
Cortactin Promotes Effective AGS Cell Scattering by Helicobacter pylori CagA, but Not Cellular Vacuolization and Apoptosis Induced by the Vacuolating Cytotoxin VacA

 ,
, 
 and
and 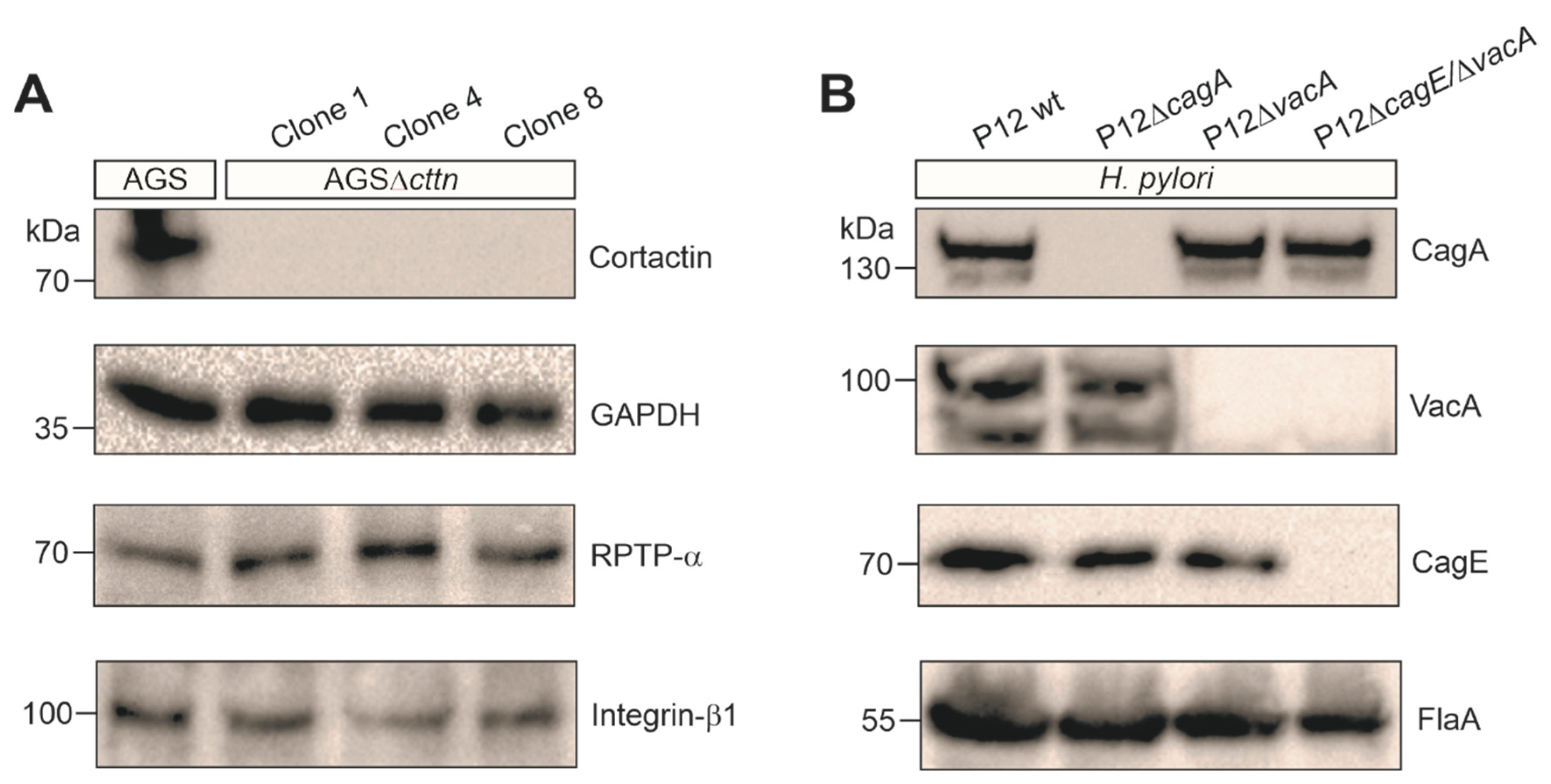{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
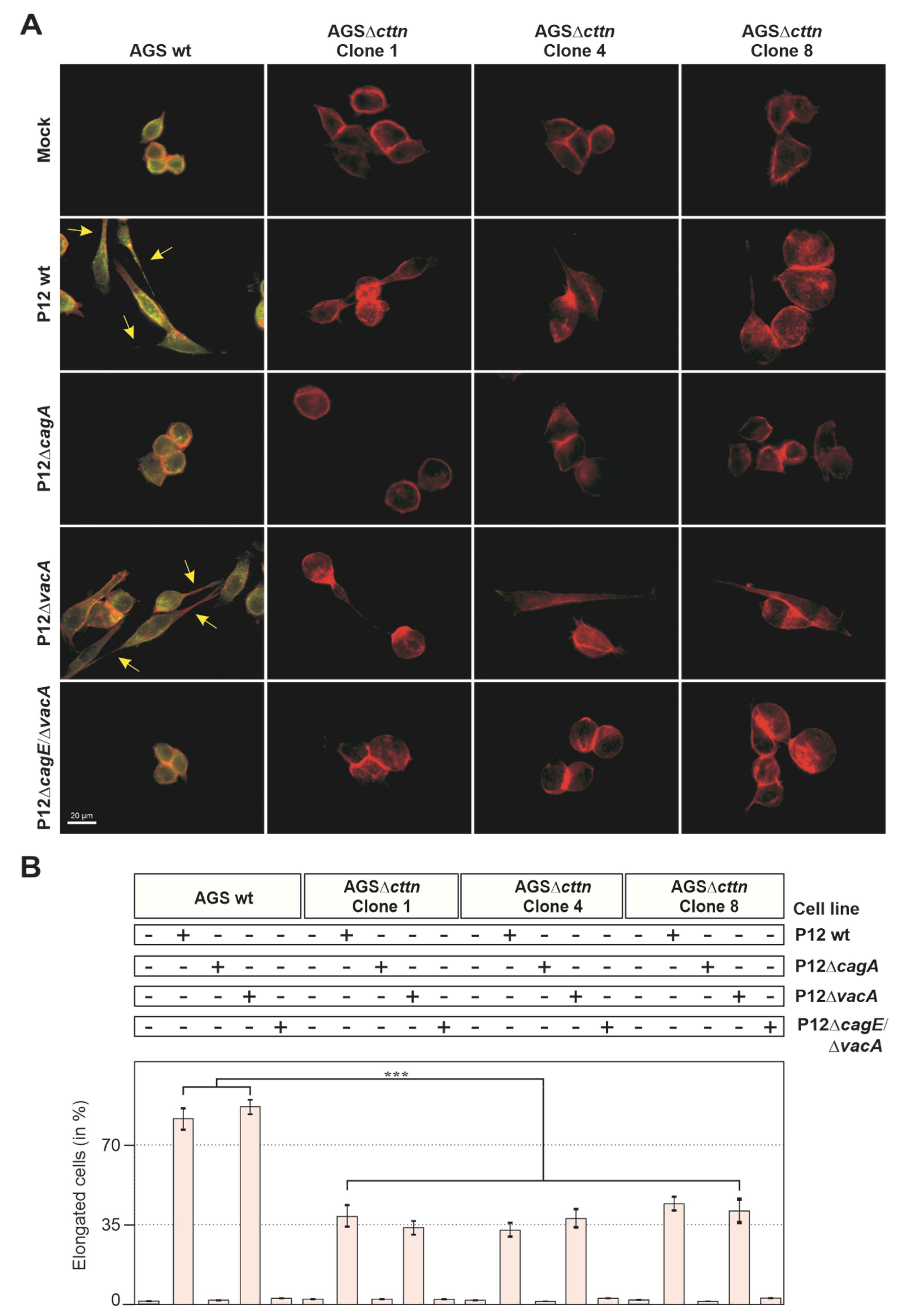
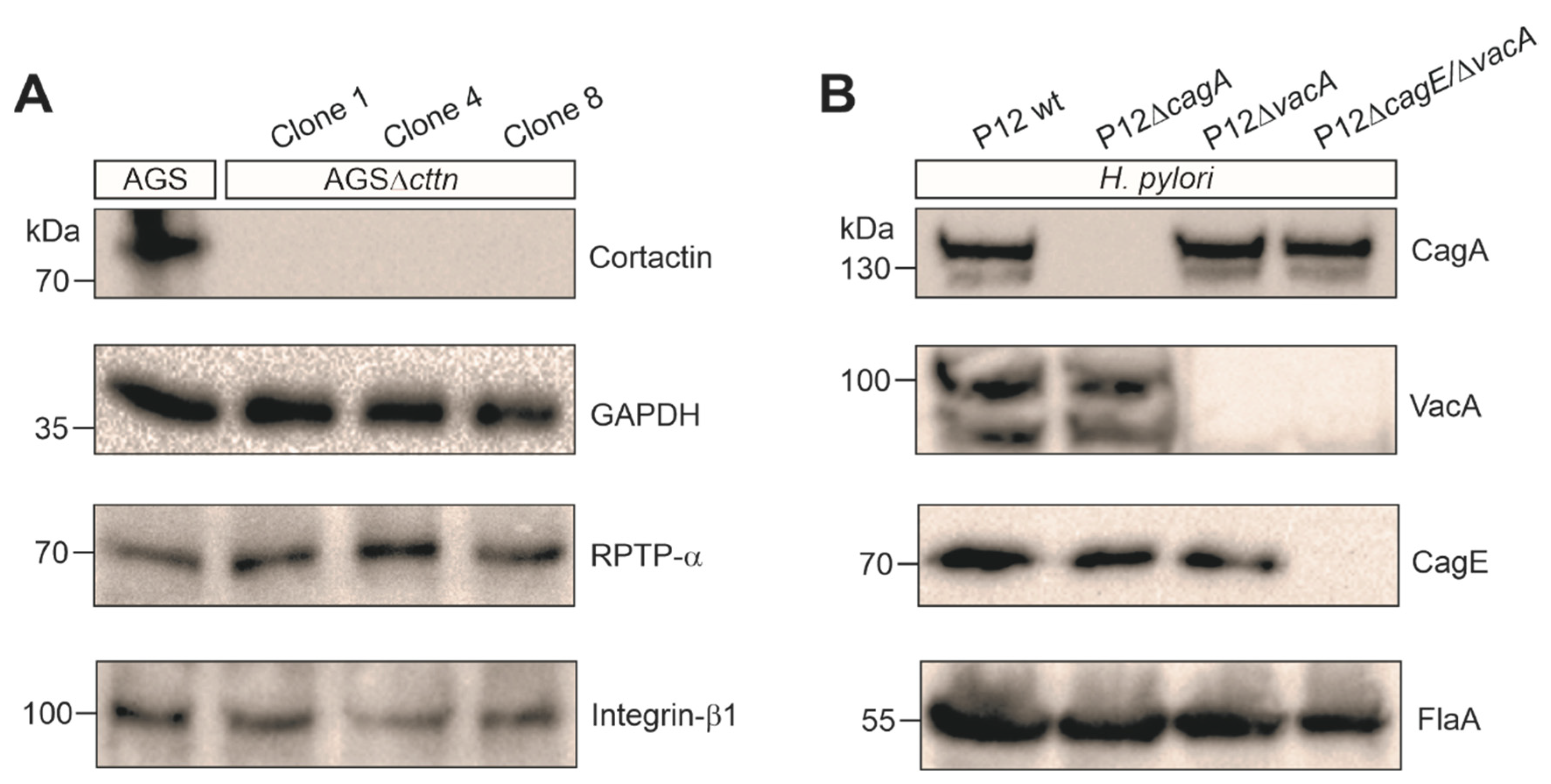
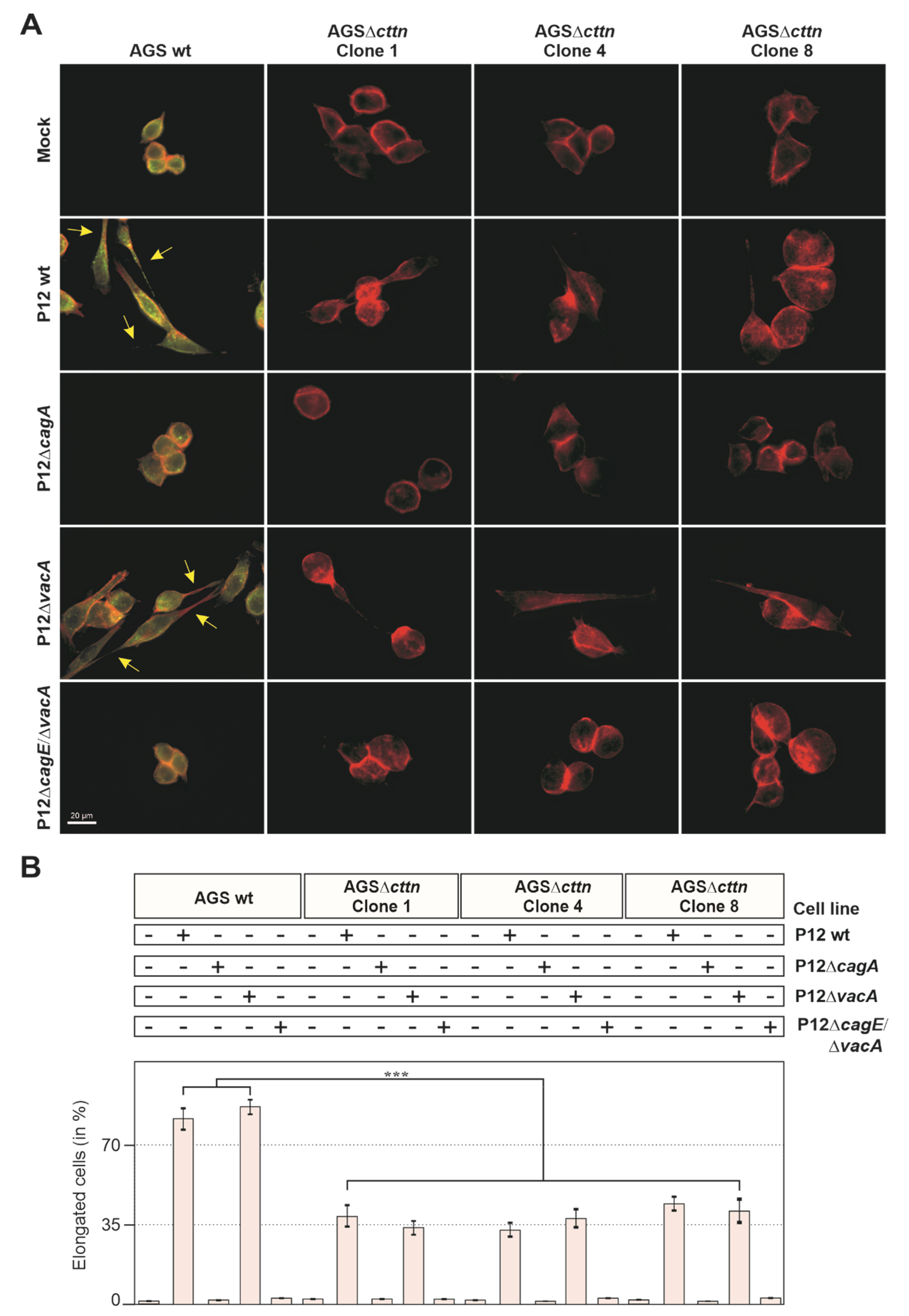
2.1. Cortactin Knockout Is Associated with Diminished AGS Cell Scattering and Elongation Phenotype
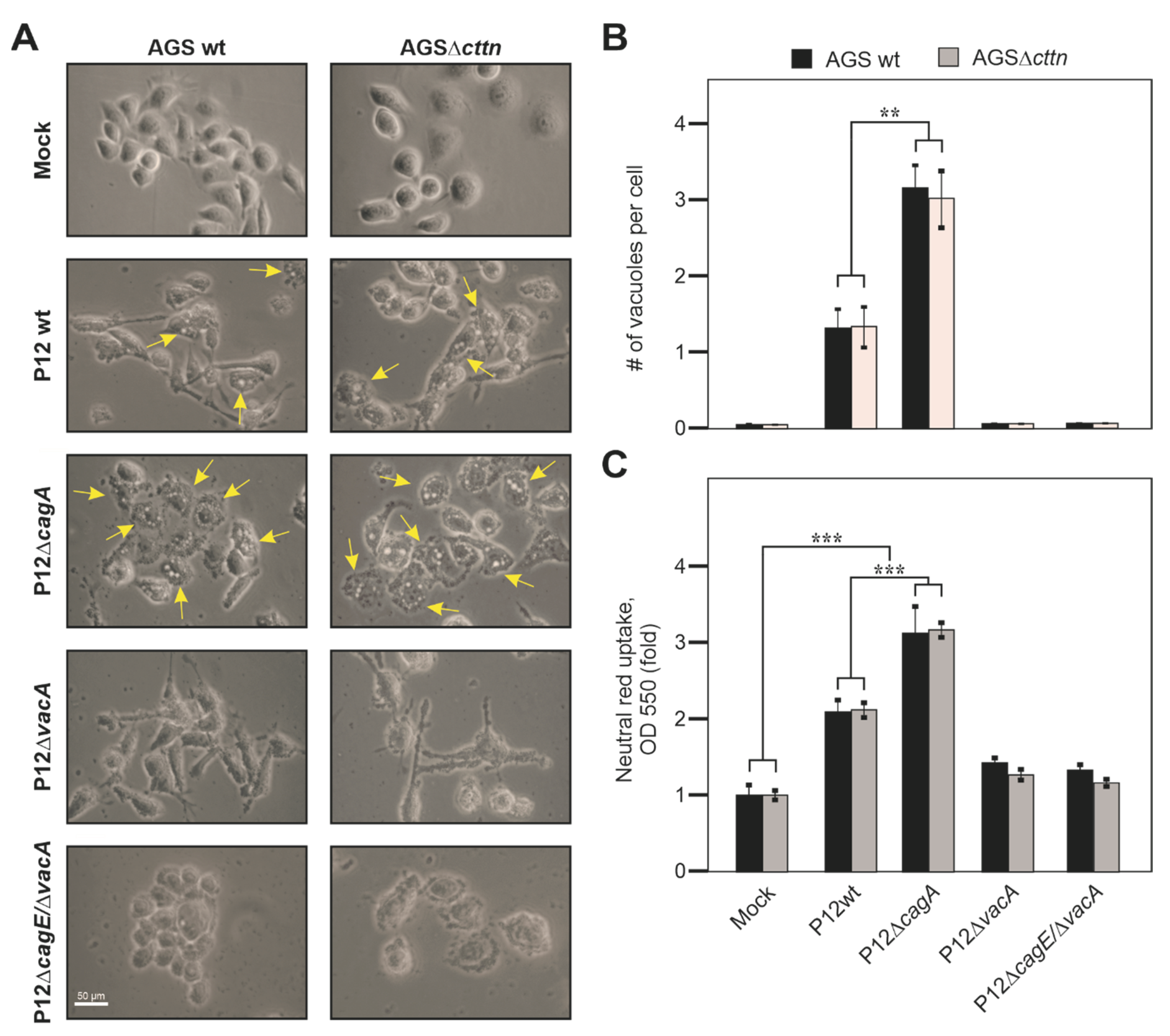
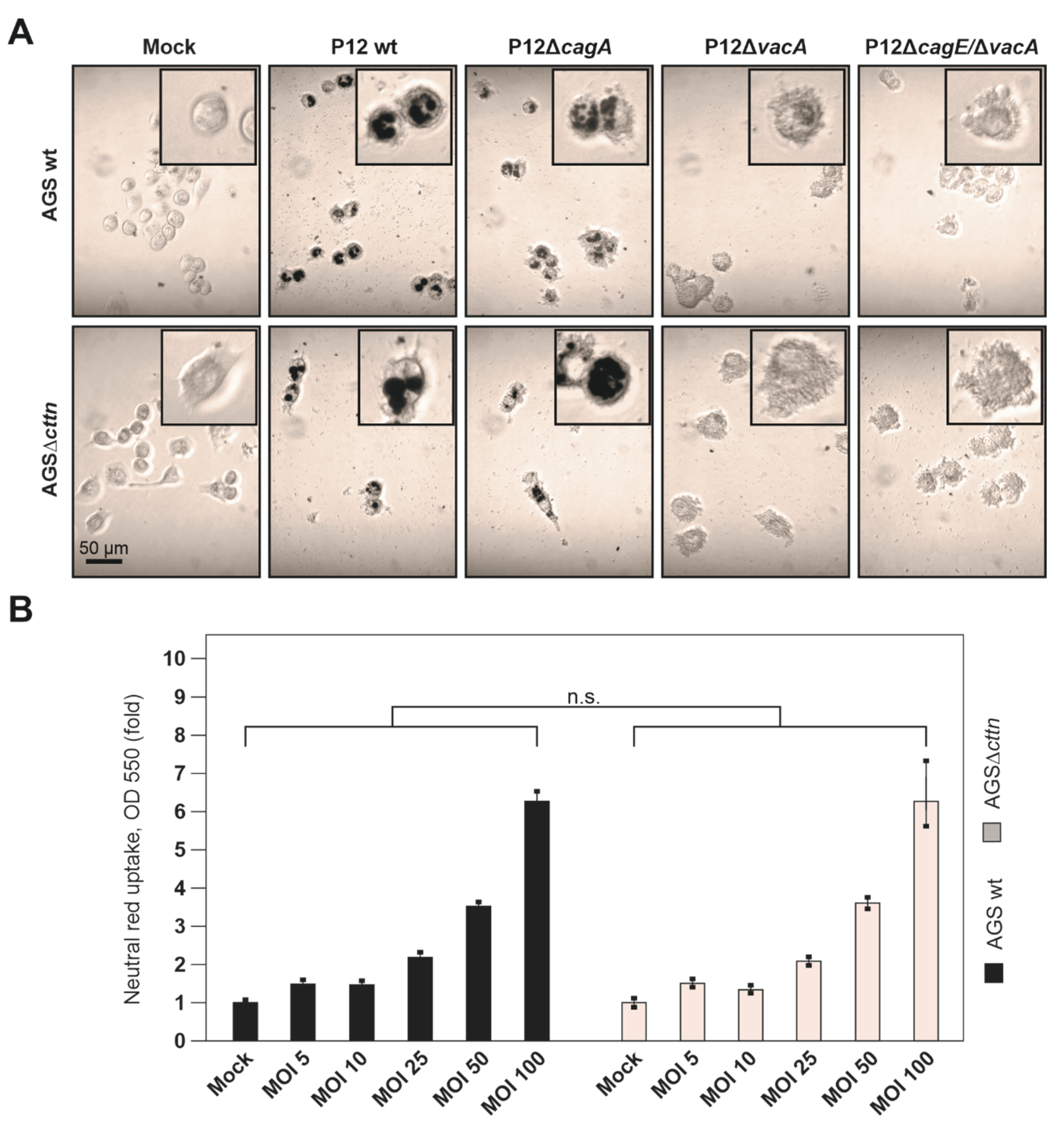
2.2. VacA-Dependent Vacuole Formation Is Not Hampered in Infected AGSΔcttn Cells
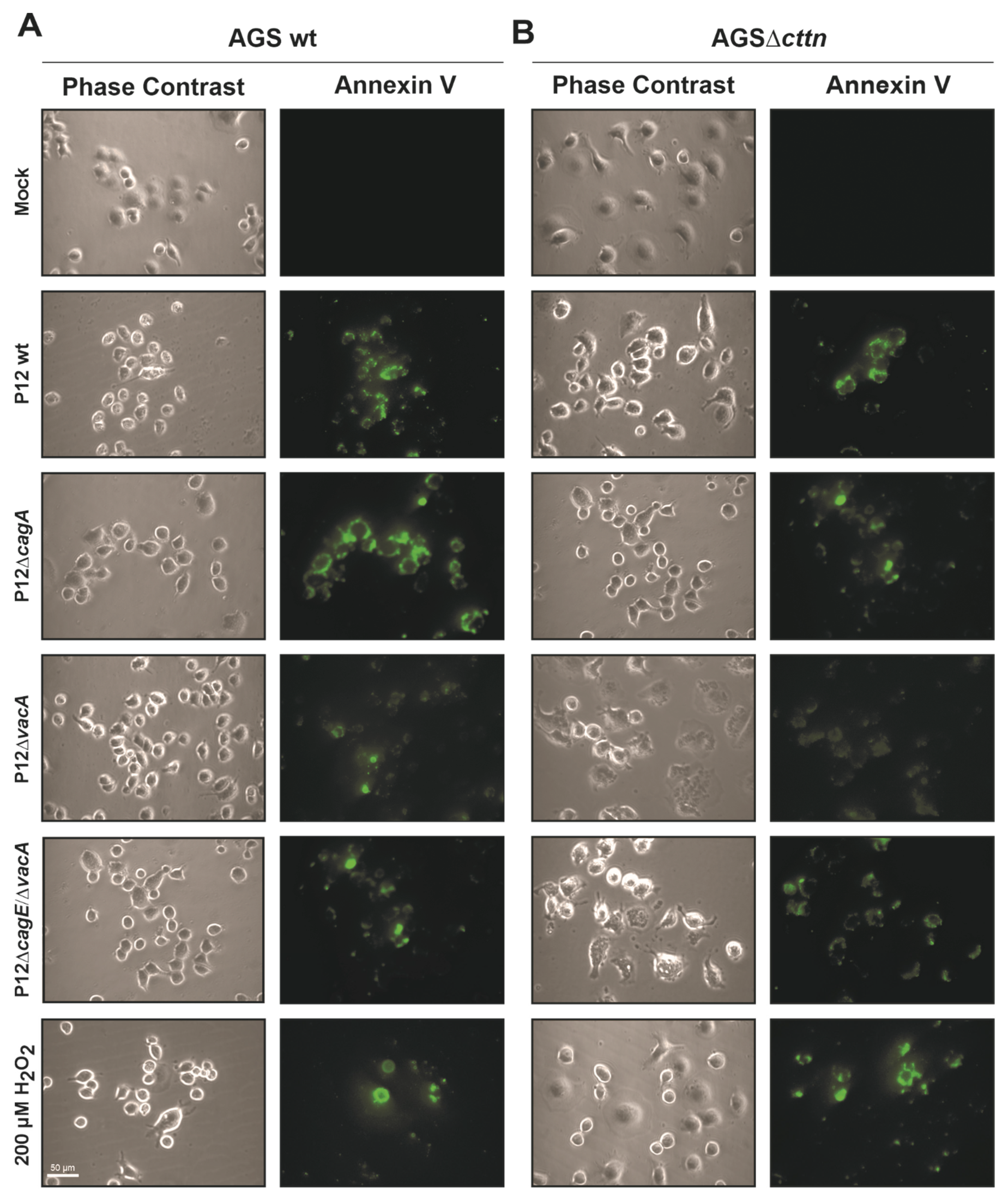
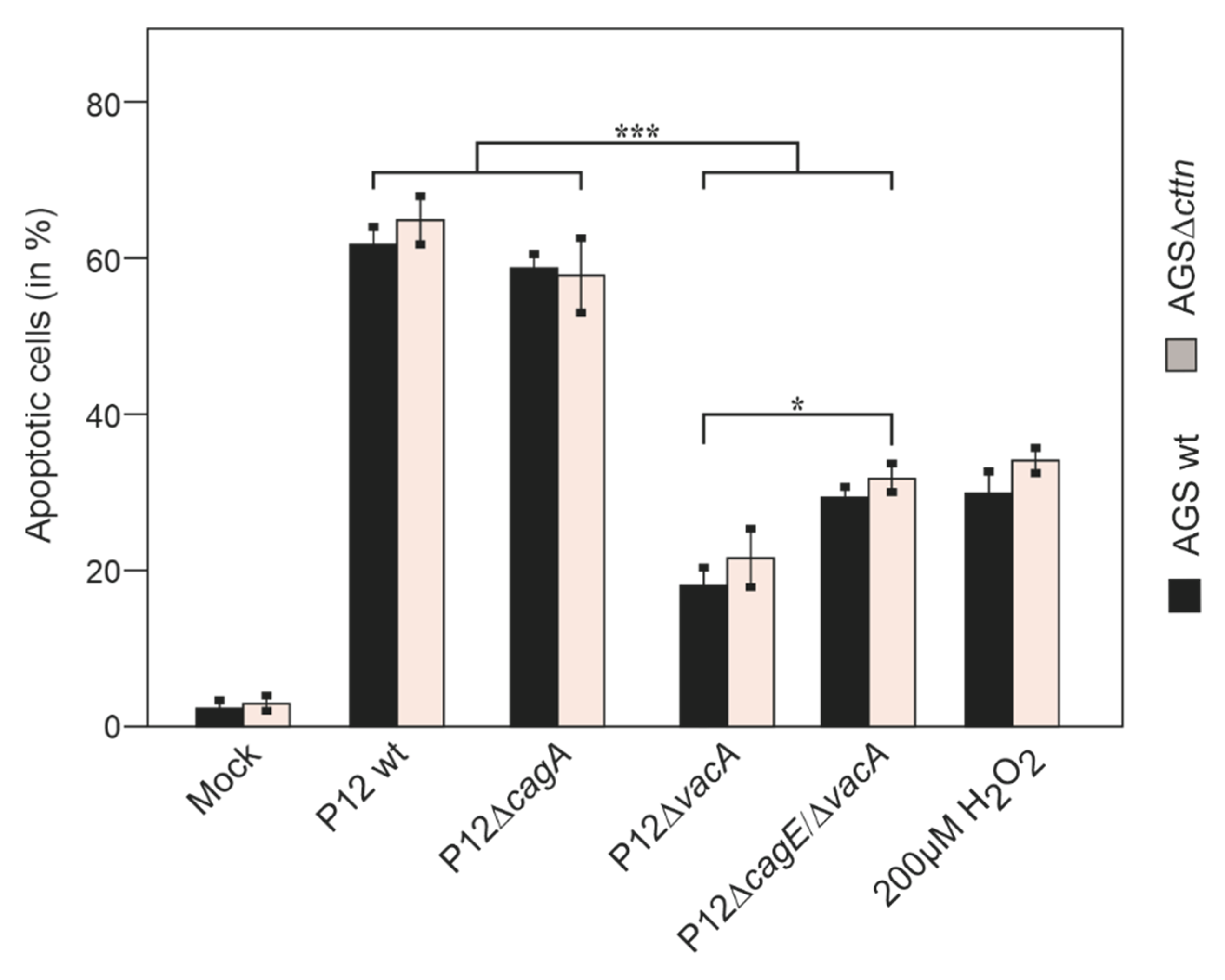
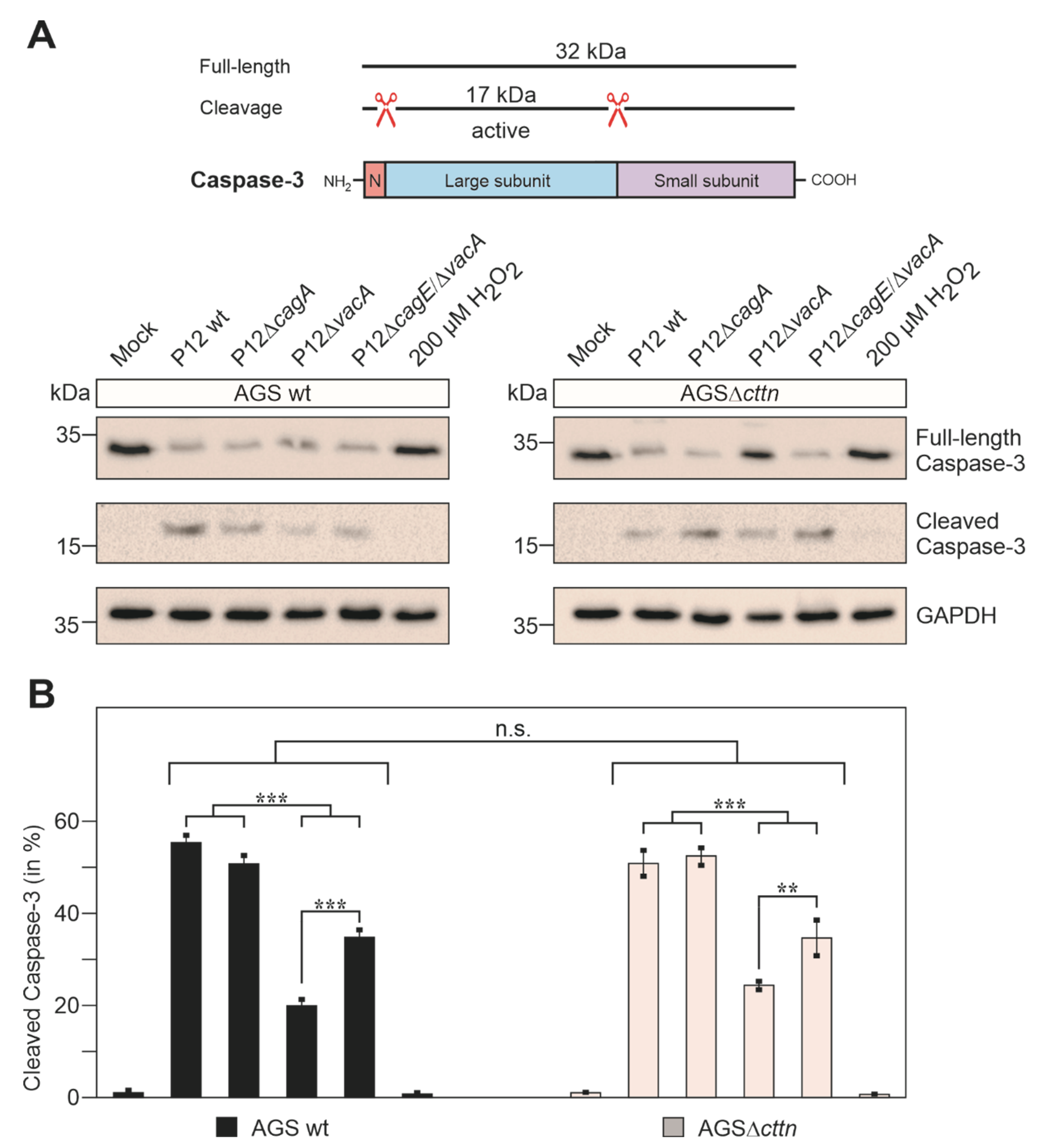
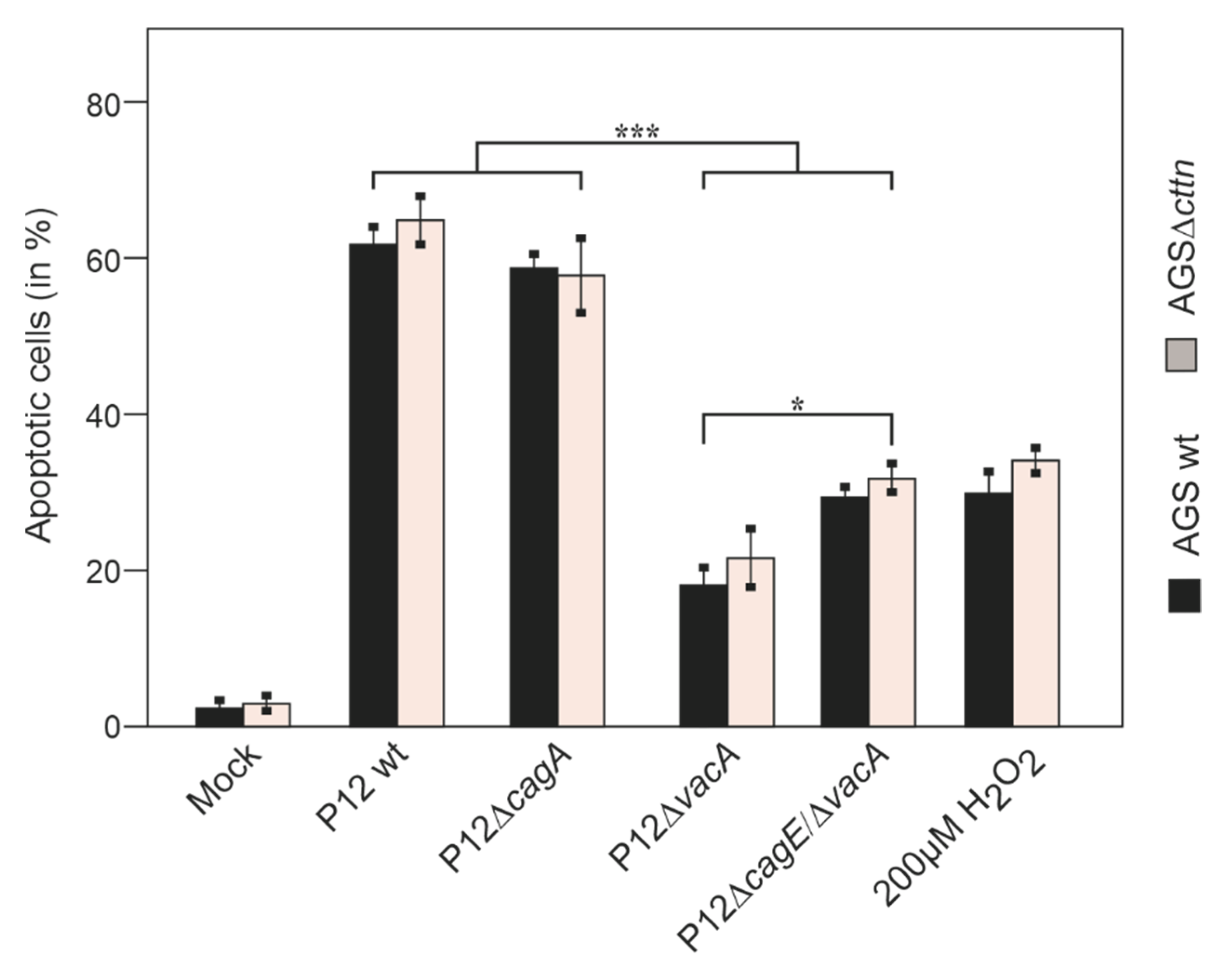
2.3. Cortactin Deficiency Does Not Impact Induction of Apoptosis Provoked by H. pylori
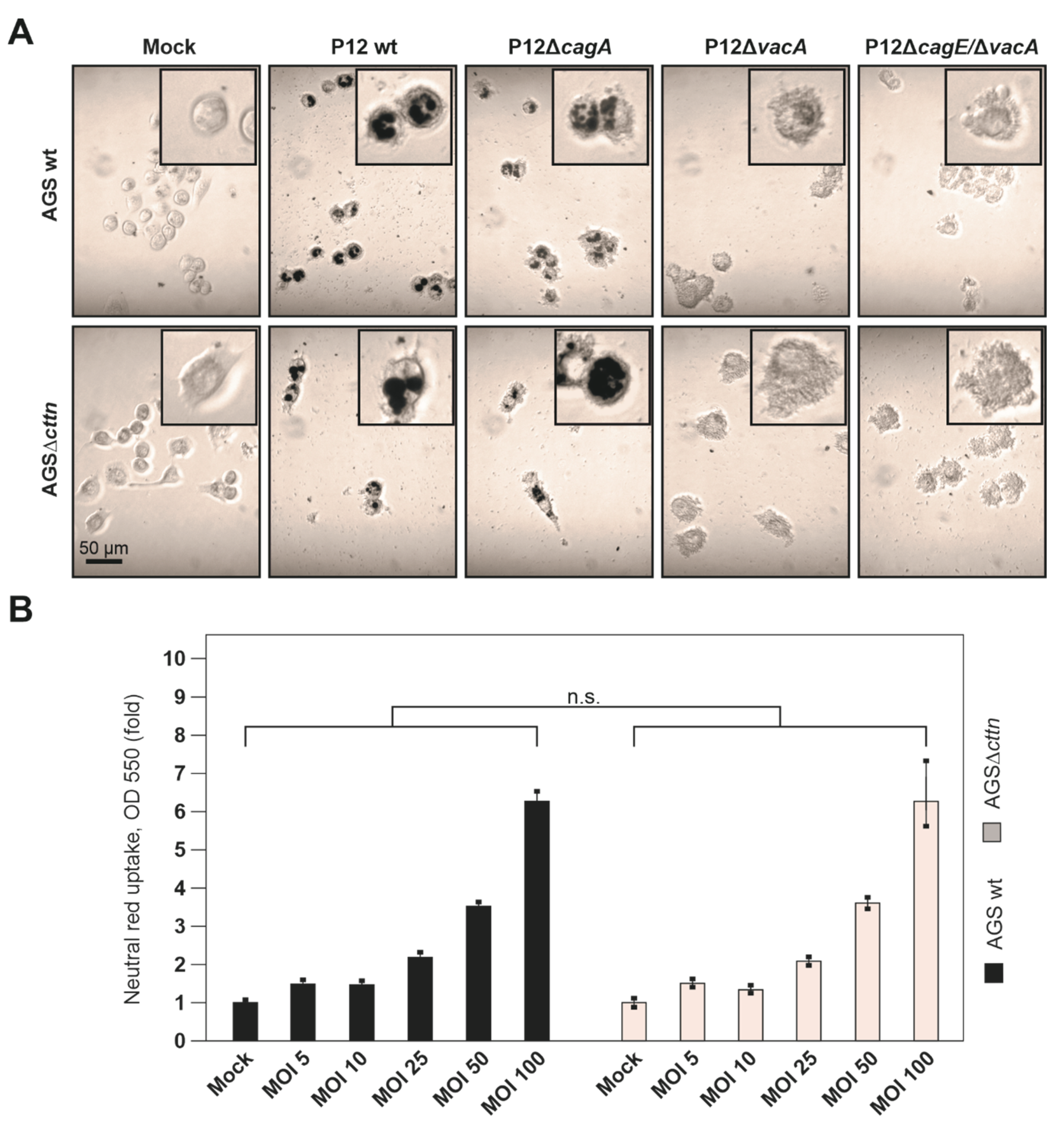
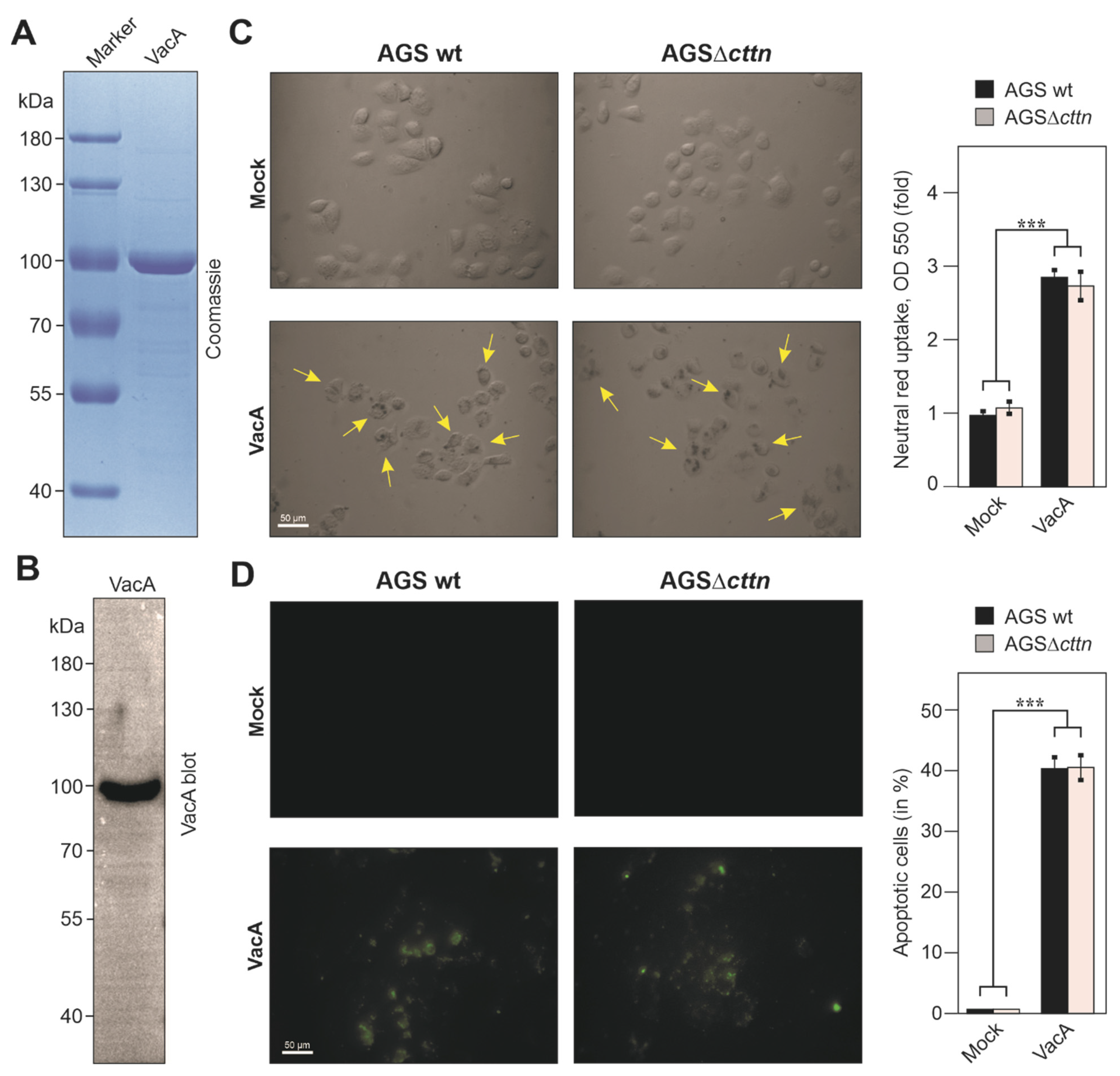
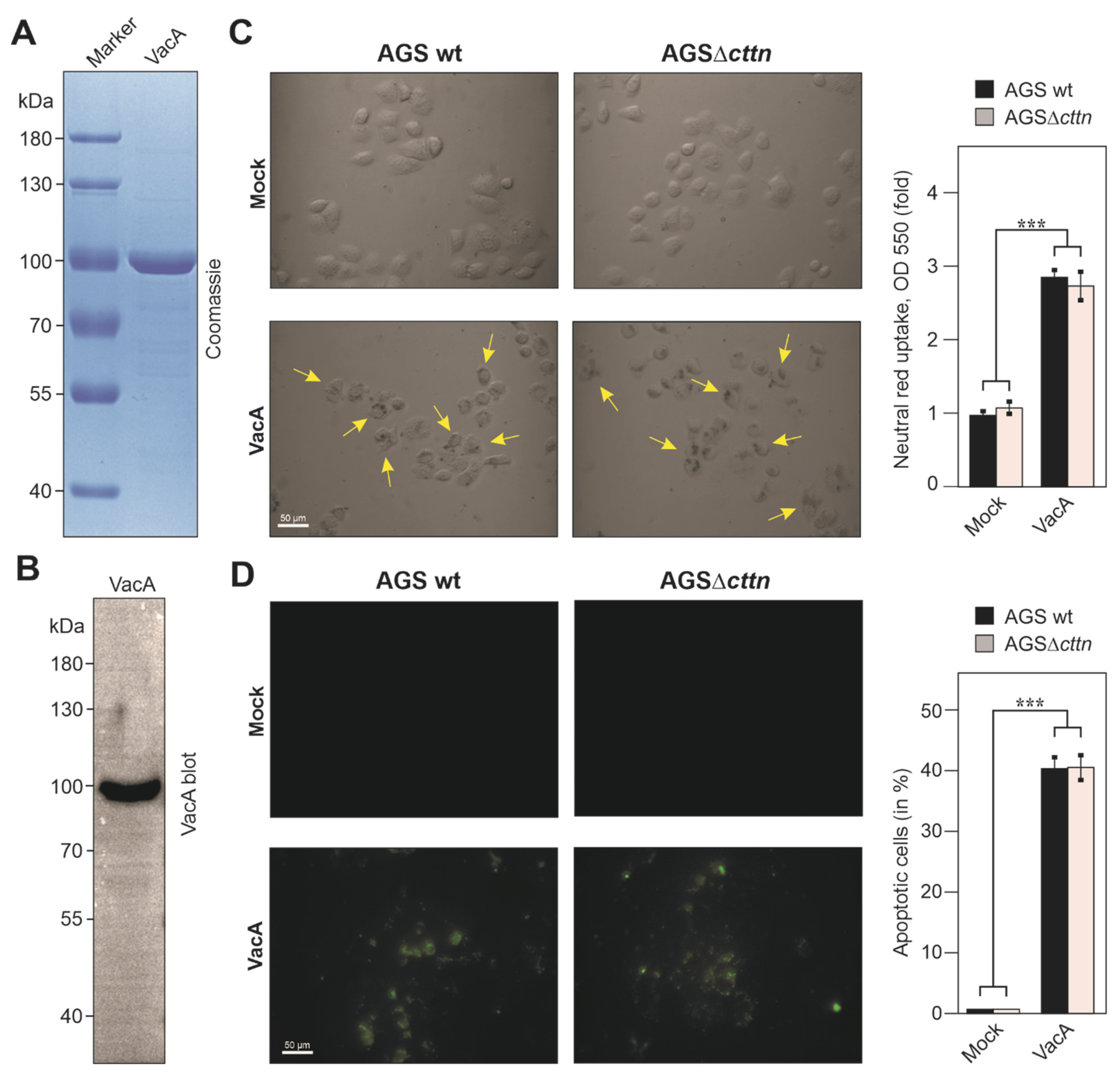
2.4. Vacuole Formation and Apoptosis by Purified VacA Is Not Affected in AGSΔcttn Cells
3. Discussion
4. Material and Methods
4.1. Cultivation of Eukaryotic Cells
4.2. Cultivation of H. pylori Strains
4.3. Infection of Eukaryotic Cells with H. pylori and Quantification of the Elongation Phenotype
4.4. SDS-PAGE and Immunoblot Analysis
4.5. Light and Fluorescence Microscopy
4.6. Annexin V Apoptosis Assay
4.7. Quantification of Vacuolating Activity
4.8. Neutral Red Staining
4.9. Live Cell Imaging
4.10. In Vitro VacA Preparation and Acid-Activation
4.11. Statistics
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hooi, J.K.Y.; Lai, W.Y.; Ng, W.K.; Suen, M.M.Y.; Underwood, F.E.; Tanyingoh, D.; Malfertheiner, P.; Graham, D.Y.; Wong, V.W.S.; Wu, J.C.Y.; et al. Global Prevalence of Helicobacter pylori Infection: Systematic Review and Meta-Analysis. Gastroenterology 2017, 153, 420–429. [Google Scholar] [CrossRef] [Green Version]
- Parsonnet, J.; Friedman, G.D.; Vandersteen, D.P.; Chang, Y.; Vogelman, J.H.; Orentreich, N.; Sibley, R.K. Helicobacter pylori infection and the risk of gastric carcinoma. N. Engl. J. Med. 1991, 325, 1127–1131. [Google Scholar] [CrossRef]
- Peek, R.M.; Blaser, M.J. Helicobacter pylori and gastrointestinal tract adenocarcinomas. Nat. Rev. Cancer 2002, 2, 28–37. [Google Scholar] [CrossRef]
- Cover, T.L.; Blanke, S.R. Helicobacter pylori VacA, a paradigm for toxin multifunctionality. Nat. Rev. Microbiol. 2005, 3, 320–332. [Google Scholar] [CrossRef]
- Yamaoka, Y. Mechanisms of disease: Helicobacter pylori virulence factors. Nat. Rev. Gastroenterol. Hepatol. 2010, 7, 629–641. [Google Scholar] [CrossRef] [Green Version]
