
Leishmaniasis Vaccines: Applications of RNA Technology and Targeted Clinical Trial Designs


 and
and 
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
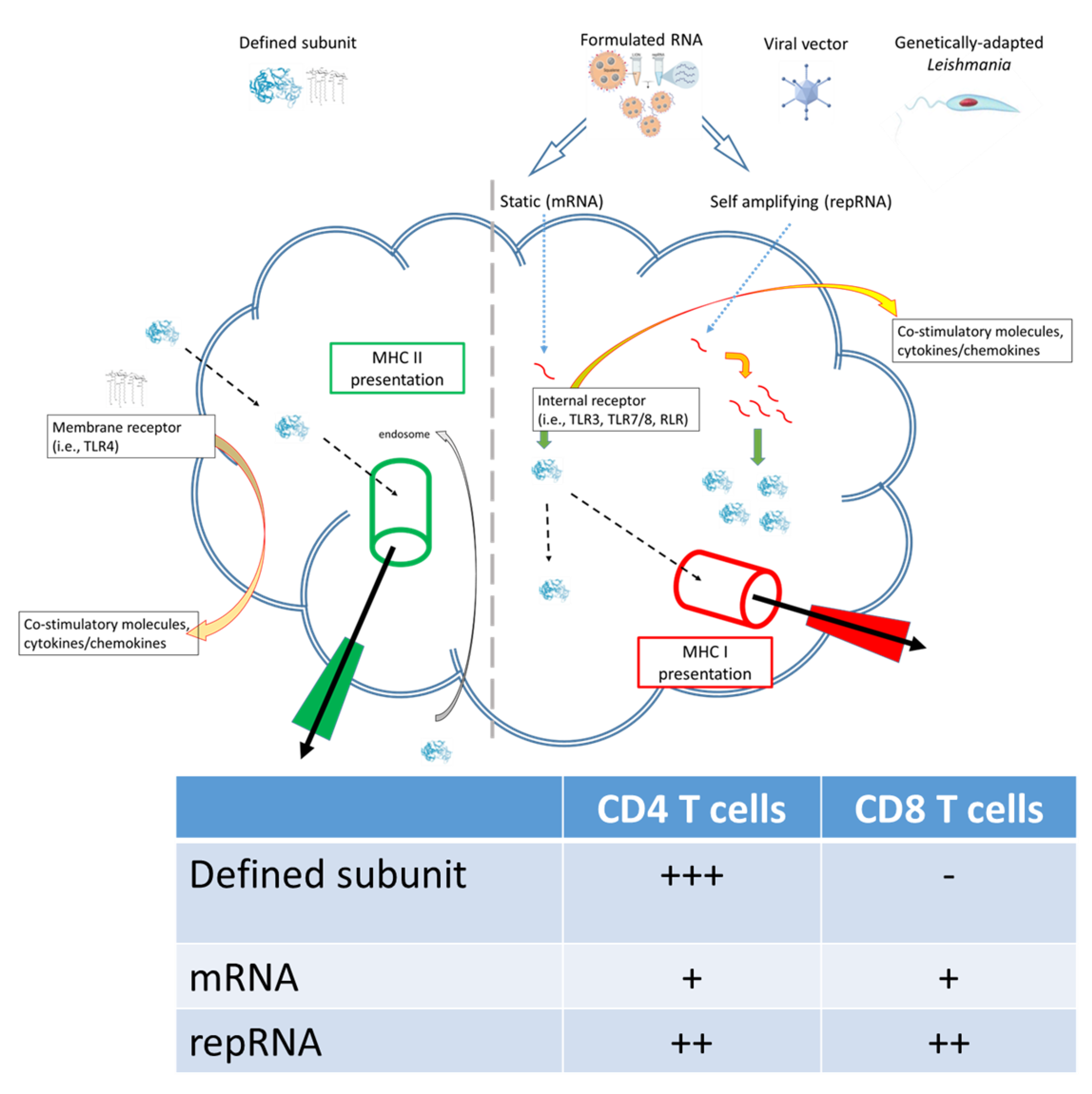
2. Potential Application of RNA Technology for Leishmania Vaccines
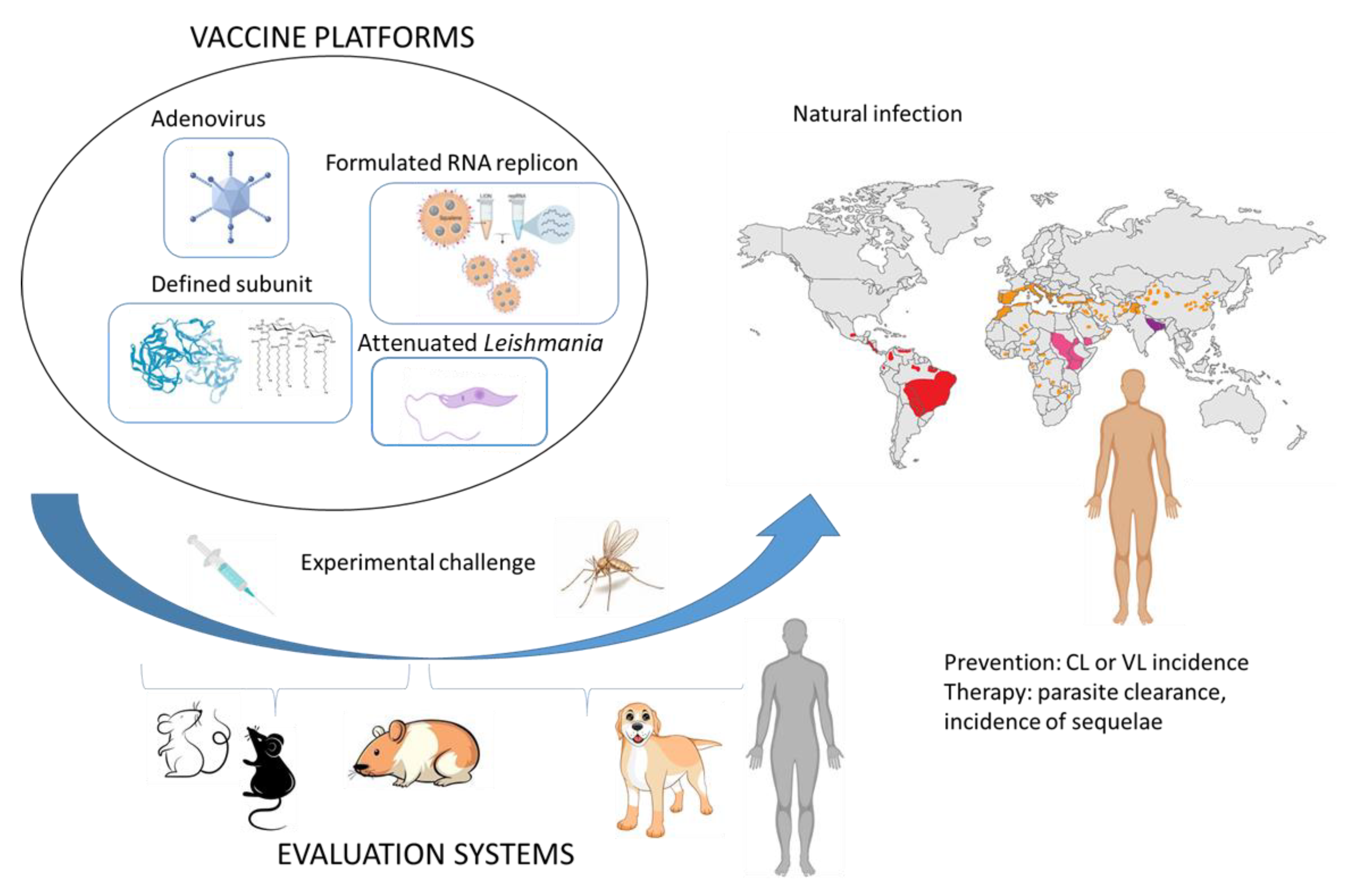
3. Current Challenges for Leishmania Vaccines
3.1. Development
3.2. Clinical Evaluation
4. Leverage of a Profitable Veterinary Application?
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Brito, M.E.F.; Mendonça, M.G.; Gomes, Y.M.; Jardim, M.L.; Abath, F.G. Dynamics of the antibody response in patients with therapeutic or spontaneous cure of American cutaneous leishmaniasis. Trans. R. Soc. Trop. Med. Hyg. 2001, 95, 203–206. [Google Scholar] [CrossRef]
- Carvalho, E.M.; Correia Filho, D.; Bacellar, O.; Almeida, R.P.; Lessa, H.; Rocha, H. Characterization of the immune response in subjects with self-healing cutaneous leishmaniasis. Am. J. Trop. Med. Hyg. 1995, 53, 273–277. [Google Scholar] [CrossRef] [PubMed]
- Heinzel, F.P.; Schoenhaut, D.S.; Rerko, R.M.; Rosser, L.E.; Gately, M.K. Recombinant interleukin 12 cures mice infected with Leishmania major. J. Exp. Med. 1993, 177, 1505–1509. [Google Scholar] [CrossRef] [PubMed]
- Mosmann, T.R.; Cherwinski, H.; Bond, M.W.; Giedlin, M.A.; Coffman, R.L. Two types of murine helper T cell clone. I. Definition according to profiles of lymphokine activities and secreted proteins. J. Immunol. 1986, 136, 2348–2357. [Google Scholar]
- Reed, S.G.; Scott, P. T-cell and cytokine responses in leishmaniasis. Curr. Opin. Immunol. 1993, 5, 524–531. [Google Scholar] [CrossRef]
- Hutchins, A.S.; Artis, D.; Hendrich, B.D.; Bird, A.P.; Scott, P.; Reiner, S.L. Cutting Edge: A Critical Role for Gene Silencing in Preventing Excessive Type 1 Immunity. J. Immunol. 2005, 175, 5606–5610. [Google Scholar] [CrossRef] [Green Version]
- Alexander, J.; Brombacher, F. T Helper1/T Helper2 Cells and Resistance/Susceptibility to Leishmania Infection: Is This Paradigm Still Relevant? Front. Immunol. 2012, 3, 80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gautam, S.; Kumar, R.; Singh, N.; Singh, A.K.; Rai, M.; Sacks, D.; Sundar, S.; Nylén, S. CD8 T cell exhaustion in human visceral leishmaniasis. J. Infect. Dis. 2014, 209, 290–299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaushal, H.; Bras-Gonçalves, R.; Negi, N.S.; Lemesre, J.L.; Papierok, G.; Salotra, P. Role of CD8(+) T cells in protection against Leishmania donovani infection in healed Visceral Leishmaniasis individuals. BMC Infect. Dis. 2014, 14, 653. [Google Scholar] [CrossRef] [Green Version]
- Den Boer, M.L.; Alvar, J.; Davidson, R.N.; Ritmeijer, K.; Balasegaram, M. Developments in the treatment of visceral leishmaniasis. Expert Opin. Emerg. Drugs 2009, 14, 395–410. [Google Scholar] [CrossRef] [Green Version]
- Salari, S.; Bamorovat, M.; Sharifi, I.; Almani, P.G.N. Global distribution of treatment resistance gene markers for leishmaniasis. J. Clin. Lab. Anal. 2022, 36, e24599. [Google Scholar] [CrossRef] [PubMed]
