
Immune Responses to IAV Infection and the Roles of L-Selectin and ADAM17 in Lymphocyte Homing


Abstract
1. Influenza Virus
2. Tissue Tropism
3. Mouse Models for Influenza Virus Research
4. Innate Immune Responses to IAV Infection and Immunopathology
5. Adaptive Immune Responses to IAV Infection
5.1. Roles of the Draining Lymph Nodes
5.2. Roles of Lymphocytes
6. The Role of Lymphocyte Homing in Adaptive Immune Responses to IAV
6.1. The Multistep Adhesion Cascade of Leukocyte Homing
6.2. T Lymphocyte Homing to Lymph Nodes and Lungs during IAV Infection
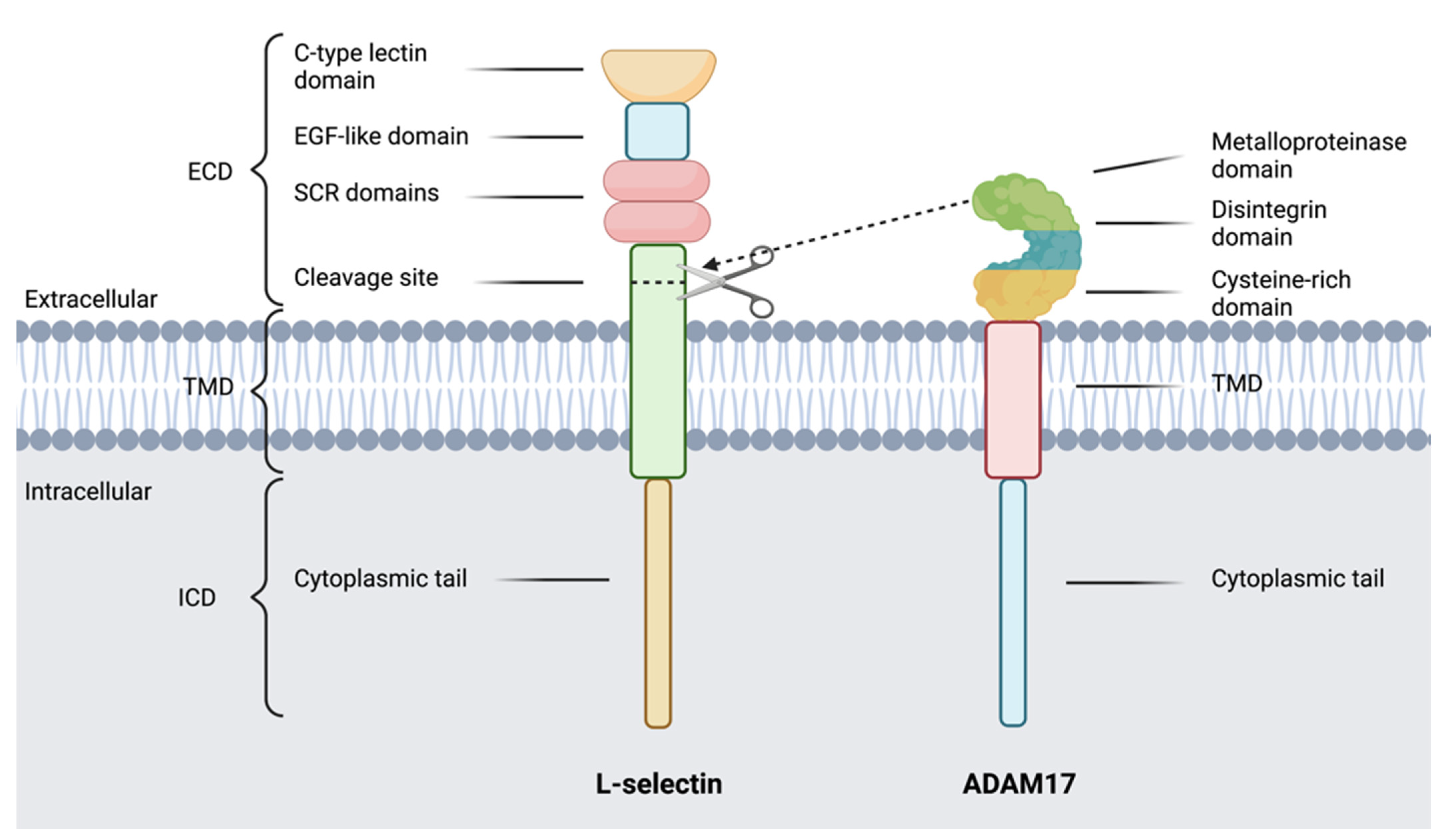
7. L-Selectin
7.1. L-Selectin Ligands and Roles in Lymphocyte Homing
7.2. L-Selectin in IAV Infection
8. ADAMs
8.1. ADAM17 in Immunity
8.1.1. ADAM17 Regulation of L-Selectin Expression by T cells
8.1.2. ADAM17 in Infection
8.1.3. Pharmaceutical Agents to Block ADAM17
9. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Influenza (Seasonal). Available online: https://www.who.int/news-room/fact-sheets/detail/influenza-(seasonal) (accessed on 29 September 2020).
- Mohammed, R.N.; Watson, H.A.; Vigar, M.; Ohme, J.; Thomson, A.; Humphreys, I.R.; Ager, A. L-selectin Is Essential for Delivery of Activated CD8+ T Cells to Virus-Infected Organs for Protective Immunity. Cell Rep. 2016, 14, 760–771. [Google Scholar] [CrossRef] [PubMed]
- Stevens, J.; Blixt, O.; Glaser, L.; Taubenberger, J.K.; Palese, P.; Paulson, J.C.; Wilson, I.A. Glycan Microarray Analysis of the Hemagglutinins from Modern and Pandemic Influenza Viruses Reveals Different Receptor Specificities. J. Mol. Biol. 2006, 355, 1143–1155. [Google Scholar] [CrossRef] [PubMed]
- Van Riel, D.; Munster, V.J.; De Wit, E.; Rimmelzwaan, G.F.; Fouchier, R.A.M.; Osterhaus, A.D.M.E.; Kuiken, T. Human and Avian Influenza Viruses Target Different Cells in the Lower Respiratory Tract of Humans and Other Mammals. Am. J. Pathol. 2007, 171, 1215–1223. [Google Scholar] [CrossRef] [PubMed]
- Weinheimer, V.K.; Becher, A.; Tönnies, M.; Holland, G.; Knepper, J.; Bauer, T.T.; Schneider, P.; Neudecker, J.; Rückert, J.C.; Szymanski, K.; et al. Influenza A Viruses Target Type II Pneumocytes in the Human Lung. J. Infect. Dis. 2012, 206, 1685–1694. [Google Scholar] [CrossRef] [PubMed]
- Travanty, E.; Zhou, B.; Zhang, H.; Di, Y.P.; Alcorn, J.F.; Wentworth, D.E.; Mason, R.; Wang, J. Differential Susceptibilities of Human Lung Primary Cells to H1N1 Influenza Viruses. J. Virol. 2015, 89, 11935. [Google Scholar] [CrossRef] [PubMed]
- Ettensohn, D.B.; Frampton, M.W.; Nichols, J.E.; Roberts, N.J. Human Alveolar Macrophages May Not Be Susceptible to Direct Infection by a Human Influenza Virus. J. Infect. Dis. 2016, 214, 1658–1665. [Google Scholar] [CrossRef] [PubMed]
- Londrigan, S.L.; Short, K.R.; Ma, J.; Gillespie, L.; Rockman, S.P.; Brooks, A.G.; Reading, P.C. Infection of Mouse Macrophages by Seasonal Influenza Viruses Can Be Restricted at the Level of Virus Entry and at a Late Stage in the Virus Life Cycle. J. Virol. 2015, 89, 12319–12329. [Google Scholar] [CrossRef]
- Hou, W.; Gibbs, J.S.; Lu, X.; Brooke, C.B.; Roy, D.; Modlin, R.L.; Bennink, J.R.; Yewdell, J.W. Viral infection triggers rapid differentiation of human blood monocytes into dendritic cells. Blood 2012, 119, 3128–3131. [Google Scholar] [CrossRef]
- Österlund, P.; Pirhonen, J.; Ikonen, N.; Rönkkö, E.; Strengell, M.; Mäkelä, S.M.; Broman, M.; Hamming, O.J.; Hartmann, R.; Ziegler, T.; et al. Pandemic H1N1 2009 Influenza A Virus Induces Weak Cytokine Responses in Human Macrophages and Dendritic Cells and Is Highly Sensitive to the Antiviral Actions of Interferons. J. Virol. 2010, 84, 1414–1422. [Google Scholar] [CrossRef]
- Zhao, Y.; Lu, M.; Lau, L.T.; Lu, J.; Gao, Z.; Liu, J.; Yu, A.C.H.; Cao, Q.; Ye, J.; McNutt, M.A.; et al. Neutrophils may be a vehicle for viral replication and dissemination in human h5n1 avian influenza. Clin. Infect. Dis. 2008, 47, 1575–1578. [Google Scholar] [CrossRef]
- Mao, H.; Tu, W.; Qin, G.; Law, H.K.W.; Sia, S.F.; Chan, P.-L.; Liu, Y.; Lam, K.-T.; Zheng, J.; Peiris, M.; et al. Influenza Virus Directly Infects Human Natural Killer Cells and Induces Cell Apoptosis. J. Virol. 2009, 83, 9215–9222. [Google Scholar] [CrossRef] [PubMed]
- Dougan, S.K.; Ashour, J.; Karssemeijer, R.A.; Popp, M.W.; Avalos, A.M.; Barisa, M.; Altenburg, A.F.; Ingram, J.R.; Cragnolini, J.J.; Guo, C.; et al. Antigen-specific B-cell receptor sensitizes B cells to infection by influenza virus. Nature 2013, 503, 406–409. [Google Scholar] [CrossRef]
- Hao, X.; Kim, T.S.; Braciale, T.J. Differential Response of Respiratory Dendritic Cell Subsets to Influenza Virus Infection. J. Virol. 2008, 82, 4908–4919. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Kumar, P.; Moran, T.M.; Garcia-Sastre, A.; Zhou, Y.; Malarkannan, S. The functional impairment of natural killer cells during influenza virus infection. Immunol. Cell Biol. 2009, 87, 579–589. [Google Scholar] [CrossRef] [PubMed]
- Samarasinghe, A.E.; Melo, R.C.N.; Duan, S.; LeMessurier, K.S.; Liedmann, S.; Surman, S.L.; Lee, J.J.; Hurwitz, J.L.; Thomas, P.G.; McCullers, J.A. Eosinophils Promote Antiviral Immunity in Mice Infected with Influenza A Virus. J. Immunol. 2017, 198, 3214–3226. [Google Scholar] [CrossRef] [PubMed]
- Nguyen-Van-Tam, J.S. 2009 Pandemic Influenza A (H1N1): Pathology and Pathogenesis of 100 Fatal Cases in the United States. Am. J. Pathol. 2010, 177, 166–175. [Google Scholar] [CrossRef]
- De Jong, M.D.; Simmons, C.P.; Thanh, T.T.; Hien, V.M.; Smith, G.J.D.; Chau, T.N.B.; Hoang, D.M.; Van Vinh Chau, N.; Khanh, T.H.; Dong, V.C.; et al. Fatal outcome of human influenza A (H5N1) is associated with high viral load and hypercytokinemia. Nat. Med. 2006, 12, 1203–1207. [Google Scholar] [CrossRef]
- Govorkova, E.A.; Rehg, J.E.; Krauss, S.; Yen, H.-L.; Guan, Y.; Peiris, M.; Nguyen, T.D.; Hanh, T.H.; Puthavathana, P.; Long, H.T.; et al. Lethality to Ferrets of H5N1 Influenza Viruses Isolated from Humans and Poultry in 2004. J. Virol. 2005, 79, 2191–2198. [Google Scholar] [CrossRef]
- Sellers, S.A.; Hagan, R.S.; Hayden, F.G.; Fischer, W.A. The hidden burden of influenza: A review of the extra-pulmonary complications of influenza infection. Influenza Other Respi. Viruses 2017, 11, 372–393. [Google Scholar] [CrossRef]
- Shope, R.E. The infection of mice with swine influenza virus. J. Exp. Med. 1935, 62, 561–572. [Google Scholar] [CrossRef]
- Tumpey, T.M.; García-Sastre, A.; Taubenberger, J.K.; Palese, P.; Swayne, D.E.; Pantin-Jackwood, M.J.; Schultz-Cherry, S.; Solórzano, A.; Van Rooijen, N.; Katz, J.M.; et al. Pathogenicity of Influenza Viruses with Genes from the 1918 Pandemic Virus: Functional Roles of Alveolar Macrophages and Neutrophils in Limiting Virus Replication and Mortality in Mice. J. Virol. 2005, 79, 14933–14944. [Google Scholar] [CrossRef] [PubMed]