- International Agency for Research on Cancer. Schistosomes, Liver Flukes and Helicobacter pylori; IARC Working Group on the Evaluation of Carcinogenic Risks to Humans, IARC: Lyon, Spain, 1994; Volume 61, 241p. [Google Scholar]
- Mobley, H.L.T. The role of Helicobacter pylori urease in the pathogenesis of gastritis and peptic ulceration. Aliment. Pharmacol. Ther. 1996, 10, 57–64. [Google Scholar] [CrossRef]
- Schreiber, S.; Konradt, M.; Groll, C.; Scheid, P.; Hanauer, G.; Werling, H.O.; Josenhans, C.; Suerbaum, S. The spatial orientation of H. pylori in the gastric mucus. Proc. Natl. Acad. Sci. USA 2004, 101, 5024–5029. [Google Scholar] [CrossRef] [Green Version]
- Huang, J.Y.; Sweeney, E.G.; Guillemin, K.; Amieva, M.R. Multiple Acid Sensors Control Helicobacter pylori Colonization of the Stomach. PLoS Pathog. 2017, 13, e1006118. [Google Scholar] [CrossRef] [Green Version]
- Odenbreit, S. Adherence properties of Helicobacter pylori: Impact on pathogenesis and adaptation to the host. Int. Med. Microbiol. 2005, 295, 317–324. [Google Scholar] [CrossRef]
- Aspholm, M.; Kalia, A.; Ruhl, S.; Schedin, S.; Arnqvist, A.; Linden, S.; Sjostrom, R.; Gerhard, M.; Semino-Mora, C.; Dubois, A.; et al. Helicobacter pylori adhesion to carbohydrates. In Methods in Enzymology; Elsevier: San Diego, CA, USA, 2006; Volume 417, pp. 293–339. [Google Scholar]
- Moonens, K.; Hamway, Y.; Neddermann, M.; Reschke, M.; Tegtmeyer, N.; Kruse, T.; Kammerer, R.; Mejías- Luque, R.; Singer, B.B.; Backert, S.; et al. Helicobacter pylori adhesin HopQ disrupts trans dimerization in human CEACAMs. EMBO J. 2018, 37, e98665. [Google Scholar] [CrossRef]
- Schmidt, T.P.; Perna, A.M.; Fugmann, T.; Böhm, M.; Hiss, J.; Hallerm, S.; Götz, C.; Tegtmeyer, N.; Hoy, B.; Rau, T.T.; et al. Identification of E-cadherin signature motifs functioning as cleavage sites for Helicobacter pylori HtrA. Sci. Rep. 2016, 6, 23264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tegtmeyer, N.; Moodley, Y.; Yamaoka, Y.; Pernitzsch, S.R.; Schmidt, V.; Traverso, F.R.; Schmidt, T.P.; Rad, R.; Yeoh, K.G.; Bow, H.; et al. Characterisation of worldwide Helicobacter pylori strains reveals genetic conservation and essentiality of serine protease HtrA. Mol. Microbiol. 2016, 99, 925–944. [Google Scholar] [CrossRef] [PubMed]
- Tegtmeyer, N.; Wessler, S.; Necchi, V.; Rohde, M.; Harrer, A.; Rau, T.T.; Asche, C.I.; Boehm, M.; Loessner, H.; Figueiredo, C.; et al. Helicobacter pylori Employs a Unique Basolateral Type IV Secretion Mechanism for CagA Delivery. Cell Host Microbe 2017, 22, 552–560.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fischer, W.; Tegtmeyer, N.; Stingl, K.; Backert, S. Four Chromosomal Type IV Secretion Systems in Helicobacter pylori: Composition, Structure and Function. Front. Microbiol. 2020, 11, 1592. [Google Scholar] [CrossRef] [PubMed]
