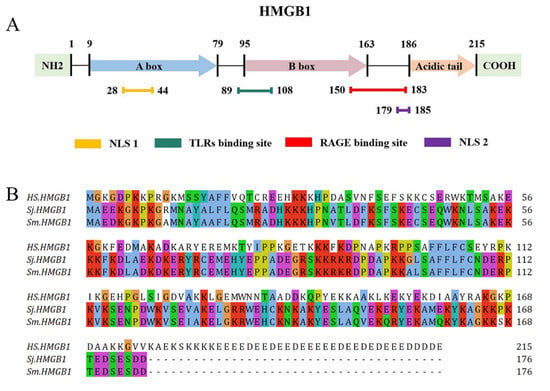
Figure 1.
Structure of human HMGB1 and the complete HMGB1 protein sequence of Homo sapiens, S. japonicum and S. mansoni. (A) Human HMGB1 consists of 215 aa residues, which can be divided into the A box (aa 9–79), B box (aa 95–163), an acidic tail (aa 186–215), two NLSs (aa 28–44 and aa 179–185), a TLR binding site (aa 89–108), and a RAGE binding site (aa 150–183). (B) The complete protein sequence of H. sapiens HMGB1 (GenBank accession no. NP_002119) in relation to the orthologues of S. japonicum (DQ005528) and S. mansoni (DQ005529).
2.2. HMGB1 Receptors
RAGE, a transmembrane protein of the immunoglobulin superfamily, is expressed on the surfaces of monocytes, macrophages, endothelial cells, vascular smooth muscle cells, and neurons [27]. RAGE expression is higher in most cell types and tissues in a disease state as compared with a healthy state [28,29,30]. In addition to HMGB1, RAGE interacts with many other ligands, such as advanced glycation end products, members of the S100 protein family, and beta-amyloid peptide [28], which further triggers activation of the Ras-extracellular signal-regulated kinase 1/2 (ERK1/2) pathway [31], stress-activated protein kinase/c-Jun-NH2-terminal kinase (SAPK/JNK) pathway, Cdc42/Rac pathway [32], and p38 mitogen-activated protein kinase (MAPK) pathway [30]. Finally, the proinflammatory effects of HMBG1 can be amplified through signaling cascades of transcription factors, such as nuclear factor (NF)-κB [33], cAMP response element-binding protein [31], and members of the signal transducers and activators of transcription family with the involvement of the RAGE ligand [34]. Accumulating evidence suggests that activation of RAGE signaling in the liver can lead to the development of numerous types of hepatic disorders [35]. For example, Gaens et al. [36] found that fatty acids could stimulate Nε-(carboxymethyl) lysine accumulation in hepatocytes and subsequently elicit inflammatory reactions and hepatic steatosis via the induction of RAGE, while Nomota et al. [37] suggested that the lack of galectin-3 was related to the progression of non-alcoholic fatty liver disease in mice, which was also associated with up-regulated RAGE expression in hepatocytes.
TLRs are type I transmembrane pattern recognition receptors that can identify diverse signals, such as damage-associated molecular patterns and pathogen-associated molecular patterns [38]. TLRs have been identified as intermediary players linking the innate and adaptive immune responses in favor of initial enhancement of inflammation [39,40,41]. In gastric ischemia-reperfusion injury, HMGB1 interacts with TLR2, which leads to significant organ damage after reperfusion [42]. Meanwhile, Azam et al. [43] found that myeloid differentiation primary response 88 (MyD88)- or non-MyD88-dependent signaling could be activated by HMGB1 to trigger downstream signals via activation of TLR4, which enhances the secretion of cytokines via the NF-κB pathway. In addition, HMGB1 has been associated with activation of the TLR9/MyD88 pathway and the release of proinflammatory cytokines in autoimmune thyroiditis [44]. Besides, the lack of HMGB1 leads to increased susceptibility to hepatocyte death through TLR9 in response to oxidative stress [22]. In summary, TLRs participate in the signal transduction of HMGB1, which further promotes the transcription of cytokines and chemokines via activation of inflammatory signaling pathways.
3. Schistosome-Induced Liver Damage
3.1. Life Cycles of Schistosomes
The entire life cycles of schistosomes are key to the occurrence and spread of schistosomiasis. The life cycles of S. japonicum, S. mansoni, and S. haematobium, the three main species of schistosomes that infect humans, slightly differ according to distinct egg-laying sites, as well as migration inside the host [45,46].
The release of S. japonicum, S. mansoni, and S. haematobium cercariae from the intermediate host snails (freshwater Oncomelania, Biomphalaria, and Bulinus spp., respectively) breach the epidermis as the first step of infection [6]. Almost 90% of S. japonicum can penetrate the dermal vessels within 2 h after infection and reach the dermis within 24 h. However, the migratory pattern of S. mansoni and S. haematobium significantly differ from that of S. japonicum. At 72 h after infection, the majority of S. mansoni and S. haematobium schistosomula reach the dermal vessels [47,48].
As the second step of the migration process, the schistosomula travel through the circulation to the lungs. Due to the small diameters of pulmonary capillaries, the young parasites elongate to facilitate penetration [49]. S. japonicum and S. mansoni schistosomula are detected in the lungs between 2–3 days post infection (dpi), with S. japonicum peaking at 3 dpi and S. mansoni at 7 dpi [50,51]. On the other hand, S. haematobium can reach the lungs at around 7 dpi and remain for up to 25 dpi [52].
As the third step of the migration process, schistosomula can travel from the lungs to the systemic organs through the pulmonary veins. The descending (abdominal) aorta, gastric arteries, gastroduodenal artery, or splenic artery may be the direct or indirect route for schistosomula to reach the hepatic portal vein [5]. To facilitate maturation, schistosomula use ventral suckers to crawl on the branches of the hepatic portal vein to feed on blood [53]. Upon maturation of the young parasites to adults, which occurs in about 5–7 weeks, the worms form mating pairs and migrate from the liver of the host to the preferred sites to lay eggs [6].
As the fourth stage of the migration process, adult mating pairs of S. japonicum and S. mansoni migrate to the venous vasculature close to the intestines and lay eggs [50,51]. The eggs penetrate the blood vessel wall to reach the intestinal lumen and are excreted in feces to complete the migration process in the host [5]. Previous autopsy studies of humans infected with S. japonicum or S. mansoni reported that most parasites were widely distributed in the liver, hepatic portal vein, and mesenteries, which were considered their natural habitat [54,55]. Due to the inexorable exploratory nature of intravascular schistosomes, eggs have also been discovered in nonproductive sites, which include the spleen [54], stomach [56], pancreas [57], gall bladder [57], ovary [58], fallopian tube [59], prostate [60], brain [61], and spinal cord [62]. Interestingly, SjHMGB1 is abundantly expressed in the adult and egg phases, but barely detectable in the cercaria stage [63]. However, further studies are needed to elucidate the involvement of HMGB1 in the development of S. japonicum.
Unlike S. japonicum and S. mansoni, S. haematobium primarily migrates from the hepatic portal vein to the vesicle venous plexus surrounding the bladder and induces urinary schistosomiasis [5]. In women, S. haematobium eggs have been found in the cervix, uterus, vagina, fallopian tubes, and ovaries, which all contribute to female genital schistosomiasis [64,65]. In men, eggs released by S. haematobium have been found in the epididymis, prostate, seminal vesicles, vas deferens, and testes, leading to male genital schistosomiasis [66]. Eggs in the vesicle venous plexus can penetrate the blood vessel and the wall of the bladder, then enter the bladder lumen and finally exit the body in urine [5,6].
Once eggs are excreted from the host (S. japonicum or S. mansoni in feces and S. haematobium in urine), miracidia hatch from the eggs in freshwater and look for a suitable snail as an intermediate host to complete and repeat the life cycle.
Nevertheless, about one-third to one-half of eggs released by schistosomes fail to reach the external environment but rather become trapped in various tissues of the host, such as the liver, where the eggs induce a potent inflammatory response, leading to the formation of granulomas, the most basic and crucial pathologic form of schistosomiasis [67]. The life cycle of S. japonicum and the process of egg-induced granuloma formation are illustrated in Figure 2.
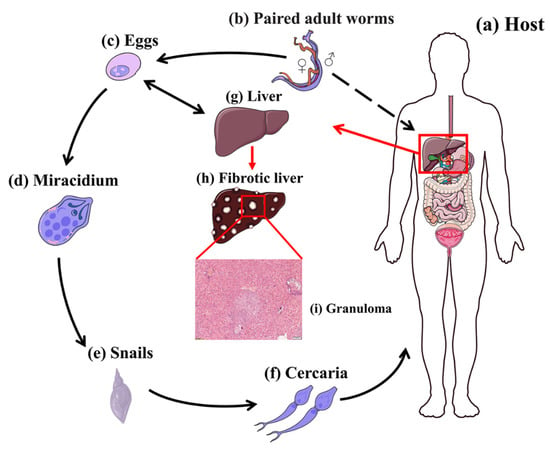
Figure 2.