- Bouvier, N.M.; Lowen, A.C. Animal Models for Influenza Virus Pathogenesis and Transmission. Viruses 2010, 2, 1530–1563. [Google Scholar] [CrossRef] [PubMed]
- Ibricevic, A.; Pekosz, A.; Walter, M.; Newby, C.; Battaile, J.; Brown, E.; Holtzman, M.; Brody, S. Influenza virus receptor specificity and cell tropism in mouse and human airway epithelial cells. J. Virol. 2006, 80, 7469–7480. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, B.; Błażejewska, P.; Heßmann, M.; Bruder, D.; Geffers, R.; Mauel, S.; Gruber, A.D.; Schughart, K. Host Genetic Background Strongly Influences the Response to Influenza A Virus Infections. PLoS ONE 2009, 4, e4857. [Google Scholar] [CrossRef]
- Rosshart, S.P.; Herz, J.; Vassallo, B.G.; Hunter, A.; Wall, M.K.; Badger, J.H.; McCulloch, J.A.; Anastasakis, D.G.; Sarshad, A.A.; Leonardi, I.; et al. Laboratory mice born to wild mice have natural microbiota and model human immune responses. Science 2019, 365, 6452. [Google Scholar] [CrossRef]
- Fiege, J.K.; Block, K.E.; Pierson, M.J.; Nanda, H.; Shepherd, F.K.; Mickelson, C.K.; Stolley, J.M.; Matchett, W.E.; Wijeyesinghe, S.; Meyerholz, D.K.; et al. Mice with diverse microbial exposure histories as a model for preclinical vaccine testing. Cell Host Microbe 2021, 29, 1815–1827.e6. [Google Scholar] [CrossRef]
- Kollmus, H.; Pilzner, C.; Leist, S.R.; Heise, M.; Geffers, R.; Schughart, K. Of mice and men: The host response to influenza virus infection. Mamm. Genome 2018, 29, 446. [Google Scholar] [CrossRef]
- Davies, W.L.; Grunert, R.R.; Haff, R.F.; McGahen, J.W.; Neumayer, E.M.; Paulshock, M.; Watts, J.C.; Wood, T.R.; Hermann, E.C.; Hoffmann, C.E. Antiviral Activity of 1-Adamantanamine (Amantadine). Science 1964, 144, 862–863. [Google Scholar] [CrossRef]
- Rabinovich, S. Rimantadine therapy of influenza A infection in mice. Antimicrob. Agents Chemother. 1972, 1, 408–411. [Google Scholar] [CrossRef]
- Mendel, D.B.; Tai, C.Y.; Escarpe, P.A.; Li, W.; Sidwell, R.W.; Huffman, J.H.; Sweet, C.; Jakeman, K.J.; Merson, J.; Lacy, S.A.; et al. Oral administration of a prodrug of the influenza virus neuraminidase inhibitor GS 4071 protects mice and ferrets against influenza infection. Antimicrob. Agents Chemother. 1998, 42, 640–646. [Google Scholar] [CrossRef] [PubMed]
- Von Itzstein, M.; Wu, W.-Y.; Kok, G.B.; Pegg, M.S.; Dyason, J.C.; Jin, B.; Van Phan, T.; Smythe, M.L.; White, H.F.; Oliver, S.W.; et al. Rational design of potent sialidase-based inhibitors of influenza virus replication. Nature 1993, 363, 418–423. [Google Scholar] [CrossRef] [PubMed]
- Sidwell, R.W.; Smee, D.F.; Huffman, J.H.; Barnard, D.L.; Bailey, K.W.; Morrey, J.D.; Babu, Y.S. In vivo influenza virus-inhibitory effects of the cyclopentane neuraminidase inhibitor RWJ-270201. Antimicrob. Agents Chemother. 2001, 45, 749–757. [Google Scholar] [CrossRef] [PubMed]
- Zanin, M.; Baviskar, P.; Webster, R.; Webby, R. The Interaction between Respiratory Pathogens and Mucus. Cell Host Microbe 2016, 19, 159–168. [Google Scholar] [CrossRef]
- Dou, D.; Revol, R.; Östbye, H.; Wang, H.; Daniels, R. Influenza A virus cell entry, replication, virion assembly and movement. Front. Immunol. 2018, 9, 1581. [Google Scholar] [CrossRef]
- Lin, K.L.; Suzuki, Y.; Nakano, H.; Ramsburg, E.; Gunn, M.D. CCR2+ Monocyte-Derived Dendritic Cells and Exudate Macrophages Produce Influenza-Induced Pulmonary Immune Pathology and Mortality. J. Immunol. 2008, 180, 2562–2572. [Google Scholar] [CrossRef] [PubMed]
- Sprenger, H.; Meyer, R.G.; Kaufmann, A.; Bußfeld, D.; Rischkowsky, E.; Gemsa, D. Selective induction of monocyte and not neutrophil-attracting chemokines after influenza A virus infection. J. Exp. Med. 1996, 184, 1191–1196. [Google Scholar] [CrossRef]
- Lin, S.-J.; Lo, M.; Kuo, R.-L.; Shih, S.-R.; Ojcius, D.M.; Lu, J.; Lee, C.-K.; Chen, H.-C.; Lin, M.Y.; Leu, C.-M.; et al. The pathological effects of CCR2+ inflammatory monocytes are amplified by an IFNAR1-triggered chemokine feedback loop in highly pathogenic influenza infection. J. Biomed. Sci. 2014, 21, 99. [Google Scholar] [CrossRef] [PubMed]
- Nakanishi Yumi Hashimoto, Y.; Moki, T.; Takizawa, T. Neutrophils and Macrophages in Mice Virus-Infected, Apoptotic Cells by Evidence for Phagocytosis of Influenza. J. Immunol. Ref. 2007, 178, 2448–2457. [Google Scholar] [CrossRef]
- Cardani, A.; Boulton, A.; Kim, T.S.; Braciale, T.J. Alveolar Macrophages Prevent Lethal Influenza Pneumonia By Inhibiting Infection Of Type-1 Alveolar Epithelial Cells. PLoS Pathog. 2017, 13, e1006140. [Google Scholar] [CrossRef]
- Snelgrove, R.J.; Goulding, J.; Didierlaurent, A.M.; Lyonga, D.; Vekaria, S.; Edwards, L.; Gwyer, E.; Sedgwick, J.D.; Barclay, A.N.; Hussell, T. A critical function for CD200 in lung immune homeostasis and the severity of influenza infection. Nat. Immunol. 2008, 9, 1074–1083. [Google Scholar] [CrossRef]
- Cella, M.; Jarrossay, D.; Faccheth, F.; Alebardi, O.; Nakajima, H.; Lanzavecchia, A.; Colonna, M. Plasmacytoid monocytes migrate to inflamed lymph nodes and produce large amounts of type I interferon. Nat. Med. 1999, 5, 919–923. [Google Scholar] [CrossRef] [PubMed]
- Aldridge, J.R.; Moseley, C.E.; Boltz, D.A.; Negovetich, N.J.; Reynolds, C.; Franks, J.; Brown, S.A.; Doherty, P.C.; Webster, R.G.; Thomas, P.G. TNF/iNOS-producing dendritic cells are the necessary evil of lethal influenza virus infection. Proc. Natl. Acad. Sci. USA 2009, 106, 5306–5311. [Google Scholar] [CrossRef] [PubMed]
- Tate, M.; Brooks, A.; Reading, P. The role of neutrophils in the upper and lower respiratory tract during influenza virus infection of mice. Respir. Res. 2008, 9, 57. [Google Scholar] [CrossRef]
- Tripathi, S.; Verma, A.; Kim, E.-J.; White, M.R.; Hartshorn, K.L. LL-37 modulates human neutrophil responses to influenza A virus. J. Leukoc. Biol. 2014, 96, 931–938. [Google Scholar] [CrossRef]
- Lim, K.; Hyun, Y.M.; Lambert-Emo, K.; Capece, T.; Bae, S.; Miller, R.; Topham, D.J.; Kim, M. Neutrophil trails guide influenzaspecific CD8+ T cells in the airways. Science 2015, 349, aaa4352. [Google Scholar] [CrossRef] [PubMed]
- Akaike, T.; Ando, M.; Oda, T.; Doi, T.; Ijiri, S.; Araki, S.; Maeda, H. Dependence on O2− generation by xanthine oxidase of pathogenesis of influenza virus infection in mice. J. Clin. Investig. 1990, 85, 739–745. [Google Scholar] [CrossRef]
- Brandes, M.; Klauschen, F.; Kuchen, S.; Germain, R.N. A Systems Analysis Identifies a Feedforward Inflammatory Circuit Leading to Lethal Influenza Infection. Cell 2013, 154, 197–212. [Google Scholar] [CrossRef]