- Odenbreit, S.; Puls, J.; Sedlmaier, B.; Gerland, E.; Fischer, W.; Haas, R. Translocation of Helicobacter pylori CagA into gastric epithelial cells by type IV secretion. Science 2000, 287, 1497–1500. [Google Scholar] [CrossRef] [PubMed]
- Backert, S.; Tegtmeyer, N. Type IV Secretion and Signal Transduction of Helicobacter pylori CagA through Interactions with Host Cell Receptors. Toxins 2017, 9, 115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parsonnet, J.; Friedman, G.D.; Orentreich, N.; Vogelman, H. Risk for gastric cancer in people with CagA positive or CagA negative Helicobacter pylori infection. Gut 1997, 40, 297–301. [Google Scholar] [CrossRef]
- Selbach, M.; Moese, S.; Hauck, C.R.; Meyer, T.F.; Backert, S. Src is the kinase of the Helicobacter pylori CagA protein in vitro and in vivo. J. Biol. Chem. 2002, 277, 6775–6778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Backert, S.; Müller, E.C.; Jungblut, P.R.; Meyer, T.F. Tyrosine phosphorylation patterns and size modification of the Helicobacter pylori CagA protein after translocation into gastric epithelial cells. Proteomics 2001, 1, 608–617. [Google Scholar] [CrossRef]
- Backert, S.; Blaser, M.J. The Role of CagA in the Gastric Biology of Helicobacter pylori. Cancer Res. 2016, 76, 4028–4031. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Selbach, M.; Paul, F.E.; Brandt, S.; Guye, P.; Daumke, O.; Backert, S.; Dehio, C.; Mann, M. Host Cell Interactome of Tyrosine-Phosphorylated Bacterial Proteins. Cell Host Microbe 2009, 5, 397–403. [Google Scholar] [CrossRef] [Green Version]
- Tegtmeyer, N.; Neddermann, M.; Asche, C.I.; Backert, S. Subversion of host kinases: A key network in cellular signaling hijacked by Helicobacter pylori CagA. Mol. Microbiol. 2017, 105, 358–372. [Google Scholar] [CrossRef] [Green Version]
- Segal, E.D.; Cha, J.; Lo, J.; Falkow, S.; Tompkins, L.S. Altered states: Involvement of phosphorylated CagA in the induction of host cellular growth changes by H. pylori. Proc. Natl. Acad. Sci. USA 1999, 96, 14559–14564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mimuro, H.; Suzuki, T.; Nagai, S.; Rieder, G.; Suzuki, M.; Nagai, T.; Fujita, Y.; Nagamatsu, K.; Ishijima, N.; Koyasu, S.; et al. Helicobacter pylori dampens gut epithelial self-renewal by inhibiting apoptosis, a bacterial strategy to enhance colonization of the stomach. Cell Host Microbe 2007, 2, 250–263. [Google Scholar] [CrossRef] [Green Version]
- Tegtmeyer, N.; Zabler, D.; Schmidt, D.; Hartig, R.; Brandt, S.; Backert, S. Importance of EGF receptor, HER2/Neu and Erk1/2 kinase signalling for host cell elongation and scattering induced by the Helicobacter pylori CagA protein: Antagonistic effects of the vacuolating cytotoxin VacA. Cell. Microbiol. 2009, 11, 488–505. [Google Scholar] [CrossRef]