Lifecycle of S. japonicum. (a) Host. (b) Paired adult worms (larger male enfolding slender female). (c) Eggs. (d) Miracidium. (e) Intermediate host Oncomelania. (f) Cercariae. (g) Healthy liver. (h) Fibrotic liver with granuloma. (i) Representative histology of S. japonicum-induced granuloma formation in BALB/c mice at 6 weeks post-infection. Formalin-fixed, paraffin-embedded liver sections stained with hematoxylin and eosin (H&E). Scale bars, 100 mm.
3.2. Formation of Liver Granulomas
Schistosome eggs have highly antigenic structures that constantly secrete various types of toxic or antigenic substances into the tissues of host [68]. Evidence suggests that schistosome eggs, but not adult worms, induce the pathogenicity of schistosome infections [2,6]. Granulomas and subsequent hepatic fibrosis occurring in the middle and late stages of infection result from the host immune response to the soluble egg antigen of schistosome eggs [69,70].
The dynamic development of liver granulomas can be segmented into acute and chronic phases based on the interactions of various cell types, including macrophages, neutrophils, eosinophils, and hepatic stellate cells (HSCs) [9,71]. Generally, the early acute phase of schistosome infection is regulated through T-helper (Th)1 responses via the up-regulated expression of the Th1 proinflammatory cytokines interleukin (IL)-1, IL-12, interferon (IFN)-γ, and tumor necrosis factor (TNF)-α, which targets the migrating schistosomula and mature adult worms [72]. In the chronic phase (6–8 weeks post infection), the immune response gradually switches to a Th2 response as the schistosome eggs are deposited [5,72]. The switch from the Th1- to Th2-mediated immune response can protect the host against the lethal outcomes of chronic, unregulated Th1 proinflammatory responses [73].
The composition of schistosome-induced granulomas varies among species, as neutrophils are the predominant cell type of S. japonicum-induced hepatic granulomas, while S. mansoni targets eosinophils, neutrophils, and macrophages, with eosinophils accounting for 50–70% of all granulomas [74] (Figure 3). The difference in the proportions of immune cells in S. japonicum- and S. mansoni-induced hepatic granulomas is due to a glycoprotein released by S. mansoni eggs, known as the IL-4-inducing principle of schistosome eggs/α-1, which is not expressed by S. japonicum eggs [75]. The function of this protein is activated upon binding to chemokines, such as C-X-C motif chemokine ligand 8, which is associated with the chemotaxis of neutrophil, giving rise to the inhibition of neutrophil infiltration to the site of infection [76].
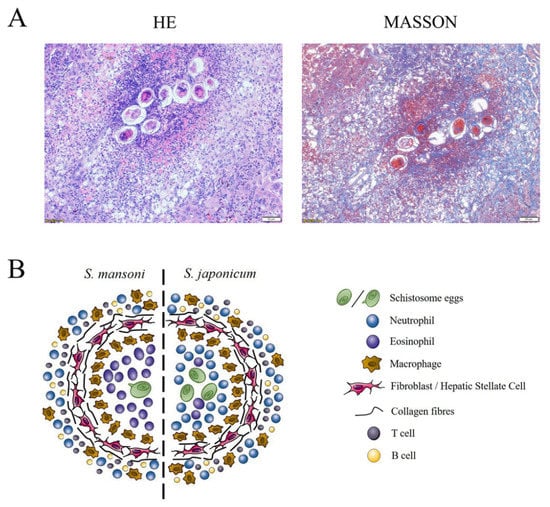
Figure 3.
Representative histological images of hepatic granulomas of S. japonicum infection and illustration of granuloma composition of S. mansoni and S. japonicum infection. (A) Representative histology of S. japonicum-induced granuloma in BALB/c mice at 6 weeks post-infection. Formalin-fixed, paraffin-embedded liver sections stained with H&E and Masson. Scale bars, 50 mm. (B) Major cell populations of S. mansoni and S. japonicum infection. In S. mansoni infection, a granuloma is formed by a single egg and has an eosinophil dominant core. In S japonicum infection, a granuloma is formed by a cluster of eggs and the neutrophil as the dominate cell type. Abbreviation: HSC, hepatic stellate cell.
Generally, after infection, granulomas begin to engage with eosinophils, macrophages, and neutrophils around the trapped egg, which further induces inflammatory responses [77]. In response to tissue injury, HSCs are then stimulated to generate ECM to induce fibrotic responses that contribute to the formation of an extra fibrotic zone [74]. Macrophages, eosinophils, and neutrophils accumulate at the periphery of the granuloma prior to infiltration [71]. At a later stage, several granule proteins involved with collagen degradation and reabsorption are released, which leads to remodeling of the impaired tissues and modulation of the pathology of granulomas [9,77]. During granuloma formation, neutrophils not only express proinflammatory cytokines, such as TNF- α, in the granuloma core, but also factors involved in collagen degradation and remodeling, such as matrix metalloproteinase-9 [77]. Besides, macrophages and HSCs also play pivotal roles in granuloma formation. HSCs are the main source of collagen in several hepatic fibrotic diseases other than schistosome-induced hepatic fibrosis, such as alcoholic liver disease, chronic viral hepatitis, and nonalcoholic fatty liver disease [71,78,79]. Storage of vitamin A is the primary function of quiescent HSCs, while activated HSCs in the fibrotic zone serve as an indicator of collagen deposition in hepatic schistosomiasis, which can be detected by immunostaining for the HSCs markers glial fibrillary acidic protein and α-smooth muscle actin (α-SMA) [80]. Furthermore, subsequent associations of activated HSCs with the NF-κB and transforming growth factor (TGF)/Smads pathways promote hepatic inflammation and fibrosis, which are discussed in greater detail later in this review [81,82]. Macrophages are vital to the pathogenesis of schistosomiasis and account for more than 30% of cells in liver granulomas induced by S. mansoni [83]. However, information on the proportion of macrophages in S. japonicum-induced granulomas is limited, thus warranting further exploration. Macrophages can be classified into two groups with distinct biological features: alternatively activated macrophages, which enhance fibrogenesis of fibroblasts by providing profibrogenic factors, and classically activated macrophages, which promote inflammation in the early stage of liver injury and inhibit fibrogenesis of fibroblasts by releasing antifibrogenic or fibrolytic factors in the late stage of hepatic fibrosis [84].
The dynamic development of liver granuloma is regulated by neutrophils as the primary cell type and serves a dual purpose, as well as HSCs and macrophages [9,85]. The collaboration of various cell types can lead to the formation of hepatic fibrosis and subsequent hepatosplenomegaly, portal hypertension, ascites, and esophageal varices, which can be fatal to the schistosomiasis host [2,6]. An illustration of the granuloma composition and representative histological images of murine hepatic granulomas due to S. japonicum infection are shown in Figure 3.
4. HMGB1 and Schistosome-Induced Liver Damage
Chronic hepatic inflammation is closely related to fibrosis in virtually all liver diseases and experimental models of fibrogenesis. However, the molecular relationship between inflammation and hepatic fibrosis remains unclear. Following the formation of hepatic granulomas induced by chronic schistosomiasis, HMGB1 is released from stressed or activated cells and exhibits pleiotropic functions in hepatic inflammation and fibrosis. In the following sections, the role of HMGB1 in schistosome-induced liver damage is discussed in more detail.
4.1. HMGB1 and Liver Inflammation
Animal studies have found that HMGB1 triggered systemic inflammatory diseases, including mastitis, sepsis, arthritis, epileptogenesis, necrotizing enterocolitis, acute lung injury, traumatic brain injury, and liver injury [86]. Elevation of HMGB1 levels in plasma and extracellular tissues is consider to promote inflammation. Structurally, the TLR-binding site of the B-box domain of HMGB1 initiates the HMGB1-induced inflammatory activities, whereas the A-box antagonizes this function [15]. Previous studies have demonstrated the role of the HMGB1/TLR4 signaling pathway in several HMGB1-mediated inflammatory diseases. For example, Lian et al. [87] reported that upon release, HMGB1 acted as a late-phase mediator of lipopolysaccharide-induced depression, which could be abrogated by a TLR4 inhibitor, while Yu et al. [88] found out that the application of glycyrrhizin (GL), a natural antagonist of HMGB1 extracted from licorice root, inhibited activation of the TLR4/NF-κB signaling pathway in necrotizing enterocolitis. In ischemia-reperfusion injury, inhibition of HMGB1 attenuates the expression of downstream proinflammatory cytokines activated by the TLR4/NF-κB signaling pathway [42].