- Leung, K.N.; Leung, G.L. Induction of natural killer cells during murine influenza virus infection. Immunobiology 1981, 160, 352–366. [Google Scholar] [CrossRef]
- Carlin, L.E.; Hemann, E.A.; Zacharias, Z.R.; Heusel, J.W.; Legge, K.L. Natural killer cell recruitment to the lung during influenza A virus infection is dependent on CXCR3, CCR5, and virus exposure dose. Front. Immunol. 2018, 9, 781. [Google Scholar] [CrossRef]
- Monteiro, J.M.; Harvey, C.; Trinchieri, G. Role of Interleukin-12 in Primary Influenza Virus Infection. J. Virol. 1998, 72, 4825–4831. [Google Scholar] [CrossRef]
- Ge, M.Q.; Ho, A.W.S.; Tang, Y.; Wong, K.H.S.; Chua, B.Y.L.; Gasser, S.; Kemeny, D.M. NK Cells Regulate CD8+ T Cell Priming and Dendritic Cell Migration during Influenza A Infection by IFN-γ and Perforin-Dependent Mechanisms. J. Immunol. 2012, 189, 2099–2109. [Google Scholar] [CrossRef] [PubMed]
- Kos, F.J.; Engleman, E.G. Role of Natural Killer Cells in the Generation of Influenza Virus-Specific Cytotoxic T Cells. Cell. Immunol. 1996, 173, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Abdul-Careem, M.F.; Mian, M.F.; Yue, G.; Gillgrass, A.; Chenoweth, M.J.; Barra, N.G.; Chew, M.V.; Chan, T.; Al-Garawi, A.A.; Jordana, M.; et al. Critical Role of Natural Killer Cells in Lung Immunopathology During Influenza Infection in Mice. J. Infect. Dis. 2012, 206, 167–177. [Google Scholar] [CrossRef] [PubMed]
- Coro, E.S.; Chang, W.L.W.; Baumgarth, N. Type I IFN Receptor Signals Directly Stimulate Local B Cells Early following Influenza Virus Infection. J. Immunol. 2006, 176, 4343–4351. [Google Scholar] [CrossRef] [PubMed]
- Förster, R.; Schubel, A.; Breitfeld, D.; Kremmer, E.; Renner-Müller, I.; Wolf, E.; Lipp, M. CCR7 Coordinates the Primary Immune Response by Establishing Functional Microenvironments in Secondary Lymphoid Organs. Cell 1999, 99, 23–33. [Google Scholar] [CrossRef]
- Luther, S.A.; Tang, H.L.; Hyman, P.L.; Farr, A.G.; Cyster, J.G. Coexpression of the chemokines ELC and SLC by T zone stromal cells and deletion of the ELC gene in the plt/plt mouse. Proc. Natl. Acad. Sci. USA 2000, 97, 12694–12699. [Google Scholar] [CrossRef]
- Mobley, J.L.; Dailey, M.O. Regulation of adhesion molecule expression by CD8 T cells in vivo. I. Differential regulation of gp90MEL-14 (LECAM-1), Pgp-1, LFA-1, and VLA-4 alpha during the differentiation of cytotoxic T lymphocytes induced by allografts. J. Immunol. 1992, 148, 2348–2356. [Google Scholar]
- Benechet, A.P.; Menon, M.; Xu, D.; Samji, T.; Maher, L.; Murooka, T.T.; Mempel, T.R.; Sheridan, B.S.; Lemoine, F.M.; Khanna, K.M. T cell-intrinsic S1PR1 regulates endogenous effector T-cell egress dynamics from lymph nodes during infection. Proc. Natl. Acad. Sci. USA 2016, 113, 2182–2187. [Google Scholar] [CrossRef]
- Bromley, S.K.; Mempel, T.R.; Luster, A.D. Orchestrating the orchestrators: Chemokines in control of T cell traffic. Nat. Immunol. 2008, 9, 970–980. [Google Scholar] [CrossRef]
- Topham, D.J.; Tripp, R.A.; Doherty, P.C. CD8+ T cells Clear Influenza Virus by Perforin or Fas-dependent processes. J. Immunol. 1997, 159, 5197–5200. [Google Scholar]
- Bender, B.S.; Croghan, T.; Zhang, L.; Small, P.A. Transgenic mice lacking class I major histocompatibility complex-restricted T cells have delayed viral clearance and increased mortality after influenza virus challenge. J. Exp. Med. 1992, 175, 1143–1145. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Madan, R.; Karp, C.L.; Braciale, T.J. Effector T cells control lung inflammation during acute influenza virus infection by producing IL-10. Nat. Med. 2009, 15, 277–284. [Google Scholar] [CrossRef] [PubMed]
- Moskophidis, D.; Kioussis, D. Contribution of Virus-specific CD8+ Cytotoxic T Cells to Virus Clearance or Pathologic Manifestations of Influenza Virus Infection in a T Cell Receptor Transgenic Mouse Model. J. Exp. Med. 1998, 188, 223. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Haist, V.; Baumgärtner, W.; Schughart, K. Sustained viral load and late death in Rag2−/− mice after influenza A virus infection. Virol. J. 2010, 7, 172. [Google Scholar] [CrossRef] [PubMed]
- Szabo, S.J.; Kim, S.T.; Costa, G.L.; Zhang, X.; Fathman, C.G.; Glimcher, L.H. A Novel Transcription Factor, T-bet, Directs Th1 Lineage Commitment. Cell 2000, 100, 655–669. [Google Scholar] [CrossRef]
- Kumamoto, Y.; Mattei, L.M.; Sellers, S.; Payne, G.W.; Iwasaki, A. CD4+ T cells support cytotoxic T lymphocyte priming by controlling lymph node input. Proc. Natl. Acad. Sci. USA 2011, 108, 8749–8754. [Google Scholar] [CrossRef] [PubMed]
- Moser, B. CXCR5, the Defining Marker for Follicular B Helper T (TFH) Cells. Front. Immunol. 2015, 6, 296. [Google Scholar] [CrossRef] [PubMed]
- Hornick, E.E.; Zacharias, Z.R.; Legge, K.L. Kinetics and Phenotype of the CD4 T Cell Response to Influenza Virus Infections. Front. Immunol. 2019, 10, 2351. [Google Scholar] [CrossRef]
- Antunes, I.; Kassiotis, G. Suppression of Innate Immune Pathology by Regulatory T Cells during Influenza A Virus Infection of Immunodeficient Mice. J. Virol. 2010, 84, 12564–12575. [Google Scholar] [CrossRef]
- Rangel-Moreno, J.; Carragher, D.M.; Misra, R.S.; Kusser, K.; Hartson, L.; Moquin, A.; Lund, F.E.; Randall, T.D. B Cells Promote Resistance to Heterosubtypic Strains of Influenza via Multiple Mechanisms. J. Immunol. 2008, 180, 454–463. [Google Scholar] [CrossRef]
- Padilla-Quirarte, H.O.; Lopez-Guerrero, D.V.; Gutierrez-Xicotencatl, L.; Esquivel-Guadarrama, F. Protective Antibodies Against Influenza Proteins. Front. Immunol. 2019, 10, 1677. [Google Scholar] [CrossRef] [PubMed]
- Huang, K.-Y.A.; Li, C.K.-F.; Clutterbuck, E.; Chui, C.; Wilkinson, T.; Gilbert, A.; Oxford, J.; Lambkin-Williams, R.; Lin, T.-Y.; McMichael, A.J.; et al. Virus-Specific Antibody Secreting Cell, Memory B-cell, and Sero-Antibody Responses in the Human Influenza Challenge Model. J. Infect. Dis. 2014, 209, 1354–1361. [Google Scholar] [CrossRef] [PubMed]
- Lam, J.H.; Baumgarth, N. The Multifaceted B Cell Response to Influenza Virus. J. Immunol. 2019, 202, 351. [Google Scholar] [CrossRef] [PubMed]
- Springer, T.A. Traffic signals for lymphocyte recirculation and leukocyte emigration: The multistep paradigm. Cell 1994, 76, 301–314. [Google Scholar] [CrossRef]
- Ager, A. ADAMs and Ectododomain Proteolytic Shedding in Leukocyte Migration: Focus on L-Selectin and ADAM17. Curr. Immunol. Rev. 2012, 8, 103–117. [Google Scholar] [CrossRef]