- Atherton, J.C.; Cao, P.; Peek, R.M.; Tummuru, M.K.R.; Blaser, M.J.; Cover, T.L. Mosaicism in vacuolating cytotoxin alleles of Helicobacter pylori. J. Biol. Chem. 1995, 270, 17771–17777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leunk, R.D.; Johnson, P.T.; David, B.C.; Kraft, W.G.; Morgan, D.R. Cytotoxic activity in broth-culture filtrates of Campylobacter pylori. J. Med. Microbiol. 1988, 26, 93–99. [Google Scholar] [CrossRef] [Green Version]
- van Doorn, L.J.; Figueiredo, C.; Sanna, R.; Plaisier, A.; Schneeberger, P.; De Boer, W.; Quint, W. Clinical relevance of the cagA, vacA, and iceA status of Helicobacter pylori. Gastroenterology 1998, 115, 58–66. [Google Scholar] [CrossRef]
- Rhead, J.L.; Letley, D.P.; Mohammadi, M.; Hussein, N.; Mohagheghi, M.A.; Eshagh Hosseini, M.; Atherton, J.C. A new Helicobacter pylori vacuolating cytotoxin determinant, the intermediate region, is associated with gastric cancer. Gastroenterology 2007, 133, 926–936. [Google Scholar] [CrossRef] [PubMed]
- Szabo, I.; Brutsche, S.; Tombola, F.; Moschioni, M.; Satin, B.; Telford, J.L.; Rappuoli, R.; Montecucco, C.; Papini, E.; Zoratti, M. Formation of anion-selective channels in the cell plasma membrane by the toxin VacA of Helicobacter pylori is required for its biological activity. EMBO J. 1999, 18, 5517–5527. [Google Scholar] [CrossRef] [Green Version]
- Galmiche, A.; Rassow, J.; Doye, A.; Cagnol, S.; Chambard, J.C.; Contamin, S.; de Thillot, V.; Just, I.; Ricci, V.; Solcia, E.; et al. The N-terminal 34 kDa fragment of Helicobacter pylori vacuolating cytotoxin targets mitochondria and induces cytochrome c release. EMBO J. 2000, 19, 6361–6370. [Google Scholar] [CrossRef] [PubMed]
- Cover, T.L.; Krishna, U.S.; Israel, D.A.; Peek, R.M. Induction of gastric epithelial cell apoptosis by Helicobacter pylori vacuolating cytotoxin. Cancer Res. 2003, 63, 951–957. [Google Scholar] [PubMed]
- Oldani, A.; Cormont, M.; Hofman, V.; Chiozzi, V.; Oregioni, O.; Canonici, A.; Sciullo, A.; Sommi, P.; Fabbri, A.; Ricci, V.; et al. Helicobacter pylori counteracts the apoptotic action of its VacA toxin by injecting the CagA protein into gastric epithelial cells. PLoS Pathog. 2009, 5, e1000603. [Google Scholar] [CrossRef] [Green Version]
- Knorr, J.; Ricci, V.; Hatakeyama, M.; Backert, S. Classification of Helicobacter pylori Virulence Factors: Is CagA a Toxin or Not? Trends Microbiol. 2019, 27, 731–738. [Google Scholar] [CrossRef]