- Kaye, P.M.; Mohan, S.; Mantel, C.; Malhame, M.; Revill, P.; Le Rutte, E.; Pakash, V.; Layton, A.M.; Lacey, C.J.N.; Malvolti, S. Overcoming roadblocks in the development of vaccines for leishmaniasis. Expert Rev. Vaccines 2021, 20, 1419–1430. [Google Scholar] [CrossRef] [PubMed]
- Bottazzi, M.E.; Hotez, P.J. “Running the Gauntlet”: Formidable challenges in advancing neglected tropical diseases vaccines from development through licensure, and a “Call to Action”. Hum. Vacc. Immunother. 2019, 15, 2235–2242. [Google Scholar] [CrossRef] [Green Version]
- Versteeg, L.; Almutairi, M.M.; Hotez, P.J.; Pollet, J. Enlisting the mRNA Vaccine Platform to Combat Parasitic Infections. Vaccines 2019, 7, 122. [Google Scholar] [CrossRef] [Green Version]
- Mohan, S.; Revill, P.; Malvolti, S.; Malhame, M.; Sculpher, M.; Kaye, P.M. Estimating the global demand curve for a leishmaniasis vaccine: A generalisable approach based on global burden of disease estimates. PLoS Negl. Trop. Dis. 2022, 16, e0010471. [Google Scholar] [CrossRef]
- Corbett, K.S.; Edwards, D.K.; Leist, S.R.; Abiona, O.M.; Boyoglu-Barnum, S.; Gillespie, R.A.; Himansu, S.; Schäfer, A.; Ziwawo, C.T.; DiPiazza, A.T.; et al. SARS-CoV-2 mRNA vaccine design enabled by prototype pathogen preparedness. Nature 2020, 586, 567–571. [Google Scholar] [CrossRef]
- Branche, A.R.; Rouphael, N.G.; Diemert, D.D.; Falsey, A.R.; Losada, C.; Baden, L.R.; Frey, S.E.; Whitaker, J.A.; Little, S.J.; Anderson, E.J.; et al. SARS-CoV-2 Variant Vaccine Boosters Trial: Preliminary Analyses. MedRxiv 2022. [Google Scholar] [CrossRef]
- Ying, B.; Scheaffer, S.M.; Whitener, B.; Liang, C.Y.; Dmytrenko, O.; Mackin, S.; Wu, K.; Lee, D.; Avena, L.E.; Chong, Z.; et al. Boosting with variant-matched or historical mRNA vaccines protects against Omicron infection in mice. Cell 2022, 185, 1572–1587.e11. [Google Scholar] [CrossRef] [PubMed]
- Hawman, D.W.; Meade-White, K.; Archer, J.; Leventhal, S.; Wilson, D.; Shaia, C.; Randall, S.; Khandhar, A.P.; Hsiang, T.-Y.; Gale, M., Jr.; et al. SARS-CoV2 variant-specific replicating RNA vaccines protect from disease and pathology and reduce viral shedding following challenge with heterologous SARS-CoV2 variants of concern. BioRxiv 2021. [Google Scholar] [CrossRef]
- Brasu, N.; Elia, I.; Russo, V.; Montacchiesi, G.; Stabile, S.A.; De Intinis, C.; Fesi, F.; Gizzi, K.; Macagno, M.; Montone, M.; et al. Memory CD8+ T cell diversity and B cell responses correlate with protection against SARS-CoV-2 following mRNA vaccination. Nat. Immunol. 2022, 23, 1445–1456. [Google Scholar] [CrossRef] [PubMed]
- Wherry, E.J.; Barouch, D.H. T cell immunity to COVID-19 vaccines. Science 2022, 377, 821–822. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, L.S.; Barreto, A.S.; Bomfim, L.G.S.; Gomes, M.C.; Ferreira, N.L.C.; da Cruz, G.S.; Magalhães, L.; de Jesus, A.R.; Palatnik de Sousa, C.B.; Correa, C.B.; et al. Multifunctional, TNF-α and IFN-γ-Secreting CD4 and CD8 T Cells and CD8High T Cells Are Associated With the Cure of Human Visceral Leishmaniasis. Front. Immunol. 2021, 12, 773983. [Google Scholar] [CrossRef]