- Ley, K.; Laudanna, C.; Cybulsky, M.I.; Nourshargh, S. Getting to the site of inflammation: The leukocyte adhesion cascade updated. Nat. Rev. Immunol. 2007, 7, 678–689. [Google Scholar] [CrossRef]
- Moss, M.L.; Minond, D. Review Article Recent Advances in ADAM17 Research: A Promising Target for Cancer and Inflammation. Mediat. Inflamm. 2017, 2017, 9673537. [Google Scholar] [CrossRef]
- Faveeuw, C.; Preece, G.; Ager, A. Transendothelial migration of lymphocytes across high endothelial venules into lymph nodes is affected by metalloproteinases. Blood 2001, 98, 688–695. [Google Scholar] [CrossRef]
- Hafezi-Moghadam, A.; Thomas, K.L.; Prorock, A.J.; Huo, Y.; Ley, K. L-Selectin Shedding Regulates Leukocyte Recruitment. J. Exp. Med. 2001, 193, 863–872. [Google Scholar] [CrossRef]
- Rzeniewicz, K.; Newe, A.; Gallardo, A.R.; Daviesa, J.; Holt, M.R.; Patel, A.; Charras, G.T.; Stramer, B.; Molenaar, C.; Tedder, T.F.; et al. L-selectin shedding is activated specifically within transmigrating pseudopods of monocytes to regulate cell polarity in vitro. Proc. Natl. Acad. Sci. USA 2015, 112, E1461–E1470. [Google Scholar] [CrossRef]
- Gallatin, W.M.; Weissman, I.L.; Butcher, E.C. Pillars Article: A Cell-Surface Molecule Involved in Organ-Specific Homing of Lymphocytes. Nature 1983, 304, 30–34. [Google Scholar] [CrossRef]
- Streeter, P.R.; Rouse, B.T.; Butcher, E.C. Immunohistologic and functional characterization of a vascular addressin involved in lymphocyte homing into peripheral lymph nodes. J. Cell Biol. 1988, 107, 1853–1862. [Google Scholar] [CrossRef] [PubMed]
- Baekkevold, E.S.; Yamanaka, T.; Palframan, R.T.; Carlsen, H.S.; Reinholt, F.P.; von Andrian, U.H.; Brandtzaeg, P.; Haraldsen, G. The Ccr7 Ligand ELC (Ccl19) Is Transcytosed in High Endothelial Venules and Mediates T Cell Recruitment. J. Exp. Med. 2001, 193, 1105–1112. [Google Scholar] [CrossRef] [PubMed]
- Stein, J.V.; Rot, A.; Luo, Y.; Narasimhaswamy, M.; Nakano, H.; Gunn, M.D.; Matsuzawa, A.; Quackenbush, E.J.; Dorf, M.E.; von Andrian, U.H. The Cc Chemokine Thymus-Derived Chemotactic Agent 4 (Tca-4, Secondary Lymphoid Tissue Chemokine, 6ckine, Exodus-2) Triggers Lymphocyte Function–Associated Antigen 1–Mediated Arrest of Rolling T Lymphocytes in Peripheral Lymph Node High Endothelial Venules. J. Exp. Med. 2000, 191, 61–76. [Google Scholar] [CrossRef] [PubMed]
- Warnock, R.A.; Askari, S.; Butcher, E.C.; Andrian, U.H. von Molecular Mechanisms of Lymphocyte Homing to Peripheral Lymph Nodes. J. Exp. Med. 1998, 187, 205. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, J.C.U.; Jablonski-Westrich, D.; Haubold, U.; Gutierrez-Ramos, J.-C.; Springer, T.; Hamann, A. Overlapping and Selective Roles of Endothelial Intercellular Adhesion Molecule-1 (ICAM-1) and ICAM-2 in Lymphocyte Trafficking. J. Immunol. 2003, 171, 2588–2593. [Google Scholar] [CrossRef]
- Berg, E.L.; McEvoy, L.M.; Berlin, C.; Bargatze, R.F.; Butcher, E.C. L-selectin-mediated lymphocyte rolling on MAdCAM-1. Nature 1993, 366, 695–698. [Google Scholar] [CrossRef]
- Gunn, M.D.; Tangemann, K.; Tam, C.; Cyster, J.G.; Rosen, S.D.; Williams, L.T. A chemokine expressed in lymphoid high endothelial venules promotes the adhesion and chemotaxis of naive T lymphocytes. Immunology 1998, 95, 258–263. [Google Scholar] [CrossRef]
- Wagner, N.; Löhler, J.; Tedder, T.F.; Rajewsky, K.; Müller, W.; Steeber, D.A. L-selectin and I 7 integrin synergistically mediate lymphocyte migration to mesenteric lymph nodes. Eur. J. Immunol. 1998, 28, 3832–3839. [Google Scholar] [CrossRef]
- Berlin-Rufenach, C.; Otto, F.; Mathies, M.; Westermann, J.; Owen, M.J.; Hamann, A.; Hogg, N. Lymphocyte Migration in Lymphocyte Function-associated Antigen (LFA)-1–deficient Mice. J. Exp. Med. 1999, 189, 1478. [Google Scholar] [CrossRef]
- Kunkel, E.J.; Ramos, C.L.; Steeber, D.A.; Müller, W.; Wagner, N.; Tedder, T.F.; Ley, K. Patches in High Endothelial Venules of Peyer’s P-Selectin in Leukocyte Rolling and Adhesion Integrins, and 7 β The Roles of L-Selectin. J. Immunol. 1998, 161, 2449–2456. [Google Scholar] [PubMed]
- Haddad, W.; Cooper, C.J.; Zhang, Z.; Brown, J.B.; Zhu, Y.; Issekutz, A.; Fuss, I.; Lee, H.; Kansas, G.S.; Barrett, T.A. P-Selectin and P-Selectin Glycoprotein Ligand 1 Are Major Determinants for Th1 Cell Recruitment to Nonlymphoid Effector Sites in the Intestinal Lamina Propria. J. Exp. Med. 2003, 198, 369–377. [Google Scholar] [CrossRef] [PubMed]
- Warnock, R.A.; Campbell, J.J.; Dorf, M.E.; Matsuzawa, A.; McEvoy, L.M.; Butcher, E.C. The Role of Chemokines in the Microenvironmental Control of T versus B Cell Arrest in Peyer’s Patch High Endothelial Venules. J. Exp. Med. 2000, 191, 77–88. [Google Scholar] [CrossRef] [PubMed]
- Berlin, C.; Berg, E.L.; Briskin, M.J.; Andrew, D.P.; Kilshaw, P.J.; Holzmann, B.; Weissman, I.L.; Hamann, A.; Butcher, E.C. α4β7 integrin mediates lymphocyte binding to the mucosal vascular addressin MAdCAM-1. Cell 1993, 74, 185–195. [Google Scholar] [CrossRef]
- Alon, R.; Sportiello, M.; Kozlovski, S.; Kumar, A.; Reilly, E.C.; Zarbock, A.; Garbi, N.; Topham, D.J. Leukocyte trafficking to the lungs and beyond: Lessons from influenza for COVID-19. Nat. Rev. Immunol. 2020, 21, 49–64. [Google Scholar] [CrossRef]
- Doerschuk, C.M.; Beyers, N.; Coxson, H.O.; Wiggs, B.; Hogg, J.C. Comparison of neutrophil and capillary diameters and their relation to neutrophil sequestration in the lung. J. Appl. Physiol. 1993, 74, 3040–3045. [Google Scholar] [CrossRef] [PubMed]
- Ivetic, A.; Hoskins Green, H.L.; Hart, S.J. L-selectin: A Major Regulator of Leukocyte Adhesion, Migration and Signaling. Front. Immunol. 2019, 10, 1068. [Google Scholar] [CrossRef]
- Watson, M.L.; Kingsmore, S.F.; Johnston, G.I.; Siegelman, M.H.; Le Beau, M.M.; Lemons, R.S.; Bora, N.S.; Howard, T.A.; Weissman, I.L.; McEver, R.P.; et al. Genomic organization of the selectin family of leukocyte adhesion molecules on human and mouse chromosome 1. J. Exp. Med. 1990, 172, 263–272. [Google Scholar] [CrossRef]
- Spertini, O.; Cordey, A.S.; Monai, N.; Giuffrè, L.; Schapira, M. P-selectin glycoprotein ligand 1 is a ligand for L-selectin on neutrophils, monocytes, and CD34+ hematopoietic progenitor cells. J. Cell Biol. 1996, 135, 523–531. [Google Scholar] [CrossRef]
- Puri, K.D.; Finger, E.B.; Gaudernack, G.; Springer, T.A. Sialomucin CD34 is the major L-selectin ligand in human tonsil high endothelial venules. J. Cell Biol. 1995, 131, 261–270. [Google Scholar] [CrossRef]
- Derry, C.; Mordsley, K.; Preece, G.; Ager, A. Purification of L-selectin ligands synthesised by rat peripheral lymph nodes and cultured high endothelial cells. Biochem. Soc. Trans. 1997, 1, 260. [Google Scholar] [CrossRef] [PubMed]