- Tegtmeyer, N.; Wittelsberger, R.; Hartig, R.; Wessler, S.; Martinez-Quiles, N.; Backert, S. Serine Phosphorylation of Cortactin Controls Focal Adhesion Kinase Activity and Cell Scattering Induced by Helicobacter pylori. Cell Host Microbe 2011, 9, 520–531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, H.; Chen, D.F.; Ni, B.S.; Zuo, Q.F.; Wang, C.H.; Han, R.; Lan, C.H. Cortactin Mediates Apoptosis of Gastric Epithelial Cells Induced by VacA Protein of Helicobacter pylori. Dig. Dis. Sci. 2016, 61, 80–90. [Google Scholar] [CrossRef] [PubMed]
- Schuuring, E.; Verhoeven, E.; Litvinov, S.; Michalides, R. The product of the ems1 gene, amplified and overexpressed in human carcinomas, is homologous to a v-Src substrate and is located in cell-substratum contact sites. Mol. Cell. Biol. 1993, 13, 2891–2898. [Google Scholar] [CrossRef]
- Schnoor, M.; Stradal, T.E.; Rottner, K. Cortactin: Cell Functions of A Multifaceted Actin-Binding Protein. Trends Cell Biol. 2018, 28, 79–98. [Google Scholar] [CrossRef]
- Uruno, T.; Liu, J.L.; Zhang, P.J.; Fan, Y.X.; Egile, C.; Li, P.; Mueller, S.C.; Zhan, X. Activation of Arp2/3 complex-mediated actin polymerization by cortactin. Nat. Cell Biol. 2001, 3, 259–266. [Google Scholar] [CrossRef]
- Selbach, M.; Backert, S. Cortactin: An Achilles’ heel of the actin cytoskeleton targeted by pathogens. Trends Microbiol. 2005, 13, 181–189. [Google Scholar] [CrossRef]
- Ammer, A.G.; Weed, S.A. Cortactin branches out: Roles in regulating protrusive actin dynamics. Cell Motil. Cytoskelet. 2008, 65, 687–707. [Google Scholar] [CrossRef] [Green Version]
- Kelley, L.C.; Hayes, K.E.; Ammer, A.G.; Martin, K.H.; Weed, S.A. Cortactin Phosphorylated by ERK1/2 Localizes to Sites of Dynamic Actin Regulation and Is Required for Carcinoma Lamellipodia Persistence. PLoS ONE 2010, 5, e13847. [Google Scholar] [CrossRef]
- Katsube, T.; Takahisa, M.; Ueda, R.; Hashimoto, N.; Kobayashi, M.; Togashi, S. Cortactin associates with the cell-cell junction protein ZO-1 in both Drosophila and mouse. J. Biol. Chem. 1998, 273, 29672–29677. [Google Scholar] [CrossRef] [Green Version]
- Kinley, A.W.; Weed, S.A.; Weaver, A.M.; Karginov, A.V.; Bissonette, E.; Cooper, J.A.; Parsons, J.T. Cortactin interacts with WIP in regulating Arp2/3 activation and membrane protrusion. Curr. Biol. 2003, 13, 384–393. [Google Scholar] [CrossRef] [Green Version]
- Sharafutdinov, I.; Backert, S.; Tegtmeyer, N. Cortactin: A Major Cellular Target of the Gastric Carcinogen Helicobacter pylori. Cancers 2020, 12, 159. [Google Scholar] [CrossRef] [Green Version]
- Knorr, J.; Backert, S.; Tegtmeyer, N. SHP2-Independent Tyrosine Dephosphorylation of Cortactin and Vinculin during Infection with Helicobacter pylori. Eur. J. Microbiol. Immunol. 2020, 10, 20–27. [Google Scholar] [CrossRef] [PubMed]
- Knorr, J.; Sharafutdinov, I.; Fiedler, F.; Esmaeili, S.D.; Rohde, M.; Rottner, K.; Backert, S.; Tegtmeyer, N. Cortactin is required for efficient Fak, Src and Abl tyrosine kinase activation and phosphorylation of Helicobacter pylori CagA. Int. J. Mol. Sci. 2021, 22, 6045. [Google Scholar] [CrossRef] [PubMed]