In liver injury, activated HSCs can express TLR2, TLR3, TLR4, and TLR9. The interaction between TLR4 and liver inflammation has been widely reported [42,43,44]. For instance, TLR4 promotes early alcohol-induced liver injury by responding to increasing levels of circulating endotoxins [89]. The ability of TLR4 to inhibit the replication of hepatitis B virus (HBV) suggests that TLR4 is associated with chronic HBV infection [90]. Besides, TLR4 can induce hepatocellular carcinoma by increasing the number of lymphocytes and follicular helper-like cells residing in the liver and the production of pro-inflammatory cytokines [91]. In an animal model of carbon tetrachloride-induced hepatic fibrosis, TLR4 was reported to activate the Myd88/NF-κB pathway in HSCs [92]. The NF–κB complex consists of p50 and p65 dimers, which are retained in the cytoplasm by binding to the inhibitory protein IκB [93]. Once activated, NF-κB is translocated from the cytoplasm to the nucleus, where it binds to the κB site of target genes and induces the stimulation of inflammatory cytokines, which can result in liver damage [94]. Seki et al. [92] found that as compared with wild-type mice, the expression of the proinflammatory cytokines TNF-α and IL-6 was significantly reduced in Myd88-deficient mice. An interesting recent study reported that inhibition of HMGB1/TLR4 intracellular signaling decreased production of NF-κB, TNF-α, and IL-6 in cholestatic liver inflammation, which further confirms the close interactions among HMGB1, TLR4, and NF-κB in the promotion of liver inflammation [95].
HMGB1 and the NF-κB pathway are tightly linked through TLR4. Activation of HMGB1 and downstream signaling pathways contribute to inflammation and play pivotal roles in schistosome infection of various organs, including the liver. However, the direct connection between HMGB1 and schistosome-induced liver inflammation is poorly understood. In a study conducted by Vicentino et al. [19], high levels of HMGB1 were observed in the sera of mice and patients infected with S. mansoni. In order to comfirm the involvement of HMGB1 in schistosome-induced liver inflammation, mice were treated with the HMGB1 inhibitors GL and the synthetic compound 3-chloro-5-(4-pyridyl)-4,5-dihydroisoxazole (DIC) [19]. The results show that inhibition of HMGB1 leads to healthier livers and higher survival rates with down-regulation of the proinflammatory cytokines IL-6, IL4, IL-5, IL-13, and IL-17A and up-regulation of the anti-inflammatory cytokine IL-10 [19], demonstrating the important role of the HMGB1/TLR4 signaling pathway in schistosome-induced liver inflammation. In this regard, due to the contributing role of HMGB1 in various inflammatory diseases, toxic and antigenic substances released by schistosome eggs may directly or indirectly trigger the proinflammatory function of HMGB1, which leads to downstream activation of the TLR4/NF-κB signaling pathway.
4.2. HMGB1 and Liver Fibrosis
Liver fibrosis is a wound-repair process in response to liver injury and inflammation, which can be induced by a variety of persistent factors such as toxins, chronic viral infection, long-term alcohol abuse, and chronic parasitic disease [96,97,98]. Despite the complex etiology, liver fibrosis is characterized by the increased deposition of ECM involving activated HSCs [99]. Damaged HSCs exhibits a proliferative and fibrogenic myofibroblast-like phenotype, which can be confirmed by the expression of α-SMA [100]. Recent studies have suggested that activated HSCs accumulate in the periphery of murine and human granulomas induced by S. japonicum egg infections and are identified as effector cells that further contribute to granuloma-associated fibrosis [80]. Therefore, it is important to understand the physiological relationship between granuloma formation and down-modulation of HSCs.
HMGB1 is a contributing factor in hepatic fibrosis. Damage to the liver parenchyma triggers the release of HMGB1, resulting in the transdifferentiation of HSCs into scar-forming liver myofibroblasts [101]. Activated HSCs then secrete matrix proteins, such as α-SMA and collagen type I, into the extracellular space, leading to liver scar formation [102]. Structurally, HMGB1-driven α-SMA upregulation in cultured HSCs is reportedly dependent on RAGE [103]. Serum HMGB1 levels are significantly upregulated in patients with chronic HBV infection and in those with mild fibrosis as compared with severe fibrosis [104]. In a mouse model of carbon tetrachloride-induced liver fibrosis, the increase in serum HMGB1 was proportional to the elevation of TGF-β1 and collagen deposition during fibrogenesis [103]. More importantly, in the same study, HMGB1 stimulated the proliferation of HSCs in vitro, up-regulated the synthesis of α-SMA, and triggered Smad2 phosphorylation through the TGF-β1/Smad signaling pathway [103].
TGF-β1 is the most common TGF-β isoform in the mammalian liver [81,105]. Previous studies have demonstrated that the biological effects of TGF-β1 in cellular proliferation, differentiation, and fibrogenesis are dependent on phosphorylation of the downstream mediators Smad2 and Smad3, which existed in complexes and negatively regulated by the inhibitory mediator Smad7 [106,107]. In addition, as one of the first signals to activate quiescent HSCs during liver fibrosis, TGF-β1 is released by necrotic hepatocytes upon interactions with the cell surface receptors TβRI and TβRII, both of which are expressed by HSCs [108]. Hence, it can be concluded that the TGF-β1/Smad pathway is a key mediator of HSCs during hepatic fibrosis. Interestingly, HMGB1 and the TGF-β1/Smad pathway also seem to be relevant in renal and lung fibrosis [109,110]. However, further studies are needed to explore the interaction between HMGB1 and TGF-β1 in HSCs.
The pathogenesis of schistosome-induced liver fibrosis involves multiple pathways [71]. Studies have shown that HMGB1 was predominantly localized in the cytoplasm of hepatocytes surrounding schistosome-induced granulomas in the livers of mice with acute schistosomiasis as well as the nuclei of hepatocytes during chronic infections [19]. Interestingly, based on data of other fibrotic disease, this fibrotic signal might not be triggered by HMGB1 via TLRs. The receptor-specific selectivity of a fibrotic response appears to differ from liver inflammation, where HMGB1 interacts with both TLRs and RAGE. Arriazu et al. [111] suggested that HMGB1-elicited signaling in fibrotic disease involved not only the TGF-β1/Smad signaling pathway, but also the RAGE/phosphoinositide 3-kinase (PI3K)/AKT or RAGE-ERK pathway. As a fundamental regulator of signal transduction in protein synthesis, AKT can be transformed into a phosphorylation state via stimulation by activated PI3K [112], which then promotes the release of the inflammatory cytokines TNF-α, IL-1β, and IL-18, thereby stimulating the inflammatory cascade response resulting in damage to various organs and tissues [101].Therefore, HMGB1 seems to play a pivotal role in liver fibrosis by activating HSCs mainly through RAGE receptors to initiate collagen synthesis and deposition in liver fibrosis.
A recent report of HMGB1 in schistosome-induced liver fibrosis found that serum levels and relative mRNA expression of HMGB1 were significantly higher in the livers of the S. japonicum/S. mansoni-infection group, while the expression levels of the pro-inflammatory and fibrogenic cytokines IFN-γ, TGF-β1, and IL-6 were reduced [19,20]. Based on the close relationship between HMGB1 and TGF-β1 and the PI3K/AKT pathway in various fibrotic diseases, the HMGB1/TGF-β1 and HMGB1/RAGE/PI3K/AKT pathways may have a synergistic effect in granuloma-associated liver fibrosis, although little has been reported about any direct connections, such as whether upregulation of the TGF-β1/Smad pathway is mediated by the RAGE binding site of HMGB1. Since elevated serum and tissue levels of HMGB1 have been correlated with inflammation- and/or fibrosis-mediated pathologies in fibrosis of the liver, lung, and kidney, HMGB1 should be further investigated as a potential drug target. An illustration of the signaling pathways involved in the activities of HMGB1 during schistosome-induced liver damage is presented in Figure 4.
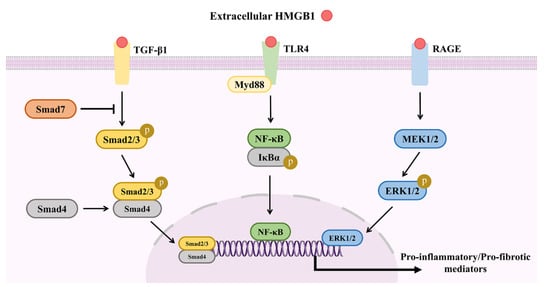
Figure 4.