- Sassetti, C.; Tangemann, K.; Singer, M.S.; Kershaw, D.B.; Rosen, S.D. Identification of Podocalyxin-like Protein as a High Endothelial Venule Ligand for L-selectin: Parallels to CD34. J. Exp. Med. 1998, 187, 1965–1975. [Google Scholar] [CrossRef] [PubMed]
- Ley, K.; Tedder, T.; Kansas, G. L-selectin can mediate leukocyte rolling in untreated mesenteric venules in vivo independent of E- or P-selectin. Blood 1993, 82, 1632–1638. [Google Scholar] [CrossRef] [PubMed]
- Arbonés, M.L.; Ord, D.C.; Ley, K.; Ratech, H.; Maynard-Curry, C.; Otten, G.; Capon, D.J.; Teddert, T.F. Lymphocyte homing and leukocyte rolling and migration are impaired in L-selectin-deficient mice. Immunity 1994, 1, 247–260. [Google Scholar] [CrossRef]
- Tedder, T.F.; Steeber, D.A.; Pizcueta, P. L-selectin-deficient mice have impaired leukocyte recruitment into inflammatory sites. J. Exp. Med. 1995, 181, 2259–2264. [Google Scholar] [CrossRef] [PubMed]
- Mobley, J.L.; Rigby, S.M.; Dailey, M.O. Regulation of adhesion molecule expression by CD8 T cells in vivo. II. Expression of L-selectin (CD62L) by memory cytolytic T cells responding to minor histocompatibility antigens. J. Immunol. 1994, 153, 5443–5452. [Google Scholar]
- Watson, H.A.; Durairaj, R.R.P.; Ohme, J.; Alatsatianos, M.; Almutairi, H.; Mohammed, R.N.; Vigar, M.; Reed, S.G.; Paisey, S.J.; Marshall, C.; et al. L-Selectin Enhanced T Cells Improve the Efficacy of Cancer Immunotherapy. Front. Immunol. 2019, 10, 1321. [Google Scholar] [CrossRef]
- Mohammed, R.N.; Wehenkel, S.C.; Galkina, E.V.; Yates, E.K.; Preece, G.; Newman, A.; Watson, H.A.; Ohme, J.; Bridgeman, J.S.; Durairaj, R.R.P.; et al. ADAM17-dependent proteolysis of L-selectin promotes early clonal expansion of cytotoxic T cells. Sci. Rep. 2019, 9, 5487. [Google Scholar] [CrossRef]
- Chao, C.C.; Jensen, R.; Dailey, M.O. Mechanisms of L-selectin regulation by activated T cells. J. Immunol. 1997, 159, 1686–1694. [Google Scholar]
- Sinclair, L.V.; Finlay, D.; Feijoo, C.; Cornish, G.H.; Gray, A.; Ager, A.; Okkenhaug, K.; Hagenbeek, T.J.; Spits, H.; Cantrell, D.A. Phosphatidylinositol-3-OH kinase and nutrient-sensing mTOR pathways control T lymphocyte trafficking. Nat. Immunol. 2008, 9, 513–521. [Google Scholar] [CrossRef]
- Galkina, E.; Tanousis, K.; Preece, G.; Tolaini, M.; Kioussis, D.; Florey, O.; Haskard, D.O.; Tedder, T.F.; Ager, A. L-selectin shedding does not regulate constitutive T cell trafficking but controls the migration pathways of antigen-activated T lymphocytes. J. Exp. Med. 2003, 198, 1323–1335. [Google Scholar] [CrossRef] [PubMed]
- Weber, S.; Saftig, P. Ectodomain shedding and ADAMs in development. Development 2012, 139, 3693–3709. [Google Scholar] [CrossRef] [PubMed]
- Schlöndorff, J.; Becherer, J.D.; Blobel, C.P. Intracellular maturation and localization of the tumour necrosis factor α convertase (TACE). Biochem. J. 2000, 347, 131–138. [Google Scholar] [CrossRef] [PubMed]
- Black, R.A.; Rauch, C.T.; Kozlosky, C.J.; Peschon, J.J.; Slack, J.L.; Wolfson, M.F.; Castner, B.J.; Stocking, K.L.; Reddy, P.; Srinivasan, S.; et al. A metalloproteinase disintegrin that releases tumour-necrosis factor-∅ from cells. Nature 1997, 385, 729–733. [Google Scholar] [CrossRef]
- Moss, M.L.; Jin, S.L.C.; Milla, M.E.; Burkhart, W.; Carter, H.L.; Chen, W.J.; Clay, W.C.; Didsbury, J.R.; Hassler, D.; Hoffman, C.R.; et al. Cloning of a disintegrin metalloproteinase that processes precursor tumour-necrosis factor-α. Nature 1997, 385, 733–736. [Google Scholar] [CrossRef]
- Peschon, J.J.; Slack, J.L.; Reddy, P.; Stocking, K.L.; Sunnarborg, S.W.; Lee, D.C.; Russell, W.E.; Castner, B.J.; Johnson, R.S.; Fitzner, J.N.; et al. An essential role for ectodomain shedding in mammalian development. Science 1998, 282, 1281–1284. [Google Scholar] [CrossRef]
- Cavadas, M.; Oikonomidi, I.; Gaspar, C.J.; Burbridge, E.; Badenes, M.; Félix, I.; Bolado, A.; Hu, T.; Bileck, A.; Gerner, C.; et al. Phosphorylation of iRhom2 Controls Stimulated Proteolytic Shedding by the Metalloprotease ADAM17/TACE. Cell Rep. 2017, 21, 745–757. [Google Scholar] [CrossRef]
- Li, N.; Wang, Y.; Forbes, K.; Vignali, K.M.; Heale, B.S.; Saftig, P.; Hartmann, D.; Black, R.A.; Rossi, J.J.; Blobel, C.P.; et al. Metalloproteases regulate T-cell proliferation and effector function via LAG-3. EMBO J. 2007, 26, 494–504. [Google Scholar] [CrossRef]
- DeBerge, M.P.; Ely, K.H.; Cheng, G.-S.; Enelow, R.I. ADAM17-Mediated Processing of TNF-α Expressed by Antiviral Effector CD8+ T Cells Is Required for Severe T-Cell-Mediated Lung Injury. PLoS ONE 2013, 8, e79340. [Google Scholar] [CrossRef][Green Version]
- DeBerge, M.P.; Ely, K.H.; Wright, P.F.; Thorp, E.B.; Enelow, R.I. Shedding of TNF receptor 2 by effector CD8 + T cells by ADAM17 is important for regulating TNF-α availability during influenza infection. J. Leukoc. Biol. 2015, 98, 423–434. [Google Scholar] [CrossRef]
- Langjahr, P.; Díaz-Jiménez, D.; De La Fuente, M.; Rubio, E.; Golenbock, D.; Bronfman, F.C.; Quera, R.; Lez, M.J.G.; Hermoso, M.A.; Benjamim, C.F. Metalloproteinase-Dependent TLR2 Ectodomain Shedding is Involved in Soluble Toll-Like Receptor 2 (sTLR2) Production. PLoS ONE 2014, 9, e104624. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.S.; Kim, J.J.; Lee, M.J.; Lee, E.K.; Park, S.K. ADAM17-Mediated Ectodomain Shedding of Toll-Like Receptor 4 as a Negative Feedback Regulation in Lipopolysaccharide-Activated Aortic Endothelial Cells. Cell. Physiol. Biochem. 2018, 45, 1851–1862. [Google Scholar] [CrossRef] [PubMed]
- Matthews, V.; Schuster, B.; Schütze, S.; Bussmeyer, I.; Ludwig, A.; Hundhausen, C.; Sadowski, T.; Saftig, P.; Hartmann, D.; Kallen, K.J.; et al. Cellular cholesterol depletion triggers shedding of the human interleukin-6 receptor by ADAM10 and ADAM17 (TACE). J. Biol. Chem. 2003, 278, 38829–38839. [Google Scholar] [CrossRef]
- Budagian, V.; Bulanova, E.; Orinska, Z.; Ludwig, A.; Rose-John, S.; Saftig, P.; Borden, E.C.; Bulfone-Paus, S. Natural Soluble Interleukin-15Rα Is Generated by Cleavage That Involves the Tumor Necrosis Factor-α-converting Enzyme (TACE/ADAM17). J. Biol. Chem. 2004, 279, 40368–40375. [Google Scholar] [CrossRef] [PubMed]
- Franke, M.; Schröder, J.; Monhasery, N.; Ackfeld, T.; Hummel, T.M.; Rabe, B.; Garbers, C.; Becker-Pauly, C.; Floss, D.M.; Scheller, J. Human and Murine Interleukin 23 Receptors Are Novel Substrates for A Disintegrin and Metalloproteases ADAM10 and ADAM17. J. Biol. Chem. 2016, 291, 10551–10561. [Google Scholar] [CrossRef]
- Uchikawa, S.; Yoda, M.; Tohmonda, T.; Kanaji, A.; Matsumoto, M.; Toyama, Y.; Horiuchi, K. ADAM17 regulates IL-1 signaling by selectively releasing IL-1 receptor type 2 from the cell surface. Cytokine 2015, 71, 238–245. [Google Scholar] [CrossRef]