- Gauthier, N.C.; Monzo, P.; Kaddai, V.; Doye, A.; Ricci, V.; Boquet, P. Helicobacter pylori VacA cytotoxin: A probe for a clathrin-independent and Cdc42-dependent pinocytic pathway routed to late endosomes. Mol. Biol. Cell. 2005, 16, 4852–4866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Backert, S.; Moese, S.; Selbach, M.; Brinkmann, V.; Meyer, T.F. Phosphorylation of tyrosine 972 of the Helicobacter pylori CagA protein is essential for induction of a scattering phenotype in gastric epithelial cells. Mol. Microbiol. 2001, 42, 631–644. [Google Scholar] [CrossRef] [PubMed]
- Yahiro, K.; Wada, A.; Nakayama, M.; Kimura, T.; Ogushi, K.; Niidome, T.; Aoyagi, H.; Yoshino, K.; Yonezawa, K.; Moss, J.; et al. Protein-tyrosine phosphatase alpha, RPTP alpha, is a Helicobacter pylori VacA receptor. J. Biol. Chem. 2003, 278, 19183–19189. [Google Scholar] [CrossRef] [Green Version]
- Tegtmeyer, N.; Backert, S. Different roles of integrin-beta1 and integrin-alphav for type IV secretion of CagA versus cell elongation phenotype and cell lifting by Helicobacter pylori. PLoS Pathog. 2020, 16, e1008135. [Google Scholar] [CrossRef]
- Lin, M.T.; Juan, C.Y.; Chang, K.J.; Chen, W.J.; Kuo, M.L. IL-6 inhibits apoptosis and retains oxidative DNA lesions in human AGS cells through up-regulation of anti-apoptotic gene mcl-1. Carcinogenesis 2001, 22, 1947–1953. [Google Scholar] [CrossRef] [Green Version]
- Ito, K.; Yamaoka, Y.; Yoffe, B.; Graham, D.Y. Disturbance of Apoptosis and DNA Synthesis by Helicobacter pylori Infection of Hepatocytes. Dig. Dis. Sci. 2008, 53, 2532–2540. [Google Scholar] [CrossRef]
- Han, Z.; Hendrickson, E.A.; Bremner, T.A.; Wyche, J.H. A sequential two-step mechanism for the production of the mature p17:p12 form of caspase-3 in vitro. J. Biol. Chem. 1997, 272, 13432–13436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bougneres, L.; Girardin, S.E.; Weed, S.A.; Karginov, A.V.; Olivo-Marin, J.C.; Parsons, J.T.; Sansonetti, P.J.; Van Nhieu, G.T. Cortactin and Crk cooperate to trigger actin polymerization during Shigella invasion of epithelial cells. J. Cell Biol. 2004, 166, 225–235. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.M.; Huang, B.Q.; Splinter, P.L.; Cao, H.; Zhu, G.; McNiven, M.A.; LaRusso, N.F. Cryptosporidium invasion of biliary epithelia requires phosphorylation of cortactin via c-Src. Gastroenterology 2003, 125, 216–228. [Google Scholar] [CrossRef]
- Bonfim-Melo, A.; Zanetti, B.F.; Ferreira, E.R.; Vandoninck, S.; Han, S.W.; Van Lint, J.; Mortara, R.A.; Bahia, D. Trypanosoma cruzi extracellular amastigotes trigger the protein kinase D1-cortactin-actin pathway during cell invasion. Cell. Microbiol. 2015, 17, 1797–1810. [Google Scholar] [CrossRef] [Green Version]