- Volpedo, G.; Bhattacharya, P.; Gannavaram, S.; Pacheco-Fernandez, T.; Oljuskin, T.; Dey, R.; Satoskar, A.R.; Nakhasi, H.L. The History of Live Attenuated Centrin Gene-Deleted Leishmania Vaccine Candidates. Pathogens 2022, 11, 431. [Google Scholar] [CrossRef]
- Oliveira BC, D.; Duthie, M.S.; Pereira, V.R.A. Vaccines for leishmaniasis and the implications of their development for American tegumentary leishmaniasis. Hum. Vaccines Immunother. 2019, 16, 919–930. [Google Scholar] [CrossRef]
- Duthie, M.S.; Reed, S.G. Not All Antigens Are Created Equally: Progress, Challenges, and Lessons Associated with Developing a Vaccine for Leishmaniasis. Clin. Vaccine Immunol. 2017, 24, e00108-17. [Google Scholar] [CrossRef] [Green Version]
- Younis, B.M.; Osman, M.; Khalil, E.A.; Santoro, F.; Furini, S.; Wiggins, R.; Keding, A.; Carraro, M.; Musa, A.E.A.; Abdarahaman, M.A.A.; et al. Safety and immunogenicity of ChAd63-KH vaccine in post-kala-azar dermal leishmaniasis patients in Sudan. Mol. Ther. 2021, 29, 2366–2377. [Google Scholar] [CrossRef] [PubMed]
- Osman, M.; Mistry, A.; Keding, A.; Gabe, R.; Cook, E.; Forrester, S.; Wiggins, R.; Di Marco, S.; Colloca, S.; Siani, L.; et al. A third generation vaccine for human visceral leishmaniasis and post kala azar dermal leishmaniasis: First-in-human trial of ChAd63-KH. PLoS Negl. Trop. Dis. 2017, 11, e0005527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumari, S.; Samant, M.; Khare, P.; Misra, P.; Dutta, S.; Kolli, B.K.; Sharma, S.; Chang, K.P.; Dube, A. Photodynamic vaccination of hamsters with inducible suicidal mutants of Leishmania amazonensis elicits immunity against visceral leishmaniasis. Eur. J. Immunol. 2009, 39, 178–191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, W.-W.; Zhang, W.W.; Karmakar, S.; Gannavaram, S.; Dey, R.; Lypaczewski, P.; Ismail, N.; Siddiqui, A.; Simonyan, V.; Oliveira, F.; et al. A second generation leishmanization vaccine with a markerless attenuated Leishmania major strain using CRISPR gene editing. Nat. Commun. 2020, 11, 3461. [Google Scholar] [CrossRef] [PubMed]
- Gomes, D.C.O.; Souza, B.L.D.S.C.; Schwedersky, R.P.; Covre, L.P.; de Matos Guedes, H.L.; Lopes, U.G.; Re, M.I.; Rossi-Bergmann, B. Intranasal immunization with chitosan microparticles enhances LACK-DNA vaccine protection and induces specific long-lasting immunity against visceral leishmaniasis. Microbes Infect. 2022, 24, 104884. [Google Scholar] [CrossRef]
- Santos, T.T.O.; Machado, A.S.; Ramos, F.F.; Oliveira-da-Silva, J.A.; Lage, D.P.; Tavares, G.S.; Mendonça, D.V.C.; Cardoso, M.S.; Siqueira, W.F.; Martins, V.T.; et al. Leishmania eukaryotic elongation Factor-1 beta protein is immunogenic and induces parasitological protection in mice against Leishmania infantum infection. Microb. Pathog. 2021, 151, 104745. [Google Scholar] [CrossRef]
- Bertholet, S.; Goto, Y.; Carter, L.; Bhatia, A.; Howard, R.F.; Carter, D.; Coler, R.N.; Vedvick, T.S.; Reed, S.G. Optimized subunit vaccine protects against experimental leishmaniasis. Vaccine 2009, 27, 7036–7045. [Google Scholar] [CrossRef] [Green Version]