Signaling pathways involved in HMGB1 activities during schistosome-induced liver damage. During schistosome-induced liver damage, extracellular HMGB1 can trigger activation of the TGF-β1/Smad, TLR4/MyD88/NF-κB and RAGE/MEK/ERK in HSCs, promoting the production of pro-inflammatory or pro-fibrotic cytokines.
5. HMGB1 as a Therapeutic Target
Increasing evidence indicates that as a multi-functional mediator, HMGB1 contributes to a variety of systemic diseases, including ischemia-reperfusion injury, enterocolitis, sepsis, and liver injury, indicating the potential of HMGB1 as a therapeutic target [42,86,88,101]. Recently, under the worldwide threat of sudden acute respiratory syndrome (SARS-CoV-2), the etiological agent of COVID-19 disease, the progression from acute respiratory failure to sepsis has been correlated with the release of HMGB1 [113]. Wei et al. [114] proposed that HMGB1 regulates the expression of angiotensin-converting enzyme 2, which is essential for the entry of SARS-CoV-1 (highly pathogenic zoonotic CoVs), SARS-CoV-2 (highly pathogenic zoonotic CoVs), and human coronavirus NL63. Emerging studies have indicated that the inhibition of HMGB1 can improve the courses of all related diseases. For instance, knockout of HMGB1 protected against cell death induced by SARS-CoV-2 and the degree of protection were correlated with the level of HMGB1 [114]. Inhibition of HMGB1 was found to improve intestinal inflammation in necrotizing enterocolitis by inhibiting NLR pyrin domain containing 3 via the TLR4 and NF-κB signaling pathways [88]. Besides, HMGB1-neutralizing antibodies relieved liver inflammation and functional injury in the early stages of nonalcoholic fatty liver disease [115]. Another study demonstrated that HMGB1-neutralizing antibodies rescued cystic fibrosis-induced macrophage dysfunction and reduced lung inflammation induced by Pseudomonas aeruginosa [116].
This article highlighted the emerging roles of HMGB1 in liver diseases. Numerous studies have also suggested HMGB1 as a therapeutic target for liver diseases by blocking the release of HMGB1 or inhibiting the activity of extracellular HMGB1. GL is a glyco-conjugated triterpene produced by Glycyrrhiza inflata, which is an anti-inflammatory used for the treatment of chronic HBV infection [117]. Masahito et al. [118] found that GL reduced HMGB1 expression in Kupffer cells, suggesting that GL has therapeutic potential to prevent liver injury. A further study reported that the interactions between GL and the A box/B box of HMGB1 antagonized the inflammatory response [119]. Recently, GL was also considered a viable therapeutic option for SARS-CoV-2 infection due to the dual ability to concomitantly inhibit virus replication and diminish the production of proinflammatory mediators [120]. In schistosome-induced liver disease, Vicentino et al. [19] used GL to treat mice infected with S. mansoni from 21 to 56 dpi, which significantly reduced the area of the granuloma as well as serum cytokine levels. However, GL had no significant effect on the egg loads in the intestine, suggesting that GL had no schistosomicidal activity [19]. Similar results were obtained with the HMGB1 inhibitor DIC [94]. Interestingly, sodium butyrate, a deacetylase inhibitor, which conveys a protective effect in certain inflammatory diseases, such as sepsis and lipopolysaccharide-induced acute lung injury, can also reduce hepatic granulomas and fibrosis induced by S. japonicum by inhibiting HMGB1 expression [20]. Ge et al. [21] used small interfering RNA to directly inhibit HMGB1 expression in rat HSCs, which inhibited activation of HSCs.
Collectively, these data indicate a pro-fibrotic role of HMGB1 in schistosome-induced liver granulomas. Inhibiting the expression of HMGB1 by interference methods or antagonists, or suppressing downstream signaling pathways, such as HMGB1/RAGE, are potential strategies to treat schistosome-induced liver fibrosis.
6. Concluding Remarks
Humans and animals with chronic schistosome infection can develop chronic liver disease, which develops over many years and often decades. Without prompt treatment, once organ function has been significantly compromised and is irreversible, patients usually present with advanced disease. Studies of early biomarkers have focused on the status of liver disease. HMGB1 is a wildly expressed and conserved protein with multiple functions, including regulation of liver inflammation and fibrosis. Treatment to inhibit the secretion and function of HMGB1 could provide significant protection against liver inflammation and fibrosis. To date, limited studies have investigated the relationship between HMGB1 and schistosome-induced liver disease. Given that the lives of hundreds of millions of people are threatened by schistosomiasis, further research efforts should focus on HMGB1 as a potential therapeutic target for schistosome-induced liver disease. Moreover, the potential collaboration of PZQ with HMGB1 inhibitors as a treatment for schistosomiasis should be explored. In addition, future studies are warranted to determine whether HMGB1 is involved in the pathogenesis of S. haematobium-induced disease and the function HMGB1 in schistosomes. Regardless, the discoveries discussed in this review describe the current knowledge of the roles of HMGB1 in liver disease, with an emphasis on schistosomiasis. Reports in this area pose profound questions regarding the exploration and treatment of liver inflammation and fibrosis, both parasitic and nonparasitic in origin.
Author Contributions
Writing—original draft preparation, H.Z.; writing—review and editing, X.G., L.H., and R.L.; supervision and writing—review and editing, Y.J. All authors have read and agreed to the published version of the manuscript.
Funding
This work was funded by the Natural Science Foundation of Shanghai (no.20ZR1469300), the Chinese National Natural Science Foundation (no. 31672245).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Conflicts of Interest
The authors declare that they have no conflict of interest.
References
- WHO. TDR Strategic Direction for Research: Schistosomiasis; World Health Organization: Geneva, Switzerland, 2002. [Google Scholar]
- Gryseels, B.; Polman, K.; Clerinx, J.; Kestens, L. Human schistosomiasis. Lancet 2006, 368, 1106–1118. [Google Scholar] [CrossRef]
- Boissier, J.; Grech-Angelini, S.; Webster, B.L.; Allienne, J.F.; Huyse, T.; Mas-Coma, S.; Toulza, E.; Barré-Cardi, H.; Rollinson, D.; Kincaid-Smith, J.; et al. Outbreak of urogenital schistosomiasis in Corsica (France): An epidemiological case study. Lancet 2016, 16, 971–979. [Google Scholar] [CrossRef]
- Chitsulo, L.; Loverde, P.; Engels, D. Focus: Schistosomiasis. Nat. Rev. Microbiol. 2004, 2, 12. [Google Scholar] [CrossRef]