- Kowalski, J.R.; Egile, C.; Gil, S.; Snapper, S.B.; Li, R.; Thomas, S.M. Cortactin regulates cell migration through activation of N-WASP. J. Cell Sci. 2005, 118, 79–87. [Google Scholar] [CrossRef] [Green Version]
- Sauvonnet, N.; Dujeancourt, A.; Dautry-Varsat, A. Cortactin and dynamin are required for the clathrin-dependent endocytosis of gamma c cytokine receptor. J. Cell Biol. 2005, 168, 155–163. [Google Scholar] [CrossRef]
- Mimuro, H.; Suzuki, T.; Tanaka, J.; Asahi, M.; Haas, R.; Sasakawa, C. Grb2 is a key mediator of Helicobacter pylori CagA protein activities. Mol. Cell 2002, 10, 745–755. [Google Scholar] [CrossRef]
- Sharafutdinov, I.; Backert, S.; Tegtmeyer, N. The Helicobacter pylori type IV secretion system upregulates epithelial cortactin expression by a CagA- and JNK-dependent pathway. Cell. Microbiol. 2021, 23, e13376. [Google Scholar] [CrossRef] [PubMed]
- Maubach, G.; Lim, M.C.C.; Sokolova, O.; Backert, S.; Meyer, T.F.; Naumann, M. TIFA has dual functions in Helicobacter pylori-induced classical and alternative NF-κB pathways. EMBO Rep. 2021, 30, e52878. [Google Scholar] [CrossRef]
- Conradi, J.; Huber, S.; Gaus, K.; Mertink, F.; Royo Gracia, S.; Strijowski, U.; Backert, S.; Sewald, N. Cyclic RGD peptides interfere with binding of the Helicobacter pylori protein CagL to integrins αVβ3 and α5β1. Amino Acids 2012, 43, 219–232. [Google Scholar] [CrossRef] [PubMed]
- Moese, S.; Selbach, M.; Zimny-Arndt, U.; Jungblut, P.R.; Meyer, T.F.; Backert, S. Identification of a tyrosine-phosphorylated 35 kDa carboxy-terminal fragment (p35CagA) of the Helicobacter pylori CagA protein in phagocytic cells: Processing or breakage? Proteomics 2001, 1, 618–629. [Google Scholar] [CrossRef]
- Tegtmeyer, N.; Neddermann, M.; Lind, J.; Pachathundikandi, S.K.; Sharafutdinov, I.; Gutierrez-Escobar, A.J.; Bronstrup, M.; Tegge, W.; Hong, M.S.; Rohde, M.; et al. Toll-like Receptor 5 Activation by the CagY Repeat Domains of Helicobacter pylori. Cell Rep. 2020, 32. [Google Scholar] [CrossRef] [PubMed]
- Pachathundikandi, S.K.; Tegtmeyer, N.; Arnold, I.C.; Lind, J.; Neddermann, M.; Falkeis-Veits, C.; Chattopadhyay, S.; Bronstrup, M.; Tegge, W.; Hong, M.S.; et al. T4SS-dependent TLR5 activation by Helicobacter pylori infection. Nat. Commun. 2019, 10, 5717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hartung, M.L.; Gruber, D.C.; Koch, K.N.; Grüter, L.; Rehrauer, H.; Tegtmeyer, N.; Backert, S.; Müller, A. H. pylori induced DNA strand breaks are introduced by nucleotide excision repair endonucleases and promote NF-κB target gene expression. Cell Rep. 2015, 13, 70–79. [Google Scholar] [CrossRef] [Green Version]