- Coler, R.N.; Duthie, M.S.; Hofmeyer, K.A.; Guderian, J.; Jayashankar, L.; Vergara, J.; Rolf, T.; Misquith, A.; Lurance, J.D.; Raman, V.S.; et al. From mouse to man: Safety, immunogenicity and efficacy of a candidate leishmaniasis vaccine LEISH-F3+GLA-SE. Clin. Transl. Immunol. 2015, 4, e35. [Google Scholar] [CrossRef]
- Duthie, M.S.; Pereira, L.; Favila, M.; Hofmeyer, K.A.; Reed, S.J.; Metangmo, S.; Townsend, S.; Laurance, J.D.; Picone, A.; Misquith, A.; et al. A defined subunit vaccine that protects against vector-borne visceral leishmaniasis. NPJ Vaccines 2017, 2, 23. [Google Scholar] [CrossRef] [Green Version]
- Pardi, N.; Hogan, M.J.; Pelc, R.S.; Muramatsu, H.; Andersen, H.; DeMaso, C.R.; Dowd, K.A.; Sutherland, L.L.; Scearce, M.; Parks, R.; et al. Zika virus protection by a single low-dose nucleoside-modified mRNA vaccination. Nature 2017, 543, 248–251. [Google Scholar] [CrossRef] [Green Version]
- Pardi, N.; Weissman, D. RNA Vaccines, Methods and Protocols. Methods Mol. Biol. 2016, 1499, 143–153. [Google Scholar]
- Richner, J.M.; Himansu, S.; Dowd, K.A.; Butler, S.L.; Salazar, V.; Fox, J.M.; Julander, J.G.; Tang, W.W.; Shresta, S.; Pierson, T.C.; et al. Modified mRNA Vaccines Protect against Zika Virus Infection. Cell 2017, 168, 1114–1125. [Google Scholar] [CrossRef] [Green Version]
- Hekele, A.; Bertholet, S.; Archer, J.; Gibson, D.G.; Palladino, G.; Brito, L.A.; Otten, G.R.; Brazolli, M.; Buccato, S.; Bonci, A.; et al. Rapidly produced SAM((R)) vaccine against H7N9 influenza is immunogenic in mice. Emerg. Microbes Infect. 2013, 2, e52. [Google Scholar] [CrossRef] [PubMed]
- Leventhal, S.S.; Meade-White, K.; Rao, D.; Haddock, E.; Leung, J.; Scott, D.; Archer, J.; Randall, S.; Erasmus, J.H.; Feldmann, H.; et al. Replicating RNA vaccination elicits an unexpected immune response that efficiently protects mice against lethal Crimean-Congo hemorrhagic fever virus challenge. Ebiomedicine 2022, 82, 104188. [Google Scholar] [CrossRef] [PubMed]
- Blakney, A.K.; Ip, S.; Geall, A.J. An Update on Self-Amplifying mRNA Vaccine Development. Vaccines 2021, 9, 97. [Google Scholar] [CrossRef]
- Lundstrom, K. Replicon RNA Viral Vectors as Vaccines. Vaccines 2016, 4, 39. [Google Scholar] [CrossRef] [PubMed]
- Vogel, A.B.; Lambert, L.; Kinnear, E.; Busse, D.; Erbar, S.; Reuter, K.C.; Wicke, L.; Perkovic, M.; Beissert, T.; Hass, H.; et al. Self-Amplifying RNA Vaccines Give Equivalent Protection against Influenza to mRNA Vaccines but at Much Lower Doses. Mol. Ther. 2018, 26, 446–455. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Berglund, P.; Rhodes, G.; Parker, S.E.; Jondal, M.; Liljeström, P. Self-replicating Semliki Forest virus RNA as recombinant vaccine. Vaccine 1994, 12, 1510–1514. [Google Scholar] [CrossRef]
- Brazzoli, M.; Magini, D.; Bonci, A.; Buccato, S.; Giovani, C.; Kratzer, R.; Zurli, V.; Mangiavacchi, S.; Casini, D.; Brito, L.M.; et al. Induction of Broad-Based Immunity and Protective Efficacy by Self-amplifying mRNA Vaccines Encoding Influenza Virus Hemagglutinin. J. Virol. 2016, 90, 332–344. [Google Scholar] [CrossRef] [Green Version]
- Martinon, F.; Krishnan, S.; Lenzen, G.; Magné, R.; Gomard, E.; Guillet, J.G.; Lévy, J.P.; Meulien, P. Induction of virus-specific cytotoxic T lymphocytes in vivo by liposome-entrapped mRNA. Eur. J. Immunol. 1993, 23, 1719–1722. [Google Scholar] [CrossRef]