- Crowe, P.D.; Walter, B.N.; Mohler, K.M.; Otten-Evans, C.; Black, R.A.; Ware, C.F. A metalloprotease inhibitor blocks shedding of the 80-kD TNF receptor and TNF processing in T lymphocytes. J. Exp. Med. 1995, 181, 1205–1210. [Google Scholar] [CrossRef]
- Young, J.; Yu, X.; Wolslegel, K.; Nguyen, A.; Kung, C.; Chiang, E.; Kolumam, G.; Wei, N.; Wong, W.L.; DeForge, L.; et al. Lymphotoxin-αβ heterotrimers are cleaved by metalloproteinases and contribute to synovitis in rheumatoid arthritis. Cytokine 2010, 51, 78–86. [Google Scholar] [CrossRef]
- Sanchez-Guerrero, E.; Chen, E.; Kockx, M.; An, S.-W.; Chong, B.H.; Khachigian, L.M. IL-1beta Signals through the EGF Receptor and Activates Egr-1 through MMP-ADAM. PLoS ONE 2012, 8, 39811. [Google Scholar] [CrossRef]
- Yacoub, D.; Benslimane, N.; Al-Zoobi, L.; Hassan, G.; Nadiri, A.; Mourad, W. CD154 Is Released from T-cells by a Disintegrin and Metalloproteinase Domain-containing Protein 10 (ADAM10) and ADAM17 in a CD40 Protein-dependent Manner. J. Biol. Chem. 2013, 288, 36083–36093. [Google Scholar] [CrossRef] [PubMed]
- Peng, M.; Guo, S.; Yin, N.; Xue, J.; Shen, L.; Zhao, Q.; Zhang, W. Ectodomain shedding of Fcα receptor is mediated by ADAM10 and ADAM17. Immunology 2010, 130, 83–91. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, M.A.; Andersen, T.; Etzerodt, A.; Kragstrup, T.W.; Rasmussen, T.K.; Stengaard-Pedersen, K.; Hetland, M.L.; Hørslev-Petersen, K.; Junker, P.; Østergaard, M.; et al. A disintegrin and metalloprotease-17 and galectin-9 are important regulators of local 4-1BB activity and disease outcome in rheumatoid arthritis. Rheumatology 2016, 55, 1871–1879. [Google Scholar] [CrossRef] [PubMed]
- Tsakadze, N.L.; Sithu, S.D.; Sen, U.; English, W.R.; Murphy, G.; D’Souza, S.E. Tumor necrosis factor-alpha-converting enzyme (TACE/ADAM-17) mediates the ectodomain cleavage of intercellular adhesion molecule-1 (ICAM-1). J. Biol. Chem. 2006, 281, 3157–3164. [Google Scholar] [CrossRef] [PubMed]
- Garton, K.J.; Gough, P.J.; Philalay, J.; Wille, P.T.; Blobel, C.P.; Whitehead, R.H.; Dempsey, P.J.; Raines, E.W. Stimulated shedding of vascular cell adhesion molecule 1 (VCAM-1) is mediated by tumor necrosis factor-alpha-converting enzyme (ADAM 17). J. Biol. Chem. 2003, 278, 37459–37464. [Google Scholar] [CrossRef] [PubMed]
- Nagano, O.; Murakami, D.; Hartmann, D.; De Strooper, B.; Saftig, P.; Iwatsubo, T.; Nakajima, M.; Shinohara, M.; Saya, H. Cell–matrix interaction via CD44 is independently regulated by different metalloproteinases activated in response to extracellular Ca2+ influx and PKC activation. J. Cell Biol. 2004, 165, 893–902. [Google Scholar] [CrossRef] [PubMed]
- Koenen, R.R.; Pruessmeyer, J.; Soehnlein, O.; Fraemohs, L.; Zernecke, A.; Schwarz, N.; Reiss, K.; Sarabi, A.; Lindbom, L.; Hackeng, T.M.; et al. Regulated release and functional modulation of junctional adhesion molecule A by disintegrin metalloproteinases. Blood 2009, 113, 4799–4809. [Google Scholar] [CrossRef] [PubMed]
- Möller-Hackbarth, K.; Dewitz, C.; Schweigert, O.; Trad, A.; Garbers, C.; Rose-John, S.; Scheller, J. A Disintegrin and Metalloprotease (ADAM) 10 and ADAM17 Are Major Sheddases of T Cell Immunoglobulin and Mucin Domain 3 (Tim-3). J. Biol. Chem. 2013, 288, 34529–34544. [Google Scholar] [CrossRef] [PubMed]
- Romee, R.; Foley, B.; Lenvik, T.; Wang, Y.; Zhang, B.; Ankarlo, D.; Luo, X.; Cooley, S.; Verneris, M.; Walcheck, B.; et al. NK cell CD16 surface expression and function is regulated by a disintegrin and metalloprotease-17 (ADAM17). Blood 2013, 121, 3599–3608. [Google Scholar] [CrossRef] [PubMed]
- Lambert, D.W.; Yarski, M.; Warner, F.J.; Thornhill, P.; Parkin, E.T.; Smith, A.I.; Hooper, N.M.; Turner, A.J. Tumor Necrosis Factor-α Convertase (ADAM17) Mediates Regulated Ectodomain Shedding of the Severe-acute Respiratory Syndrome-Coronavirus (SARS-CoV) Receptor, Angiotensin-converting Enzyme-2 (ACE2). J. Biol. Chem. 2005, 280, 30113–30119. [Google Scholar] [CrossRef]
- Schweigert, O.; Dewitz, C.; Möller-Hackbarth, K.; Trad, A.; Garbers, C.; Rose-John, S.; Scheller, J. Soluble T cell immunoglobulin and mucin domain (TIM)-1 and -4 generated by A Disintegrin And Metalloprotease (ADAM)-10 and -17 bind to phosphatidylserine. Biochim. Biophys. Acta 2014, 1843, 275–287. [Google Scholar] [CrossRef]
- Gómez-Gaviro, M.; Domínguez-Luis, M.; Canchado, J.; Calafat, J.; Janssen, H.; Lara-Pezzi, E.; Fourie, A.; Tugores, A.; Valenzuela-Fernández, A.; Mollinedo, F.; et al. Expression and Regulation of the Metalloproteinase ADAM-8 during Human Neutrophil Pathophysiological Activation and Its Catalytic Activity on L-Selectin Shedding. J. Immunol. 2007, 178, 8053–8063. [Google Scholar] [CrossRef] [PubMed]
- Le Gall, S.M.; Bobé, P.; Reiss, K.; Horiuchi, K.; Niu, X.-D.; Lundell, D.; Gibb, D.R.; Conrad, D.; Saftig, P.; Blobel, C.P. ADAMs 10 and 17 Represent Differentially Regulated Components of a General Shedding Machinery for Membrane Proteins Such as Transforming Growth Factor, L-Selectin, and Tumor Necrosis Factor. Mol. Biol. Cell 2009, 20, 1785–1794. [Google Scholar] [CrossRef] [PubMed]
- Kaldjian, E.P.; Stoolman, L.M. Regulation of L-selectin mRNA in Jurkat cells. Opposing influences of calcium- and protein kinase C-dependent signaling pathways. J. Immunol. 1995, 154, 4351–4362. [Google Scholar] [PubMed]
- Long, C.; Wang, Y.; Herrera, A.H.; Horiuchi, K.; Walcheck, B. In vivo role of leukocyte ADAM17 in the inflammatory and host responses during E. coli-mediated peritonitis. J. Leukoc. Biol. 2010, 87, 1097–1101. [Google Scholar] [CrossRef] [PubMed]
- Long, C.; Hosseinkhani, M.R.; Wang, Y.; Sriramarao, P.; Walcheck, B. ADAM17 activation in circulating neutrophils following bacterial challenge impairs their recruitment. J. Leukoc. Biol. 2012, 92, 667–672. [Google Scholar] [CrossRef] [PubMed]
- Cappenberg, A.; Margraf, A.; Thomas, K.; Bardel, B.; McCreedy, D.A.; Van Marck, V.; Mellmann, A.; Lowell, C.A.; Zarbock, A. L-selectin shedding affects bacterial clearance in the lung: A new regulatory pathway for integrin outside-in signaling. Blood 2019, 134, 1445–1457. [Google Scholar] [CrossRef]
- Richards, H.; Longhi, M.P.; Wright, K.; Gallimore, A.; Ager, A. CD62L (L-Selectin) Down-Regulation Does Not Affect Memory T Cell Distribution but Failure to Shed Compromises Anti-Viral Immunity. J. Immunol. 2008, 180, 198–206. [Google Scholar] [CrossRef] [PubMed]
- ADAM17 Inhibitor/Rituximab After Auto HCT for DLBCL—Full Text View—ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT02141451 (accessed on 12 March 2021).