- Blumenthal, B.; Hoffmann, C.; Aktories, K.; Backert, S.; Schmidt, G. The cytotoxic necrotizing factors from Yersinia pseudotuberculosis and from Escherichia coli bind to different cellular receptors but take the same route to the cytosol. Infect. Immun. 2007, 75, 3344–3353. [Google Scholar] [CrossRef] [Green Version]
- Roure, S.; Bonis, M.; Chaput, C.; Ecobichon, C.; Mattox, A.; Barrière, C.; Geldmacher, N.; Guadagnini, S.; Schmitt, C.; Prévost, M.C.; et al. Peptidoglycan maturation enzymes affect flagellar functionality in bacteria. Mol. Microbiol. 2012, 86, 845–856. [Google Scholar] [CrossRef]
- Heimesaat, M.M.; Alutis, M.; Grundmann, U.; Fischer, A.; Tegtmeyer, N.; Böhm, M.; Kühl, A.A.; Göbel, U.B.; Backert, S.; Bereswill, S. The role of serine protease HtrA in acute ulcerative enterocolitis and extra-intestinal immune responses during Campylobacter jejuni infection of gnotobiotic IL-10 deficient mice. Front. Cell. Infect. Microbiol. 2014, 4, 77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cover, T.L.; Hanson, P.I.; Heuser, J.E. Acid-induced dissociation of VacA, the Helicobacter pylori vacuolating cytotoxin, reveals its pattern of assembly. J. Cell Biol. 1997, 138, 759–769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neddermann, M.; Backert, S. How many protein molecules are secreted by single Helicobacter pylori cells: Quantification of serine protease HtrA. Cell. Microbiol. 2019, 21, e13022. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sharafutdinov, I.; Knorr, J.; Soltan Esmaeili, D.; Backert, S.; Tegtmeyer, N. Cortactin Promotes Effective AGS Cell Scattering by Helicobacter pylori CagA, but Not Cellular Vacuolization and Apoptosis Induced by the Vacuolating Cytotoxin VacA. Pathogens 2022, 11, 3. https://doi.org/10.3390/pathogens11010003
Sharafutdinov I, Knorr J, Soltan Esmaeili D, Backert S, Tegtmeyer N. Cortactin Promotes Effective AGS Cell Scattering by Helicobacter pylori CagA, but Not Cellular Vacuolization and Apoptosis Induced by the Vacuolating Cytotoxin VacA. Pathogens. 2022; 11(1):3. https://doi.org/10.3390/pathogens11010003
Chicago/Turabian StyleSharafutdinov, Irshad, Jakob Knorr, Delara Soltan Esmaeili, Steffen Backert, and Nicole Tegtmeyer. 2022. "Cortactin Promotes Effective AGS Cell Scattering by Helicobacter pylori CagA, but Not Cellular Vacuolization and Apoptosis Induced by the Vacuolating Cytotoxin VacA" Pathogens 11, no. 1: 3. https://doi.org/10.3390/pathogens11010003
APA StyleSharafutdinov, I., Knorr, J., Soltan Esmaeili, D., Backert, S., & Tegtmeyer, N. (2022). Cortactin Promotes Effective AGS Cell Scattering by Helicobacter pylori CagA, but Not Cellular Vacuolization and Apoptosis Induced by the Vacuolating Cytotoxin VacA. Pathogens, 11(1), 3. https://doi.org/10.3390/pathogens11010003