- Ljungberg, K.; Liljestrom, P. Self-replicating alphavirus RNA vaccines. Expert Rev. Vaccines 2015, 14, 177–194. [Google Scholar] [CrossRef]
- Strauss, J.H.; Strauss, E.G. The alphaviruses: Gene expression, replication, and evolution. Microbiol. Rev. 1994, 58, 491–562. [Google Scholar] [CrossRef]
- Atasheva, S.; Kim, D.Y.; Akhrymuk, M.; Morgan, D.G.; Frolova, E.I.; Frolov, I. Pseudoinfectious Venezuelan Equine Encephalitis Virus: A New Means of Alphavirus Attenuation. J. Virol. 2013, 87, 2023–2035. [Google Scholar] [CrossRef] [Green Version]
- Kowalski, P.S.; Rudra, A.; Miao, L.; Anderson, D.G. Delivering the Messenger: Advances in Technologies for Therapeutic mRNA Delivery. Mol. Ther. 2019, 27, 710–728. [Google Scholar] [CrossRef] [Green Version]
- Mintzer, M.A.; Simanek, E.E. Nonviral Vectors for Gene Delivery. Chem. Rev. 2009, 109, 259–302. [Google Scholar] [CrossRef]
- Martin, M.E.; Rice, K.G. Peptide-guided gene delivery. AAPS J. 2007, 9, E18–E29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pack, D.W.; Hoffman, A.S.; Pun, S.; Stayton, P.S. Design and development of polymers for gene delivery. Nat. Rev. Drug Discov. 2005, 4, 581–593. [Google Scholar] [CrossRef] [PubMed]
- Malone, R.W.; Felgner, P.L.; Verma, I.M. Cationic liposome-mediated RNA transfection. Proc. Natl. Acad. Sci. USA 1989, 86, 6077–6081. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Jong, S.; Chikh, G.; Sekirov, L.; Raney, S.; Semple, S.; Klimuk, S.; Yuan, N.; Hope, M.; Cullis, P.; Tam, Y. Encapsulation in liposomal nanoparticles enhances the immunostimulatory, adjuvant and anti-tumor activity of subcutaneously administered CpG ODN. Cancer Immunol. Immunother. 2007, 56, 1251–1264. [Google Scholar] [CrossRef]
- Reichmuth, A.M.; Oberli, M.A.; Jaklenec, A.; Langer, R.; Blankschtein, D. mRNA vaccine delivery using lipid nanoparticles. Ther. Deliv. 2016, 7, 319–334. [Google Scholar] [CrossRef] [Green Version]
- Baden, L.R.; El Sahly, H.M.; Essink, B.; Kotloff, K.; Frey, S.; Novak, R.; Diemert, D.; Spector, S.A.; Rouphael, N.; Creech, C.B.; et al. Efficacy and Safety of the mRNA-1273 SARS-CoV-2 Vaccine. N. Engl. J. Med. 2021, 384, 403–416. [Google Scholar] [CrossRef]
- Greenhawt, M.; Abrams, E.M.; Shaker, M.; Chu, D.K.; Khan, D.; Akin, C.; Alqurashi, W.; Arkwright, P.; Baldwin, J.L.; Ben-Shoshan, M.; et al. The Risk of Allergic Reaction to SARS-CoV-2 Vaccines and Recommended Evaluation and Management: A Systematic Review, Meta-Analysis, GRADE Assessment, and International Consensus Approach. J. Allergy Clin. Immunol. Pract. 2021, 9, 3546–3567. [Google Scholar] [CrossRef]
- Mahdiabadi, S.; Rezaei, N. Anaphylaxis and allergic reactions to COVID-19 vaccines: A narrative review of characteristics and potential obstacles on achieving herd immunity. Health Sci. Rep. 2022, 5, e787. [Google Scholar] [CrossRef]
- Li, M.; Wang, X.; Feng, J.; Feng, Z.; Li, W.; Ya, B. Myocarditis or Pericarditis Following the COVID-19 Vaccination in Adolescents: A Systematic Review. Vaccines 2022, 10, 1316. [Google Scholar] [CrossRef]