- Nation, C.S.; Da’dara, A.A.; Marchant, J.K.; Skelly, P.J. Schistosome migration in the definitive host. PLoS Negl. Trop. Dis. 2020, 14, e0007951. [Google Scholar] [CrossRef]
- Colley, D.G.; Bustinduy, A.L.; Secor, W.E.; King, C.H. Human schistosomiasis. Lancet 2014, 383, 2253–2264. [Google Scholar] [CrossRef]
- Cheever, A.W.; Macedonia, J.G.; Mosimann, J.E.; Cheever, E.A. Kinetics of egg production and egg excretion by Schistosoma mansoni and S. japonicum in mice infected with a single pair of worms. Am. J. Trop. Med. Hyg. 1994, 50, 281–295. [Google Scholar] [CrossRef]
- He, X.; Tang, R.; Sun, Y.; Wang, Y.G.; Zhen, K.Y.; Zhang, D.M.; Pan, W.Q. MicroR-146 blocks the activation of M1 macrophage by targeting signal transducer and activator of transcription 1 in hepatic schistosomiasis. EBioMedicine 2016, 13, 339–347. [Google Scholar] [CrossRef]
- Chuah, C.; Jones, M.K.; Burke, M.L.; McManus, D.P.; Gobert, G.N. Cellular and chemokine-mediated regulation in schistosome-induced hepatic pathology. Trends Parasitol. 2014, 30, 141–150. [Google Scholar] [CrossRef]
- Soares Magalhães, R.J.; Barnett, A.G.; Clements, A.C. Geographical analysis of the role of water supply and sanitation in the risk of helminth infections of children in West Africa. Proc. Natl. Acad. Sci. USA 2011, 108, 20084–20089. [Google Scholar] [CrossRef]
- Wang, L.D.; Chen, H.G.; Guo, J.G.; Zeng, X.J.; Hong, X.L.; Xiong, J.J.; Wu, X.H.; Wang, X.H.; Wang, L.Y.; Xia, G.; et al. A strategy to control transmission of Schistosoma japonicum in China. N. Engl. J. Med. 2009, 360, 121–128. [Google Scholar] [CrossRef]
- Caligiuri, A.; Gentilini, A.; Pastore, M.; Gitto, S.; Marra, F. Cellular and Molecular Mechanisms Underlying Liver Fibrosis Regression. Cells 2021, 10, 2759. [Google Scholar] [CrossRef]
- Fenwick, A.; Savioli, L.; Engels, D.; Robert Bergquist, N.; Todd, M.H. Drugs for the control of parasitic diseases: Current status and development in schistosomiasis. Trends Parasitol. 2003, 19, 509–515. [Google Scholar] [CrossRef]
- Hagen, J.; Scheerlinck, J.P.; Gasser, R.B. Knocking down schistosomes—Promise for lentiviral transduction in parasites. Trends Parasitol. 2015, 31, 324–332. [Google Scholar] [CrossRef]
- Harris, H.E.; Andersson, U.; Pisetsky, D.S. HMGB1: A multifunctional alarmin driving autoimmune and inflammatory disease. Nat. Rev. Rheumatol. 2012, 8, 195–202. [Google Scholar] [CrossRef]
- Hu, Y.; Wang, X.; Wei, Y.; Liu, H.; Zhang, J.; Shen, Y.; Cao, J. Functional Inhibition of Natural Killer Cells in a BALB/c Mouse Model of Liver Fibrosis Induced by Schistosoma japonicum Infection. Front. Cell. Infect. Microbiol. 2020, 10, 598987. [Google Scholar] [CrossRef]
- Andersson, U.; Wang, H.; Palmblad, K.; Aveberger, A.C.; Bloom, O.; Erlandsson-Harris, H.; Janson, A.; Kokkola, R.; Zhang, M.; Yang, H.; et al. High mobility group 1 protein (HMG-1) stimulates proinflammatory cytokine synthesis in human monocytes. J. Exp. Med. 2000, 192, 565–570. [Google Scholar] [CrossRef]
- Andersson, U.; Tracey, K.J. HMGB1 is a therapeutic target for sterile inflammation and infection. Annu. Rev. Immunol. 2011, 29, 139–162. [Google Scholar] [CrossRef]
- Vicentino, A.R.R.; Carneiro, V.C.; Allonso, D.; Guilherme, R.F.; Benjamim, C.F.; Dos Santos, H.A.M.; Xavier, F.; Pyrrho, A.D.S.; Gomes, J.A.S.; Fonseca, M.C.; et al. Emerging Role of HMGB1 in the Pathogenesis of Schistosomiasis Liver Fibrosis. Front. Immunol. 2018, 9, 1979. [Google Scholar] [CrossRef]
- Chen, H.; Li, G.; Zhang, J.; Zheng, T.; Chen, Q.; Zhang, Y.; Yang, F.; Wang, C.; Nie, H.; Zheng, B.; et al. Sodium butyrate ameliorates Schistosoma japonicum-induced liver fibrosis by inhibiting HMGB1 expression. Exp. Parasitol. 2021, 231, 108171. [Google Scholar] [CrossRef]
- Ge, W.-S. Inhibition of high-mobility group box 1 expression by siRNA in rat hepatic stellate cells. World J. Gastroenterol. 2011, 17, 4090. [Google Scholar] [CrossRef]
- Huang, H.; Nace, G.W.; McDonald, K.A.; Tai, S.; Klune, J.R.; Rosborough, B.R.; Ding, Q.; Loughran, P.; Zhu, X.; Beer-Stolz, D.; et al. Hepatocyte-specific high-mobility group box 1 deletion worsens the injury in liver ischemia/reperfusion: A role for intracellular high-mobility group box 1 in cellular protection. Hepatology 2014, 59, 1984–1997. [Google Scholar] [CrossRef] [PubMed]
- Goodwin, G.H.; Sanders, C.; Johns, E.W. A new group of chromatin-associated proteins with a high content of acidic and basic amino acids. Eur. J. Biochem. 1973, 38, 14–19. [Google Scholar] [CrossRef] [PubMed]
- Lotze, M.T.; Tracey, K.J. High-mobility group box 1 protein (HMGB1): Nuclear weapon in the immune arsenal. Nat. Rev. Immunol. 2005, 5, 331–342. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Antoine, D.J.; Andersson, U.; Tracey, K.J. The many faces of HMGB1: Molecular structure-functional activity in inflammation, apoptosis, and chemotaxis. J. Leukoc. Biol. 2013, 93, 865–873. [Google Scholar] [CrossRef]
- de Oliveira, F.M.; de Abreu da Silva, I.C.; Rumjanek, F.D.; Dias-Neto, E.; Guimarães, P.E.; Verjovski-Almeida, S.; Stros, M.; Fantappié, M.R. Cloning the genes and DNA binding properties of High Mobility Group B1 (HMGB1) proteins from the human blood flukes Schistosoma mansoni and Schistosoma japonicum. Gene 2006, 377, 33–45. [Google Scholar] [CrossRef]
- Neeper, M.; Schmidt, A.M.; Brett, J.; Yan, S.D.; Wang, F.; Pan, Y.C.; Elliston, K.; Stern, D.; Shaw, A. Cloning and expression of a cell surface receptor for advanced glycosylation end products of proteins. J. Biol. Chem. 1992, 267, 14998–15004. [Google Scholar] [CrossRef]
- Fritz, G. RAGE: A single receptor fits multiple ligands. Trends Biochem. Sci. 2011, 36, 625–632. [Google Scholar] [CrossRef]
- Bierhaus, A.; Nawroth, P.P. Multiple levels of regulation determine the role of the receptor for AGE (RAGE) as common soil in inflammation, immune responses and diabetes mellitus and its complications. Diabetologia 2009, 52, 2251–2263. [Google Scholar] [CrossRef]
- Taguchi, A.; Blood, D.C.; del Toro, G.; Canet, A.; Lee, D.C.; Qu, W.; Tanji, N.; Lu, Y.; Lalla, E.; Fu, C.; et al. Blockade of RAGE-amphoterin signalling suppresses tumour growth and metastases. Nature 2000, 405, 354–360. [Google Scholar] [CrossRef]
- Huttunen, H.J.; Kuja-Panula, J.; Rauvala, H. Receptor for advanced glycation end products (RAGE) signaling induces CREB-dependent chromogranin expression during neuronal differentiation. J. Biol. Chem. 2002, 277, 38635–38646. [Google Scholar] [CrossRef]
- Huttunen, H.J.; Fages, C.; Rauvala, H. Receptor for advanced glycation end products (RAGE)-mediated neurite outgrowth and activation of NF-kappaB require the cytoplasmic domain of the receptor but different downstream signaling pathways. J. Biol. Chem. 1999, 274, 19919–19924. [Google Scholar] [CrossRef] [PubMed]