- INCB7839 in Treating Children With Recurrent/Progressive High-Grade Gliomas—Full Text View—ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT04295759?term=INCB7839&draw=2&rank=1 (accessed on 12 March 2021).
- Qian, M.; Bai, S.A.; Brogdon, B.; Wu, J.T.; Liu, R.Q.; Covington, M.B.; Vaddi, K.; Newton, R.C.; Fossler, M.J.; Garner, C.E.; et al. Pharmacokinetics and pharmacodynamics of DPC 333 ((2R)-2-((3R)-3-amino-3{4-[2-methyl-4-quinolinyl) methoxy] phenyl}-2-oxopyrrolidinyl)-N-hydroxy-4-methylpentanamide, a potent and selective inhibitor of tumor necrosis factor α-converting enzyme in rodents, dogs, chimpanzees, and humans. Drug Metab. Dispos. 2007, 35, 1916–1925. [Google Scholar] [CrossRef]
- Thabet, M.M.; Huizinga, T.W.J. Drug evaluation: Apratastat, a novel TACE/MMP inhibitor for rheumatoid arthritis. Curr. Opin. Investig. Drugs 2006, 7, 1014–1019. [Google Scholar]
- Tape, C.J.; Willems, S.H.; Dombernowsky, S.L.; Stanley, P.L.; Fogarasi, M.; Ouwehand, W.; McCafferty, J.; Murphy, G. Cross-domain inhibition of TACE ectodomain. Proc. Natl. Acad. Sci. USA 2011, 108, 5578–5583. [Google Scholar] [CrossRef]
- Kwok, H.F.; Botkjaer, K.A.; Tape, C.J.; Huang, Y.; McCafferty, J.; Murphy, G. Development of a ‘mouse and human cross-reactive’ affinity-matured exosite inhibitory human antibody specific to TACE (ADAM17) for cancer immunotherapy. Protein Eng. Des. Sel. 2014, 27, 179–190. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Rios-Doria, J.; Sabol, D.; Chesebrough, J.; Stewart, D.; Xu, L.; Tammali, R.; Cheng, L.; Du, Q.; Schifferli, K.; Rothstein, R.; et al. Amonoclonal antibody to ADAM17 inhibits tumor growth by inhibiting EGFR and non-EGFR-mediated pathways. Mol. Cancer Ther. 2015, 14, 1637–1649. [Google Scholar] [CrossRef] [PubMed]
- Scherle, P.; Liu, X.; Li, J.; Fridman, J.; Li, Y.; Yao, W.; Williams, W.; Levy, R.; Vaddi, K.; Newton, R.; et al. Selective inhibition of ADAM metalloproteases blocks HER-2 extracellular domain (ECD) cleavage and potentiates the anti-tumor effects of trastuzumab. Cancer Biol. Ther. 2006, 24, 13021. [Google Scholar] [CrossRef]
- Venkatesh, H.S.; Tam, L.T.; Woo, P.J.; Lennon, J.; Nagaraja, S.; Gillespie, S.M.; Ni, J.; Duveau, D.Y.; Morris, P.J.; Zhao, J.J.; et al. Targeting neuronal activity-regulated neuroligin-3 dependency in high-grade glioma. Nature 2017, 549, 533–537. [Google Scholar] [CrossRef]
- Newton, R.C.; Bradley, E.C.; Levy, R.S.; Doval, D.; Bondarde, S.; Sahoo, T.P.; Lokanatha, D.; Julka, P.K.; Nagarkar, R.; Friedman, S.M. Clinical benefit of INCB7839, a potent and selective ADAM inhibitor, in combination with trastuzumab in patients with metastatic HER2+ breast cancer. J. Clin. Oncol. 2010, 28, 3025. [Google Scholar] [CrossRef]
- Baumgart, A.; Seidl, S.; Vlachou, P.; Michel, L.; Mitova, N.; Schatz, N.; Specht, K.; Koch, I.; Schuster, T.; Grundler, R.; et al. ADAM17 Regulates Epidermal Growth Factor Receptor Expression through the Activation of Notch1 in Non–Small Cell Lung Cancer. Cancer Res. 2010, 70, 5368–5378. [Google Scholar] [CrossRef]
- Kim, M.L.; Zhang, B.; Mills, I.P.; Milla, M.E.; Brunden, K.R.; Lee, V.M.-Y. Effects of TNFα-Converting Enzyme Inhibition on Amyloid β Production and APP Processing In Vitro and In Vivo. J. Neurosci. 2008, 28, 12052–12061. [Google Scholar] [CrossRef]
- Grootveld, M. BMS-561392. Bristol-Myers Squibb. Curr. Opin. Investig. Drugs 2003, 4, 598–602. [Google Scholar]
- Sharma, M.; Mohapatra, J.; Malik, U.; Wagh, A.; Singh, A.; Patel, H.M.; Pandey, D.; Kadam, S.; Shah, G.B.; Chatterjee, A.; et al. Selective inhibition of tumor necrosis factor-α converting enzyme attenuates liver toxicity in a murine model of concanavalin A induced auto-immune hepatitis. Int. Immunopharmacol. 2013, 17, 229–236. [Google Scholar] [CrossRef]
- Maddox, B. Tumor Necrosis Factor Alpha Converting Enzyme Inhibition during Tumor Necrosis Factor Alpha Converting Enzyme Inhibition during Acute Colitis in Mice: A Regional Analysis Acute Colitis in Mice: A Regional Analysis. MSU Graduate Thesis, Missouri State University, Springfield, MO, USA, 2015. [Google Scholar]
- Vidal, P.M.; Lemmens, E.; Avila, A.; Vangansewinkel, T.; Chalaris, A.; Rose-John, S.; Hendrix, S. ADAM17 is a survival factor for microglial cells in vitro and in vivo after spinal cord injury in mice. Cell Death Dis. 2013, 4, e954. [Google Scholar] [CrossRef]
- Gooz, M.B.; Maldonado, E.N.; Dang, Y.; Amria, M.Y.; Higashiyama, S.; Abboud, H.E.; Lemasters, J.J.; Bell, P.D. ADAM17 promotes proliferation of collecting duct kidney epithelial cells through ERK activation and increased glycolysis in polycystic kidney disease. American J. Physiol.-Renal Physiol. 2014, 307, F551–F559. [Google Scholar] [CrossRef] [PubMed]
- Lartey, N.L.; Valle-Reyes, S.; Vargas-Robles, H.; Jiménez-Camacho, K.E.; Guerrero-Fonseca, I.M.; Castellanos-Martínez, R.; Montoya-García, A.; García-Cordero, J.; Cedillo-Barrón, L.; Nava, P.; et al. ADAM17 inhibition prevents neutrophilia and lung injury in a mouse model of Covid-19. bioRxiv 2021. [Google Scholar] [CrossRef]
- Sharma, A.; Bender, S.; Zimmermann, M.; Riesterer, O.; Broggini-Tenzer, A.; Pruschy, M.N. Secretome Signature Identifies ADAM17 as Novel Target for Radiosensitization of Non–Small Cell Lung Cancer. Clin. Cancer Res. 2016, 22, 4428–4439. [Google Scholar] [CrossRef] [PubMed]
- Maekawa, M.; Tadaki, H.; Tomimoto, D.; Okuma, C.; Sano, R.; Ishii, Y.; Katsuda, Y.; Yoshiuchi, H.; Kakefuda, R.; Ohta, T.; et al. A Novel TNF-α Converting Enzyme (TACE) Selective Inhibitor JTP-96193 Prevents Insulin Resistance in KK-A y Type 2 Diabetic Mice and Diabetic Peripheral Neuropathy in Type 1 Diabetic Mice. Biol. Pharm. Bull 1906, 42, 1906–1912. [Google Scholar] [CrossRef]
- Huang, Y.; Benaich, N.; Tape, C.; Kwok, H.F.; Murphy, G. Targeting the Sheddase Activity of ADAM17 by an Anti-ADAM17 Antibody D1(A12) Inhibits Head and Neck Squamous Cell Carcinoma Cell Proliferation and Motility via Blockage of Bradykinin Induced HERs Transactivation. Int. J. Biol. Sci. 2014, 10, 702. [Google Scholar] [CrossRef]
- Richards, F.M.; Tape, C.J.; Jodrell, D.I.; Murphy, G. Anti-tumour effects of a specific anti-ADAM17 antibody in an ovarian cancer model in vivo. PLoS ONE 2012, 7, e40597. [Google Scholar] [CrossRef]
- Caiazza, F.; McGowan, P.M.; Mullooly, M.; Murray, A.; Synnott, N.; O’Donovan, N.; Flanagan, L.; Tape, C.J.; Murphy, G.; Crown, J.; et al. Targeting ADAM-17 with an inhibitory monoclonal antibody has antitumour effects in triple-negative breast cancer cells. Br. J. Cancer 2015, 112, 1895–1903. [Google Scholar] [CrossRef]
- Yang, Z.; Chan, K.I.; Kwok, H.F.; Tam, K.Y. Novel Therapeutic Anti-ADAM17 Antibody A9(B8) Enhances EGFR-TKI–Mediated Anticancer Activity in NSCLC. Transl. Oncol. 2019, 12, 1516–1524. [Google Scholar] [CrossRef]
- Ye, J.; Yuen, S.M.; Murphy, G.; Xie, R.; Kwok, H.F. Anti-tumor effects of a ‘human & mouse cross-reactive’ anti-ADAM17 antibody in a pancreatic cancer model in vivo. Eur. J. Pharm. Sci. 2017, 110, 62–69. [Google Scholar] [CrossRef]