- Massari, M.; Spila-Alegiani, S.; Morciano, C.; Spuri, M.; Marchione, P.; Felicetti, P.; Belleudi, C.; Poggi, F.R.; Lazzeretti, M.; Ercolanoni, M.; et al. Postmarketing active surveillance of myocarditis and pericarditis following vaccination with COVID-19 mRNA vaccines in persons aged 12 to 39 years in Italy: A multi-database, self-controlled case series study. PLoS Med. 2022, 19, e1004056. [Google Scholar] [CrossRef]
- Wong, H.L.; Hu, M.; Zhou, C.K.; Lloyd, P.C.; Amend, K.L.; Beachler, D.C.; Secora, A.; McMahill-Walraven, C.N.; Lu, Y.; Wu, Y.; et al. Risk of myocarditis and pericarditis after the COVID-19 mRNA vaccination in the USA: A cohort study in claims databases. Lancet 2022, 399, 2191–2199. [Google Scholar] [CrossRef]
- Erasmus, J.H.; Khandhar, A.P.; O’Connor, M.A.; Walls, A.C.; Hemann, E.A.; Murapa, P.; Archer, J.; Leventhal, S.; Fuller, J.T.; Lewis, T.B.; et al. An Alphavirus-derived replicon RNA vaccine induces SARS-CoV-2 neutralizing antibody and T cell responses in mice and nonhuman primates. Sci. Transl. Med. 2020, 12, eabc9396. [Google Scholar] [CrossRef] [PubMed]
- Duthie, M.S.; Van Hoeven, N.; MacMillen, Z.; Picone, A.; Mohamath, R.; Erasmus, J.; Hsu, F.-C.; Stinchcomb, D.T.; Reed, S.G. Heterologous Immunization with Defined RNA and Subunit Vaccines Enhances T Cell Responses That Protect against Leishmania donovani. Front. Immunol. 2018, 9, 2420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shah, S.K.; Miller, F.G.; Darton, T.C.; Duenas, D.; Emerson, C.; Lynch, H.F.; Jamrozik, E.; Jecker, N.S.; Kamuya, D.; Kapulu, M.; et al. Ethics of controlled human infection to address COVID-19. Science 2020, 368, 832–834. [Google Scholar] [CrossRef]
- Eyal, N.; Lipsitch, M.; Smith, P.G. Human challenge studies to accelerate coronavirus vaccine licensure. J. Infect. Dis. 2020, 221, 1752–1756. [Google Scholar] [CrossRef] [Green Version]
- Langenberg, M.C.; Hoogerwerf, M.A.; Koopman, J.P.R.; Janse, J.J.; Kos-van Oosterhoud, J.; Feijt, C.; Jochems, S.P.; De Dood, C.J.; van Schuijlenburg, R.; Ozir-Fazalalikhan, A.; et al. A controlled human Schistosoma mansoni infection model to advance novel drugs, vaccines and diagnostics. Nat. Med. 2020, 26, 326–332. [Google Scholar] [CrossRef]
- Kirkpatrick, B.D.; Whitehead, S.S.; Pierce, K.K.; Tibery, C.M.; Grier, P.L.; Hynes, N.A.; Larsson, C.J.; Saundayo, B.P.; Talaat, K.R.; Janiak, A.; et al. The live attenuated dengue vaccine TV003 elicits complete protection against dengue in a human challenge model. Sci. Transl. Med. 2016, 8, 330ra36. [Google Scholar] [CrossRef] [Green Version]
- Gould, V.M.; Francis, J.N.; Anderson, K.J.; Georges, B.; Cope, A.V.; Tregoning, J.S. Nasal IgA Provides Protection against Human Influenza Challenge in Volunteers with Low Serum Influenza Antibody Titre. Front. Microbiol. 2017, 8, 900. [Google Scholar] [CrossRef] [Green Version]
- Payne, R.O.; Griffin, P.M.; McCarthy, J.S.; Draper, S.J. Plasmodium vivax Controlled Human Malaria Infection—Progress and Prospects. Trends Parasitol. 2017, 33, 141–150. [Google Scholar] [CrossRef] [Green Version]