- Yan, S.D.; Schmidt, A.M.; Anderson, G.M.; Zhang, J.; Brett, J.; Zou, Y.S.; Pinsky, D.; Stern, D. Enhanced cellular oxidant stress by the interaction of advanced glycation end products with their receptors/binding proteins. J. Biol. Chem. 1994, 269, 9889–9897. [Google Scholar] [CrossRef]
- Huang, J.S.; Guh, J.Y.; Chen, H.C.; Hung, W.C.; Lai, Y.H.; Chuang, L.Y. Role of receptor for advanced glycation end-product (RAGE) and the JAK/STAT-signaling pathway in AGE-induced collagen production in NRK-49F cells. J. Cell. Biochem. 2001, 81, 102–113. [Google Scholar] [CrossRef]
- Yamagishi, S.; Matsui, T. Role of receptor for advanced glycation end products (RAGE) in liver disease. Eur. J. Med. Res. 2015, 20, 15. [Google Scholar] [CrossRef] [PubMed]
- Gaens, K.H.; Niessen, P.M.; Rensen, S.S.; Buurman, W.A.; Greve, J.W.; Driessen, A.; Wolfs, M.G.; Hofker, M.H.; Bloemen, J.G.; Dejong, C.H.; et al. Endogenous formation of Nε-(carboxymethyl)lysine is increased in fatty livers and induces inflammatory markers in an in vitro model of hepatic steatosis. J. Hepatol. 2012, 56, 647–655. [Google Scholar] [CrossRef] [PubMed]
- Nomoto, K.; Tsuneyama, K.; Abdel Aziz, H.O.; Takahashi, H.; Murai, Y.; Cui, Z.G.; Fujimoto, M.; Kato, I.; Hiraga, K.; Hsu, D.K.; et al. Disrupted galectin-3 causes non-alcoholic fatty liver disease in male mice. J. Pathol. 2006, 210, 469–477. [Google Scholar] [CrossRef]
- Akira, S.; Takeda, K. Toll-like receptor signalling. Nat. Rev. Immunol. 2004, 4, 499–511. [Google Scholar] [CrossRef]
- Meng, X.; Zhang, X.; Su, X.; Liu, X.; Ren, K.; Ning, C.; Zhang, Q.; Zhang, S. Daphnes Cortex and its licorice-processed products suppress inflammation via the TLR4/NF-κB/NLRP3 signaling pathway and regulation of the metabolic profile in the treatment of rheumatoid arthritis. J. Ethnopharmacol. 2022, 283, 114657. [Google Scholar] [CrossRef]
- Cai, J.; Li, J.; Zhou, Y.; Wang, J.; Li, J.; Cui, L.; Meng, X.; Zhu, G.; Wang, H. Staphylococcus aureus facilitates its survival in bovine macrophages by blocking autophagic flux. J. Cell. Mol. Med. 2020, 24, 3460–3468. [Google Scholar] [CrossRef]
- Liu, K.; Ding, T.; Fang, L.; Cui, L.; Li, J.; Meng, X.; Zhu, G.; Qian, C.; Wang, H.; Li, J. Organic Selenium Ameliorates Staphylococcus aureus-Induced Mastitis in Rats by Inhibiting the Activation of NF-κB and MAPK Signaling Pathways. Front. Vet. Sci. 2020, 7, 443. [Google Scholar] [CrossRef]
- Li, Y.; Xu, B.; Yang, J.; Wang, L.; Tan, X.; Hu, X.; Sun, L.; Chen, S.; Zhu, L.; Chen, X.; et al. Liraglutide protects against lethal renal ischemia-reperfusion injury by inhibiting high-mobility group box 1 nuclear-cytoplasmic translocation and release. Pharmacol. Res. 2021, 173, 105867. [Google Scholar] [CrossRef] [PubMed]
- Azam, S.; Jakaria, M.; Kim, I.S.; Kim, J.; Haque, M.E.; Choi, D.K. Regulation of Toll-Like Receptor (TLR) Signaling Pathway by Polyphenols in the Treatment of Age-Linked Neurodegenerative Diseases: Focus on TLR4 Signaling. Front. Immunol. 2019, 10, 1000. [Google Scholar] [CrossRef] [PubMed]
- Guo, Q.; Qu, H.; Zhang, H.; Zhong, X. Prunella vulgaris L. Attenuates Experimental Autoimmune Thyroiditis by Inhibiting HMGB1/TLR9 Signaling. Drug Des. Dev. Ther. 2021, 15, 4559–4574. [Google Scholar] [CrossRef] [PubMed]
- Stensgaard, A.S.; Vounatsou, P.; Sengupta, M.E.; Utzinger, J. Schistosomes, snails and climate change: Current trends and future expectations. Acta Trop. 2019, 190, 257–268. [Google Scholar] [CrossRef] [PubMed]
- Hailegebriel, T.; Nibret, E.; Munshea, A. Prevalence of Schistosoma mansoni and S. haematobium in Snail Intermediate Hosts in Africa: A Systematic Review and Meta-analysis. J. Trop. Med. 2020, 2020, 8850840. [Google Scholar] [CrossRef]
- He, Y.X.; Chen, L.; Ramaswamy, K. Schistosoma mansoni, S. haematobium, and S. japonicum: Early events associated with penetration and migration of schistosomula through human skin. Exp. Parasitol. 2002, 102, 99–108. [Google Scholar] [CrossRef]
- He, Y.X.; Salafsky, B.; Ramaswamy, K. Comparison of skin invasion among three major species of Schistosoma. Trends Parasitol. 2005, 21, 201–203. [Google Scholar] [CrossRef]
- Miller, P.; Wilson, R.A. Migration of the schistosomula of Schistosoma mansoni from the lungs to the hepatic portal system. Parasitology 1980, 80, 267–288. [Google Scholar] [CrossRef]
- Wheater, P.R.; Wilson, R.A. Schistosoma mansoni: A histological study of migration in the laboratory mouse. Parasitology 1979, 79, 49–62. [Google Scholar] [CrossRef]
- Ito, J. Studies on the host-parasite relationships of Schistosoma japonicum in common laboratory animals. Jpn. J. Med. Sci. Biol. 1955, 8, 43–62. [Google Scholar] [CrossRef][Green Version]
- Georgi, J.R.; Wade, S.E.; Dean, D.A. Attrition and temporal distribution of Schistosoma mansoni and S. haematobium schistosomula in laboratory mice. Parasitology 1986, 93 Pt 1, 55–70. [Google Scholar] [CrossRef] [PubMed]
- van Oordt, B.E.; Tielens, A.G.; van den Bergh, S.G. The energy metabolism of Schistosoma mansoni during its development in the hamster. Parasitol. Res. 1988, 75, 31–35. [Google Scholar] [CrossRef]
- Cheever, A.W. A quantitative post-mortem study of Schistosomiasis mansoni in man. Am. J. Trop. Med. Hyg. 1968, 17, 38–64. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.G. Relative distribution of Schistosoma japonicum eggs in the intestine of man: A subject of inconsistency. Acta Trop. 1991, 48, 163–171. [Google Scholar] [CrossRef]
- Gelfand, M.; Ross, W.F. I. The distribution of schistosome ova in the alimentary tract in subjects of bilharziasis. Trans. R. Soc. Trop. Med. Hyg. 1953, 47, 215–217. [Google Scholar] [CrossRef]
- Alves, W. The distribution of Schistosoma eggs in human tissues. Bull. World Health Organ. 1958, 18, 1092–1097. [Google Scholar] [PubMed]
- Lee, K.F.; Hsueh, S.; Tang, M.H. Schistosomiasis of the ovary with endometriosis and corpus hemorrhagicum: A case report. Chang. Gung Med. J. 2000, 23, 438–441. [Google Scholar] [PubMed]
- Carpenter, C.B.; Mozley, P.D.; Lewis, N.G. Schistosomiasis japonica involvement of the female genital tract. Jama 1964, 188, 647–650. [Google Scholar] [CrossRef]
- Yu, Z.; Wei, C.; Wang, Y.; Ye, Z.; Wang, Z.; Chen, Z.; Ni, L.; Yang, S.; Gui, Y.; Guan, Z.; et al. Prostatic Schistosoma japonicum with atypical immunophenotyping of individual glandular tubes: A case report and review of the literature. Southeast Asian J. Trop. Med. Public Health 2013, 44, 568–573. [Google Scholar]
- Wen, S.C.H.; Anderson, R.; Ryan, M.M.; Kumbla, S.; Wray, A.; Steer, A. Pediatric Neuroschistosomiasis: A Case Report and Review of the Literature. J. Pediatric Infect. Dis. Soc. 2019, 8, 489–491. [Google Scholar] [CrossRef]
- Dastoli, P.A.; Leite, A.L.; da Costa, M.D.S.; Nicácio, J.M.; Pinho, R.S.; Ferrarini, M.A.G.; Cavalheiro, S. Medullary neuroschistosomiasis in adolescence: Case report and literature review. Child’s Nerv. Syst. 2021, 37, 2735–2741. [Google Scholar] [CrossRef] [PubMed]