- Dosch, J.; Ziemke, E.; Welling, T.; Hardiman, K.; Sebolt-Leopold, J.; Michelotti, E.; Hollingsworth, R.; Hurt, E. Abstract 975: Monoclonal antibody targeting of ADAM17 is an effective treatment for metastatic colorectal cancer resulting in tumor growth control and reductions of cancer stem cells. Cancer Res. 2015, 75, 975. [Google Scholar] [CrossRef]
- Dosch, J.; Ziemke, E.; Wan, S.; Luker, K.; Welling, T.; Hardiman, K.; Fearon, E.; Thomas, S.; Flynn, M.; Rios-Doria, J.; et al. Targeting ADAM17 inhibits human colorectal adenocarcinoma progression and tumor-initiating cell frequency. Oncotarget 2017, 8, 65090–65099. [Google Scholar] [CrossRef] [PubMed]
- Mishra, H.K.; Pore, N.; Michelotti, E.F.; Walcheck, B. Anti-ADAM17 monoclonal antibody MEDI3622 increases IFNγ production by human NK cells in the presence of antibody-bound tumor cells. Cancer Immunol. Immunother. 2018, 67, 1407–1416. [Google Scholar] [CrossRef] [PubMed]
- Mishra, H.K.; Ma, J.; Mendez, D.; Hullsiek, R.; Pore, N.; Walcheck, B. Blocking adam17 function with a monoclonal antibody improves sepsis survival in a murine model of polymicrobial sepsis. Int. J. Mol. Sci. 2020, 21, 6688. [Google Scholar] [CrossRef] [PubMed]
- Blaydon, D.C.; Biancheri, P.; Di, W.-L.; Plagnol, V.; Cabral, R.M.; Brooke, M.A.; van Heel, D.A.; Ruschendorf, F.; Toynbee, M.; Walne, A.; et al. Inflammatory Skin and Bowel Disease Linked to ADAM17 Deletion. N. Engl. J. Med. 2011, 365, 1502–1508. [Google Scholar] [CrossRef]
- Bandsma, R.H.J.; van Goor, H.; Yourshaw, M.; Horlings, R.K.; Jonkman, M.F.; Schölvinck, E.H.; Karrenbeld, A.; Scheenstra, R.; Kömhoff, M.; Rump, P.; et al. Loss of ADAM17 is associated with severe multiorgan dysfunction. Hum. Pathol. 2015, 46, 928. [Google Scholar] [CrossRef] [PubMed]
- Sallusto, F.; Lenig, D.; Förster, R.; Lipp, M.; Lanzavecchia, A. Two subsets of memory T lymphocytes with distinct homing potentials and effector functions. Nature 1999, 401, 708–712. [Google Scholar] [CrossRef] [PubMed]
- Giuliani, E.; Vassena, L.; Galardi, S.; Michienzi, A.; Desimio, M.G.; Doria, M. Dual regulation of L-selectin (CD62L) by HIV-1: Enhanced expression by Vpr in contrast with cell-surface down-modulation by Nef and Vpu. Virology 2018, 523, 121–128. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Leukocyte | Peak Accumulation in the Lung Following IAV Infection | Immunoprotection Roles | Immunopathology Roles |
|---|---|---|---|
| Monocyte | 5 days [36] | Pro-inflammatory cytokine production [37], differentiation into dendritic cells [9] | Excessive pulmonary inflammation and tissue damage leading to mortality [38] |
| Alveolar macrophage | 2 days [39] | Phagocytosis of infected cells [39], prevention of epithelial cell infection [40] | Excessive pulmonary inflammation and tissue damage leading to mortality [41] |
| Dendritic cell | 10 days [36] | Type I IFN production [42], activation and recruitment of antigen-specific T cells [36] | Excessive pulmonary inflammation and tissue damage leading to mortality [43] |
| Neutrophil | 3–5 days [44] | Pro-inflammatory cytokine production [44], phagocytosis of infected cells [39], anti-bacterial granule release and NETosis to limit viral replication [45], recruitment of CD8+ T cells [46] | Excessive pulmonary inflammation, ROS production [47] and tissue damage leading to mortality [48] |
| Natural killer cell | 2–4 days [49], 6 days [50] | Pro-inflammatory cytokine production [51], cytotoxicity of IAV infected cells [49], recruitment of dendritic cells and T cells to mediastinal lymph node [52], enhancement of CTL responses [53] | Excessive lung inflammation, increased phagocyte and neutrophil recruitment [54] |
| Location | Selectins | Selectin Ligands | Chemokine Receptors and Ligands | Integrins | Integrin Ligands |
|---|---|---|---|---|---|
| Peripheral lymph nodes | L-selectin [82] | PNAd [83] | CCR7, CCL19, CCL21 [57,84,85] | LFA-1 [86] | ICAM-1, ICAM-2 [86,87] |
| Mesenteric lymph node | L-selectin [82] | MAdCAM-1, PNAd [83,88] | CCR7, CCL19, CCL21 [89] | α4β7, LFA-1 [90,91] | MAdCAM-1 [90] |
| Peyer’s patches in gut mucosa | P-selectin 1, L-selectin [92] | PSGL-1, MAdCAM-1 [93] | CCR7, CCL21 [94] | α4β7, LFA-1 [91,95] | MAdCAM-1 [95] |
| Lungs in IAV infection 1 | L-selectin [2] | Not known | CXCR1, CXCR3, CCR4, CCR5, CXCR6 [96] | VLA-4, LFA-1 [96] | VCAM-1, ICAM-1 [96] |
| Role within the Immune System | ADAM17 Substrate |
|---|---|
| Pattern recognition | TLR2 [122], TLR4 [123] |
| Inflammation | IL-6R [124], IL-15R [125], IL-23R [126], IL-1RII [127], TNFR-I [117], TNFR-II [128], TNFα [115], Lymphotoxin-αβ [129], IL-1β, [130], CD154 [131], CD89 [132], 4-1BB [133] |
| Leukocyte adhesion and migration | ICAM-1 [134], VCAM-1 [135], L-selectin [117], CD44 [136], JAM-A [137] |
| T cell activation, proliferation and exhaustion | LAG-3 [119], TIM-3 [138] |
| Natural killer cell toxicity | CD16 [139] |
| Viral cell entry | ACE2 [140], TIM-1, TIM-4 [141] |
| Drug Name | Formulation | In Vitro Models | Mouse Models | Human Trials |
|---|---|---|---|---|
| INCB7839/ Aderbasib | Small molecule inhibitor | HER2 + breast cancer with trastuzumab [156] | High-grade glioma xenograft tumour model [157] | Phase I; paediatric glioma [150] |
| Phase I/II; DLBCL in combination with rituximab, post autologous HCT [149] | ||||
| HER2+ breast cancer xenograft tumour model with trastuzumab [156] | Phase I; HER2+ metastatic breast cancer with trastuzumab [158] | |||
| BMS-561392/ DPC-333 | Small molecule inhibitor | NSCLC [159] | Collagen-induced arthritis (CIA) model [161] | Phase II rheumatoid arthritis [161] |
| Autoimmune hepatitis [162] | ||||
| Alzheimer’s [160] | Acute colitis [163] | |||
| Alzheimer’s model [160] | ||||
| Spinal cord injury [164] | ||||
| TMI-005/ Apratastat | Small molecule inhibitor | Polycystic kidney disease [165]. | SARS-CoV-2 [166] | Phase II clinical trial for rheumatoid arthritis [152]. |
| NSCLC [167]. | ||||
| JTP-96193 | Small molecule inhibitor | Type 2 diabetes & diabetic peripheral neuropathy [168]. | ||
| D1(A12) | Human IgG antibody | HNSCC [169]. | Ovarian xenograft tumour model [170]. | |
| Triple negative breast cancer [171] | ||||
| A9(B8) | Humanised mouse IgG2 antibody | NSCLC with erlotinib/ gefitinib [172]. | Pancreatic cancer [173]. | |
| MEDI3622 | Humanised mouse IgG1 antibody | Colorectal cancer [174]. | Head and neck & colorectal xenograft tumour models [175] | |
| Ovarian cancer & Burkitt’s lymphoma [176] | Polymicrobial sepsis [177]. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Reed, S.G.; Ager, A. Immune Responses to IAV Infection and the Roles of L-Selectin and ADAM17 in Lymphocyte Homing. Pathogens 2022, 11, 150. https://doi.org/10.3390/pathogens11020150
Reed SG, Ager A. Immune Responses to IAV Infection and the Roles of L-Selectin and ADAM17 in Lymphocyte Homing. Pathogens. 2022; 11(2):150. https://doi.org/10.3390/pathogens11020150
Chicago/Turabian StyleReed, Sophie G., and Ann Ager. 2022. "Immune Responses to IAV Infection and the Roles of L-Selectin and ADAM17 in Lymphocyte Homing" Pathogens 11, no. 2: 150. https://doi.org/10.3390/pathogens11020150
APA StyleReed, S. G., & Ager, A. (2022). Immune Responses to IAV Infection and the Roles of L-Selectin and ADAM17 in Lymphocyte Homing. Pathogens, 11(2), 150. https://doi.org/10.3390/pathogens11020150