- Roestenberg, M.; Hoogerwerf, M.-A.; Ferreira, D.M.; Mordmüller, B.; Yazdanbakhsh, M. Experimental infection of human volunteers. Lancet Infect. Dis. 2018, 18, e312–e322. [Google Scholar] [CrossRef]
- Sheehy, S.H.; Duncan, C.J.; Elias, S.C.; Choudhary, P.; Biswas, S.; Halstead, F.D.; Collins, K.A.; Edwards, N.J.; Douglas, A.D.; Anagnostou, N.A.; et al. ChAd63-MVA–vectored Blood-stage Malaria Vaccines Targeting MSP1 and AMA1: Assessment of Efficacy against Mosquito Bite Challenge in Humans. Mol. Ther. 2012, 20, 2355–2368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parkash, V.; Kaye, P.M.; Layton, A.M.; Lacey, C.J. Vaccines against leishmaniasis: Using controlled human infection models to accelerate development. Expert Rev. Vaccines 2012, 20, 1407–1418. [Google Scholar] [CrossRef] [PubMed]
- Melby, P.C. Experimental Leishmaniasis in Humans: Review. Clin. Infect. Dis. 1991, 13, 1009–1017. [Google Scholar] [CrossRef] [PubMed]
- Ashwin, H.; Sadlova, J.; Vojtkova, B.; Becvar, T.; Lypaczewski, P.; Schwartz, E.; Greensted, E.; Van Bocxlaer, K.; Pasin, M.; Lipinski, K.S.; et al. Characterization of a new Leishmania major strain for use in a controlled human infection model. Nat. Commun. 2021, 12, 215. [Google Scholar] [CrossRef]
- Parkash, V.; Ashwin, H.; Sadlova, J.; Vojtkova, B.; Jones, G.; Martin, N.; Greensted, E.; Allgar, V.; Kamhawi, S.; Valenzuela, J.G.; et al. A clinical study to optimise a sand fly biting protocol for use in a controlled human infection model of cutaneous leishmaniasis (the FLYBITE study). Wellcome Open Res. 2021, 6, 168. [Google Scholar] [CrossRef]
- Parkash, V.; Jones, G.; Martin, N.; Steigmann, M.; Greensted, E.; Kaye, P.; Layton, A.M.; Lacey, C.J. Assessing public perception of a sand fly biting study on the pathway to a controlled human infection model for cutaneous leishmaniasis. Res. Involv. Engagem. 2021, 7, 33. [Google Scholar] [CrossRef]
- Giersing, B.K.; Porter, C.K.; Kotloff, K.; Neels, P.; Cravioto, A.; MacLennan, C.A. How can controlled human infection models accelerate clinical development and policy pathways for vaccines against Shigella? Vaccine 2019, 37, 4778–4783. [Google Scholar] [CrossRef]
- Viana, K.F.; Lacerda, G.; Teixeira, N.S.; Cangussu, A.S.R.; Aguiar, R.W.S.; Giunchetti, R.C. Therapeutic vaccine of killed Leishmania amazonensis plus saponin reduced parasite burden in dogs naturally infected with Leishmania infantum. Vet. Parasitol. 2018, 254, 98–104. [Google Scholar] [CrossRef]
- Toepp, A.; Larson, M.; Wilson, G.; Grinnage-Pulley, T.; Bennett, C.; Leal-Lima, A.; Anderson, B.; Parrish, M.; Anderson, M.; Fowler, H. Randomized, controlled, double-blinded field trial to assess Leishmania vaccine effectiveness as immunotherapy for canine leishmaniosis. Vaccine 2018, 36, 6433–6441. [Google Scholar] [CrossRef]
- Roatt, B.M.; Aguiar-Soares, R.D.D.O.; Reis, L.E.S.; Cardoso, J.M.D.O.; Mathias, F.A.S.; Brito, R.C.F.D.; da Silva, S.M.; De Figueiredo Gontijo, N.; Ferreira, S.; Valenzuela, J.G.; et al. A Vaccine Therapy for Canine Visceral Leishmaniasis Promoted Significant Improvement of Clinical and Immune Status with Reduction in Parasite Burden. Front. Immunol. 2017, 8, 217. [Google Scholar] [CrossRef] [Green Version]