- Yao, Y.; Yu, C.X.; Song, L.J.; Yin, X.R.; Wang, J.; Jin, Y.; Shuan, S.; Zhang, W.; Gao, H.; Xu, Y.L.; et al. Cloning and function analysis of high mobility group box 1 (HMGB1) protein of Schistosoma japonicum (Mainland strain). Zhongguo Xue Xi Chong Bing Fang Zhi Za Zhi 2014, 26, 153–159. [Google Scholar] [PubMed]
- Gelfand, M.; Ross, W.F. II. The distribution of schistosome ova in the genito-urinary tract in subjects of bilharziasis. Trans. R. Soc. Trop. Med. Hyg. 1953, 47, 218–220. [Google Scholar] [CrossRef]
- Gelfand, M.; Ross, M.D.; Blair, D.M.; Weber, M.C. Distribution and extent of schistosomiasis in female pelvic organs, with special reference to the genital tract, as determined at autopsy. Am. J. Trop. Med. Hyg. 1971, 20, 846–849. [Google Scholar] [CrossRef] [PubMed]
- Gelfand, M.; Ross, C.M.; Blair, D.M.; Castle, W.M.; Weber, M.C. Schistosomiasis of the male pelvic organs. Severity of infection as determined by digestion of tissue and histologic methods in 300 cadavers. Am. J. Trop. Med. Hyg. 1970, 19, 779–784. [Google Scholar] [CrossRef]
- Andrade, Z.A. Schistosomiasis and liver fibrosis. Parasite Immunol. 2009, 31, 656–663. [Google Scholar] [CrossRef]
- Ashton, P.D.; Harrop, R.; Shah, B.; Wilson, R.A. The schistosome egg: Development and secretions. Parasitology 2001, 122, 329–338. [Google Scholar] [CrossRef]
- Fabre, V.; Wu, H.; PondTor, S.; Coutinho, H.; Acosta, L.; Jiz, M.; Olveda, R.; Cheng, L.; White, E.S.; Jarilla, B.; et al. Tissue inhibitor of matrix-metalloprotease-1 predicts risk of hepatic fibrosis in human Schistosoma japonicum infection. J. Infect. Dis. 2011, 203, 707–714. [Google Scholar] [CrossRef]
- Coutinho, H.M.; Acosta, L.P.; Wu, H.W.; McGarvey, S.T.; Su, L.; Langdon, G.C.; Jiz, M.A.; Jarilla, B.; Olveda, R.M.; Friedman, J.F.; et al. Th2 cytokines are associated with persistent hepatic fibrosis in human Schistosoma japonicum infection. J. Infect. Dis. 2007, 195, 288–295. [Google Scholar] [CrossRef]
- Carson, J.P.; Ramm, G.A.; Robinson, M.W.; McManus, D.P.; Gobert, G.N. Schistosome-Induced Fibrotic Disease: The Role of Hepatic Stellate Cells. Trends Parasitol. 2018, 34, 524–540. [Google Scholar] [CrossRef]
- Wilson, M.S.; Mentink-Kane, M.M.; Pesce, J.T.; Ramalingam, T.R.; Thompson, R.; Wynn, T.A. Immunopathology of schistosomiasis. Immunol. Cell Biol. 2007, 85, 148–154. [Google Scholar] [CrossRef] [PubMed]
- Brunet, L.R.; Finkelman, F.D.; Cheever, A.W.; Kopf, M.A.; Pearce, E.J. IL-4 protects against TNF-alpha-mediated cachexia and death during acute schistosomiasis. J. Immunol. 1997, 159, 777–785. [Google Scholar] [PubMed]
- Warren, K.S.; Domingo, E.O. Granuloma formation around Schistosoma mansoni, S. haematobium, and S. japonicum eggs. Size and rate of development, cellular composition, cross-sensitivity, and rate of egg destruction. Am. J. Trop. Med. Hyg. 1970, 19, 292–304. [Google Scholar] [CrossRef] [PubMed]
- Schramm, G.; Gronow, A.; Knobloch, J.; Wippersteg, V.; Grevelding, C.G.; Galle, J.; Fuller, H.; Stanley, R.G.; Chiodini, P.L.; Haas, H.; et al. IPSE/alpha-1: A major immunogenic component secreted from Schistosoma mansoni eggs. Mol. Biochem. Parasitol. 2006, 147, 9–19. [Google Scholar] [CrossRef]
- Schramm, G.; Falcone, F.H.; Gronow, A.; Haisch, K.; Mamat, U.; Doenhoff, M.J.; Oliveira, G.; Galle, J.; Dahinden, C.A.; Haas, H. Molecular characterization of an interleukin-4-inducing factor from Schistosoma mansoni eggs. J. Biol. Chem. 2003, 278, 18384–18392. [Google Scholar] [CrossRef]
- Chuah, C.; Jones, M.K.; Burke, M.L.; Owen, H.C.; Anthony, B.J.; McManus, D.P.; Ramm, G.A.; Gobert, G.N. Spatial and temporal transcriptomics of Schistosoma japonicum-induced hepatic granuloma formation reveals novel roles for neutrophils. J. Leukoc. Biol. 2013, 94, 353–365. [Google Scholar] [CrossRef]
- Washington, K.; Wright, K.; Shyr, Y.; Hunter, E.B.; Olson, S.; Raiford, D.S. Hepatic stellate cell activation in nonalcoholic steatohepatitis and fatty liver. Hum. Pathol. 2000, 31, 822–828. [Google Scholar] [CrossRef]
- Vera, M.; Nieto, N. Hepatic stellate cells and alcoholic liver disease. Rev. Esp. Enferm. Dig. 2006, 98, 674–684. [Google Scholar] [CrossRef][Green Version]
- Bartley, P.B.; Ramm, G.A.; Jones, M.K.; Ruddell, R.G.; Li, Y.; McManus, D.P. A contributory role for activated hepatic stellate cells in the dynamics of Schistosoma japonicum egg-induced fibrosis. Int. J. Parasitol. 2006, 36, 993–1001. [Google Scholar] [CrossRef]
- Xu, F.; Liu, C.; Zhou, D.; Zhang, L. TGF-beta/SMAD Pathway and Its Regulation in Hepatic Fibrosis. J. Histochem. Cytochem. 2016, 64, 157–167. [Google Scholar] [CrossRef]
- Alisi, A.; He, X.; Pu, G.; Tang, R.; Zhang, D.; Pan, W. Activation of Nuclear Factor Kappa B in the Hepatic Stellate Cells of Mice with Schistosomiasis Japonica. PLoS ONE 2014, 9, e104323. [Google Scholar] [CrossRef]
- Ragheb, S.; Boros, D.L. Characterization of granuloma T lymphocyte function from Schistosoma mansoni-infected mice. J. Immunol. 1989, 142, 3239–3246. [Google Scholar] [PubMed]
- Song, E.; Ouyang, N.; Hörbelt, M.; Antus, B.; Wang, M.; Exton, M.S. Influence of alternatively and classically activated macrophages on fibrogenic activities of human fibroblasts. Cell. Immunol. 2000, 204, 19–28. [Google Scholar] [CrossRef]
- Kelly, E.; Greene, C.M.; McElvaney, N.G. Targeting neutrophil elastase in cystic fibrosis. Expert Opin. Ther. Targets 2008, 12, 145–157. [Google Scholar] [CrossRef] [PubMed]
- Paudel, Y.N.; Angelopoulou, E.; Piperi, C.; Balasubramaniam, V.; Othman, I.; Shaikh, M.F. Enlightening the role of high mobility group box 1 (HMGB1) in inflammation: Updates on receptor signalling. Eur. J. Pharmacol. 2019, 858, 172487. [Google Scholar] [CrossRef]
- Lian, Y.J.; Gong, H.; Wu, T.Y.; Su, W.J.; Zhang, Y.; Yang, Y.Y.; Peng, W.; Zhang, T.; Zhou, J.R.; Jiang, C.L.; et al. Ds-HMGB1 and fr-HMGB induce depressive behavior through neuroinflammation in contrast to nonoxid-HMGB1. Brain Behav. Immun. 2017, 59, 322–332. [Google Scholar] [CrossRef]
- Yu, R.; Jiang, S.; Tao, Y.; Li, P.; Yin, J.; Zhou, Q. Inhibition of HMGB1 improves necrotizing enterocolitis by inhibiting NLRP3 via TLR4 and NF-kappaB signaling pathways. J. Cell. Physiol. 2019, 234, 13431–13438. [Google Scholar] [CrossRef]
- Uesugi, T.; Froh, M.; Arteel, G.E.; Bradford, B.U.; Thurman, R.G. Toll-like receptor 4 is involved in the mechanism of early alcohol-induced liver injury in mice. Hepatology 2001, 34, 101–108. [Google Scholar] [CrossRef]
- Isogawa, M.; Robek, M.D.; Furuichi, Y.; Chisari, F.V. Toll-like receptor signaling inhibits hepatitis B virus replication in vivo. J. Virol. 2005, 79, 7269–7272. [Google Scholar] [CrossRef]
- Sepehri, Z.; Kiani, Z.; Kohan, F.; Alavian, S.M.; Ghavami, S. Toll like receptor 4 and hepatocellular carcinoma; A systematic review. Life Sci. 2017, 179, 80–87. [Google Scholar] [CrossRef]