- Gradoni, L.; Manzillo, V.F.; Pagano, A.; Piantedosi, D.; De Luna, R.; Gramiccia, M.; Scalone, A.; Di Muccio, T.; Oliva, G. Failure of a multi-subunit recombinant leishmanial vaccine (MML) to protect dogs from Leishmania infantum infection and to prevent disease progression in infected animals. Vaccine 2005, 23, 5245–5251. [Google Scholar] [CrossRef] [PubMed]
- Miret, J.; Nascimento, E.; Sampaio, W.; França, J.C.; Fujiwara, R.T.; Vale, A.; Dias, E.S.; Vieira, E.; Costa, R.T.; Mayrink, W.; et al. Evaluation of an immunochemotherapeutic protocol constituted of N-methyl meglumine antimoniate (Glucantime) and the recombinant Leish-110f + MPL-SE vaccine to treat canine visceral leishmaniasis. Vaccine 2008, 26, 1585–1594. [Google Scholar] [CrossRef] [PubMed]
- Moreno, J.; Nieto, J.; Masina, S.; Cañavate, C.; Cruz, I.; Chicharro, C.; Carrillo, E.; Napp, S.; Reymond, C.; Kaye, P.M.; et al. Immunization with H1, HASPB1 and MML Leishmania proteins in a vaccine trial against experimental canine leishmaniasis. Vaccine 2007, 25, 5290–5300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esch, K.J.; Juelsgaard, R.; Martinez, P.A.; Jones, D.E.; Petersen, C.A. Programmed death 1-mediated T cell exhaustion during visceral leishmaniasis impairs phagocyte function. J. Immunol. 2013, 191, 5542–5550. [Google Scholar] [CrossRef] [Green Version]
- Schaut, R.G.; Grinnage-Pulley, T.L.; Esch, K.J.; Toepp, A.J.; Duthie, M.S.; Howard, R.F.; Reed, S.G.; Petersen, C.A. Recovery of antigen-specific T cell responses from dogs infected with Leishmania (L.) infantum by use of vaccine associated TLR-agonist adjuvant. Vaccine 2016, 34, 5225–5234. [Google Scholar] [CrossRef] [Green Version]
- Nascimento, L.F.; Miranda DF, H.; Moura, L.D.; Pinho, F.A.; Werneck, G.L.; Khouri, R.; Reed, S.G.; Duthie, M.S.; Barral, A.; Barral-Netto, M.; et al. Allopurinol therapy provides long term clinical improvement, but additional immunotherapy is required for sustained parasite clearance, in L. infantum-infected dogs. Vaccine X 2020, 4, 100048. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Duthie, M.S.; Machado, B.A.S.; Badaró, R.; Kaye, P.M.; Reed, S.G. Leishmaniasis Vaccines: Applications of RNA Technology and Targeted Clinical Trial Designs. Pathogens 2022, 11, 1259. https://doi.org/10.3390/pathogens11111259
Duthie MS, Machado BAS, Badaró R, Kaye PM, Reed SG. Leishmaniasis Vaccines: Applications of RNA Technology and Targeted Clinical Trial Designs. Pathogens. 2022; 11(11):1259. https://doi.org/10.3390/pathogens11111259
Chicago/Turabian StyleDuthie, Malcolm S., Bruna A. S. Machado, Roberto Badaró, Paul M. Kaye, and Steven G. Reed. 2022. "Leishmaniasis Vaccines: Applications of RNA Technology and Targeted Clinical Trial Designs" Pathogens 11, no. 11: 1259. https://doi.org/10.3390/pathogens11111259
APA StyleDuthie, M. S., Machado, B. A. S., Badaró, R., Kaye, P. M., & Reed, S. G. (2022). Leishmaniasis Vaccines: Applications of RNA Technology and Targeted Clinical Trial Designs. Pathogens, 11(11), 1259. https://doi.org/10.3390/pathogens11111259