- Seki, E.; De Minicis, S.; Osterreicher, C.H.; Kluwe, J.; Osawa, Y.; Brenner, D.A.; Schwabe, R.F. TLR4 enhances TGF-beta signaling and hepatic fibrosis. Nat. Med. 2007, 13, 1324–1332. [Google Scholar] [CrossRef]
- Vallabhapurapu, S.; Karin, M. Regulation and function of NF-kappaB transcription factors in the immune system. Annu. Rev. Immunol. 2009, 27, 693–733. [Google Scholar] [CrossRef]
- Sang, R.; Yu, Y.; Ge, B.; Xu, L.; Wang, Z.; Zhang, X. Taraxasterol from Taraxacum prevents concanavalin A-induced acute hepatic injury in mice via modulating TLRs/NF-κB and Bax/Bc1-2 signalling pathways. Artif. Cells Nanomed. Biotechnol. 2019, 47, 3929–3937. [Google Scholar] [CrossRef]
- Nabih, E.S.; El-Kharashi, O.A. Targeting HMGB1/TLR4 axis and miR-21 by rosuvastatin: Role in alleviating cholestatic liver injury in a rat model of bile duct ligation. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2019, 392, 37–43. [Google Scholar] [CrossRef]
- Kisseleva, T. The origin of fibrogenic myofibroblasts in fibrotic liver. Hepatology 2017, 65, 1039–1043. [Google Scholar] [CrossRef]
- Nie, Y.; Liu, Q.; Zhang, W.; Wan, Y.; Huang, C.; Zhu, X. Ursolic acid reverses liver fibrosis by inhibiting NOX4/NLRP3 inflammasome pathways and bacterial dysbiosis. Gut Microbes 2021, 13, 1972746. [Google Scholar] [CrossRef]
- Wang, L.; Liao, Y.; Yang, R.; Yu, Z.; Zhang, L.; Zhu, Z.; Wu, X.; Shen, J.; Liu, J.; Xu, L.; et al. Sja-miR-71a in Schistosome egg-derived extracellular vesicles suppresses liver fibrosis caused by schistosomiasis via targeting semaphorin 4D. J. Extracell. Vesicles 2020, 9, 1785738. [Google Scholar] [CrossRef]
- Liu, Y.; Munker, S.; Müllenbach, R.; Weng, H.L. IL-13 Signaling in Liver Fibrogenesis. Front. Immunol. 2012, 3, 116. [Google Scholar] [CrossRef]
- Hernandez-Gea, V.; Friedman, S.L. Pathogenesis of liver fibrosis. Annu. Rev. Pathol. 2011, 6, 425–456. [Google Scholar] [CrossRef]
- Khambu, B.; Yan, S.; Huda, N.; Yin, X.M. Role of High-Mobility Group Box-1 in Liver Pathogenesis. Int. J. Mol. Sci. 2019, 20, 5314. [Google Scholar] [CrossRef]
- Li, L.C.; Gao, J.; Li, J. Emerging role of HMGB1 in fibrotic diseases. J. Cell. Mol. Med. 2014, 18, 2331–2339. [Google Scholar] [CrossRef] [PubMed]
- Kao, Y.H.; Lin, Y.C.; Tsai, M.S.; Sun, C.K.; Yuan, S.S.; Chang, C.Y.; Jawan, B.; Lee, P.H. Involvement of the nuclear high mobility group B1 peptides released from injured hepatocytes in murine hepatic fibrogenesis. Biochim. Biophys. Acta 2014, 1842, 1720–1732. [Google Scholar] [CrossRef] [PubMed]
- Albayrak, A.; Uyanik, M.H.; Cerrah, S.; Altas, S.; Dursun, H.; Demir, M.; Uslu, H. Is HMGB1 a new indirect marker for revealing fibrosis in chronic hepatitis and a new therapeutic target in treatment? Viral Immunol. 2010, 23, 633–638. [Google Scholar] [CrossRef] [PubMed]
- Bi, W.R.; Yang, C.Q.; Shi, Q. Transforming growth factor-β1 induced epithelial-mesenchymal transition in hepatic fibrosis. Hepato-Gastroenterology 2012, 59, 1960–1963. [Google Scholar] [CrossRef] [PubMed]
- Schnabl, B.; Kweon, Y.O.; Frederick, J.P.; Wang, X.F.; Rippe, R.A.; Brenner, D.A. The role of Smad3 in mediating mouse hepatic stellate cell activation. Hepatology 2001, 34, 89–100. [Google Scholar] [CrossRef]
- Itatani, Y.; Kawada, K.; Sakai, Y. Transforming Growth Factor-beta Signaling Pathway in Colorectal Cancer and Its Tumor Microenvironment. Int. J. Mol. Sci. 2019, 20, 5822. [Google Scholar] [CrossRef]
- Massagué, J. TGF-beta signal transduction. Annu. Rev. biochem. 1998, 67, 753–791. [Google Scholar] [CrossRef]
- Zhang, Y. MiR-92d-3p suppresses the progression of diabetic nephropathy renal fibrosis by inhibiting the C3/HMGB1/TGF-β1 pathway. Biosci. Rep. 2021, 41, BSR20203131. [Google Scholar] [CrossRef]
- Wang, W.; Zheng, F.; Zhang, A. Arsenic-induced lung inflammation and fibrosis in a rat model: Contribution of the HMGB1/RAGE, PI3K/AKT, and TGF-β1/SMAD pathways. Toxicol. Appl. Pharmacol. 2021, 432, 115757. [Google Scholar] [CrossRef]
- Arriazu, E.; Ge, X.; Leung, T.M.; Magdaleno, F.; Lopategi, A.; Lu, Y.; Kitamura, N.; Urtasun, R.; Theise, N.; Antoine, D.J.; et al. Signalling via the osteopontin and high mobility group box-1 axis drives the fibrogenic response to liver injury. Gut 2017, 66, 1123–1137. [Google Scholar] [CrossRef]
- Ge, X.; Arriazu, E.; Magdaleno, F.; Antoine, D.J.; Dela Cruz, R.; Theise, N.; Nieto, N. High Mobility Group Box-1 Drives Fibrosis Progression Signaling via the Receptor for Advanced Glycation End Products in Mice. Hepatology 2018, 68, 2380–2404. [Google Scholar] [CrossRef] [PubMed]
- Wyganowska-Swiatkowska, M.; Nohawica, M.; Grocholewicz, K.; Nowak, G. Influence of Herbal Medicines on HMGB1 Release, SARS-CoV-2 Viral Attachment, Acute Respiratory Failure, and Sepsis. A Literature Review. Int. J. Mol. Sci. 2020, 21, 4639. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.; Alfajaro, M.M.; DeWeirdt, P.C.; Hanna, R.E.; Lu-Culligan, W.J.; Cai, W.L.; Strine, M.S.; Zhang, S.M.; Graziano, V.R.; Schmitz, C.O.; et al. Genome-wide CRISPR Screens Reveal Host Factors Critical for SARS-CoV-2 Infection. Cell 2021, 184, 76–91.e13. [Google Scholar] [CrossRef]
- Chen, X.; Ling, Y.; Wei, Y.; Tang, J.; Ren, Y.; Zhang, B.; Jiang, F.; Li, H.; Wang, R.; Wen, W.; et al. Dual regulation of HMGB1 by combined JNK1/2-ATF2 axis with miR-200 family in nonalcoholic steatohepatitis in mice. FASEB J. 2018, 32, 2722–2734. [Google Scholar] [CrossRef] [PubMed]
- Entezari, M.; Weiss, D.J.; Sitapara, R.; Whittaker, L.; Wargo, M.J.; Li, J.; Wang, H.; Yang, H.; Sharma, L.; Phan, B.D.; et al. Inhibition of high-mobility group box 1 protein (HMGB1) enhances bacterial clearance and protects against Pseudomonas Aeruginosa pneumonia in cystic fibrosis. Mol. Med. 2012, 18, 477–485. [Google Scholar] [CrossRef]
- Iino, S.; Tango, T.; Matsushima, T.; Toda, G.; Miyake, K.; Hino, K.; Kumada, H.; Yasuda, K.; Kuroki, T.; Hirayama, C.; et al. Therapeutic effects of stronger neo-minophagen C at different doses on chronic hepatitis and liver cirrhosis. Hepatol. Res. 2001, 19, 31–40. [Google Scholar] [CrossRef]
- Ogiku, M.; Kono, H.; Hara, M.; Tsuchiya, M.; Fujii, H. Glycyrrhizin prevents liver injury by inhibition of high-mobility group box 1 production by Kupffer cells after ischemia-reperfusion in rats. J. Pharmacol. Exp. Ther. 2011, 339, 93–98. [Google Scholar] [CrossRef]
- Gwak, G.Y.; Moon, T.G.; Lee, D.H.; Yoo, B.C. Glycyrrhizin attenuates HMGB1-induced hepatocyte apoptosis by inhibiting the p38-dependent mitochondrial pathway. World J. Gastroenterol. 2012, 18, 679–684. [Google Scholar] [CrossRef]
- Gowda, P.; Patrick, S.; Joshi, S.D.; Kumawat, R.K.; Sen, E. Glycyrrhizin prevents SARS-CoV-2 S1 and Orf3a induced high mobility group box 1 (HMGB1) release and inhibits viral replication. Cytokine 2021, 142, 155496. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).