
Filovirus Neutralising Antibodies: Mechanisms of Action and Therapeutic Application

 ,
, 

Abstract
1. Filoviridae Background
1.1. Filoviridae Phylogeny
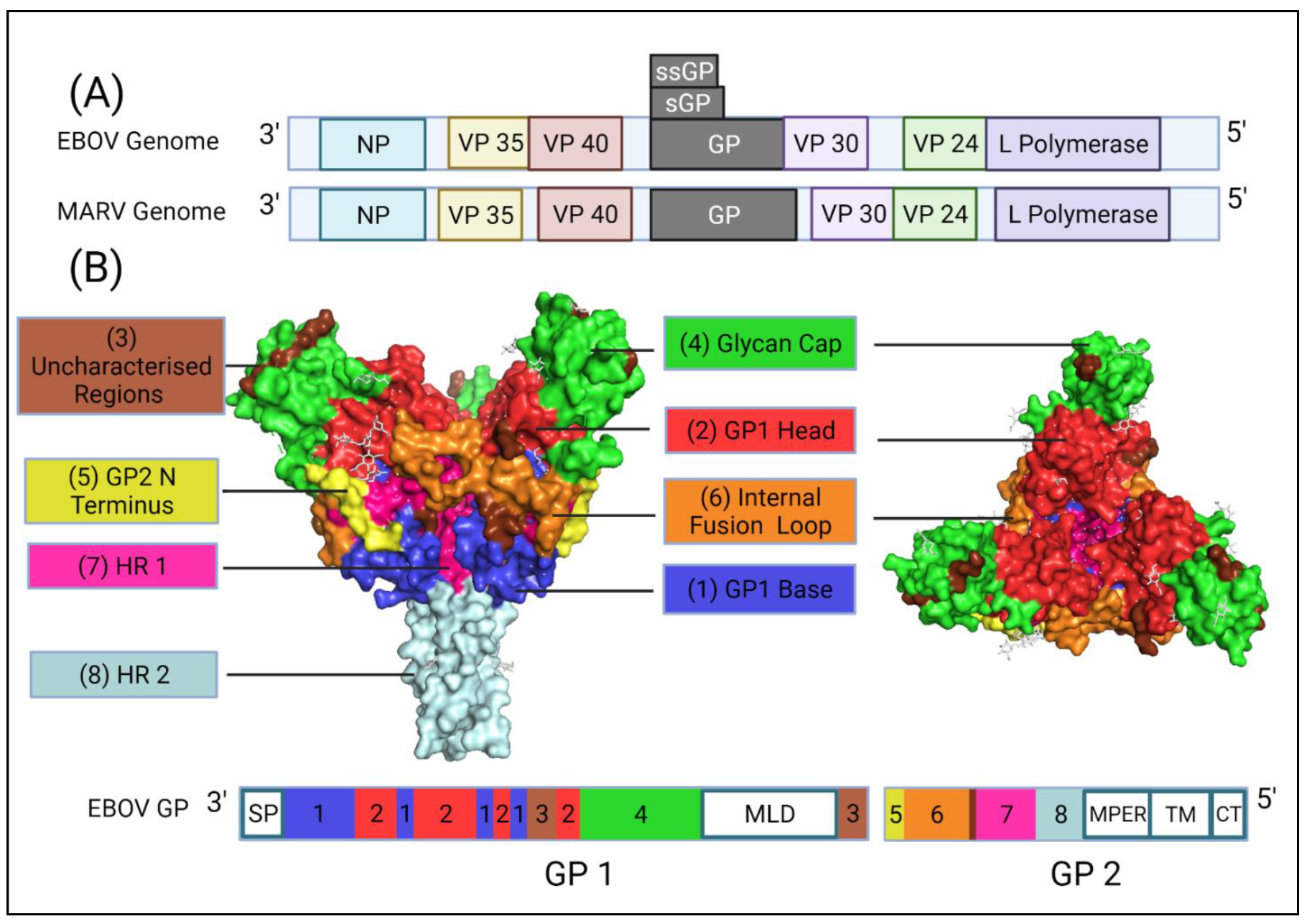
1.2. Genome Organisation of Ebolavirus
1.3. Cellular Entry of Ebola Virus
1.4. EVD and Immunity
2. Neutralising Antibodies Following a Natural Infection
2.1. Neutralising Antibodies against EBOV
2.2. Neutralising Antibodies against Non-EBOV Filoviruses
3. Antibody-Based Therapeutics and Vaccines
3.1. Monoclonal Antibodies
3.2. Convalescent Plasma
3.3. Vaccine-Induced Neutralising Responses
3.3.1. EBOV Vaccines
3.3.2. MARV Vaccine Candidates
4. Methods to Measure Neutralising Antibody Responses
5. Viral Neutralisation
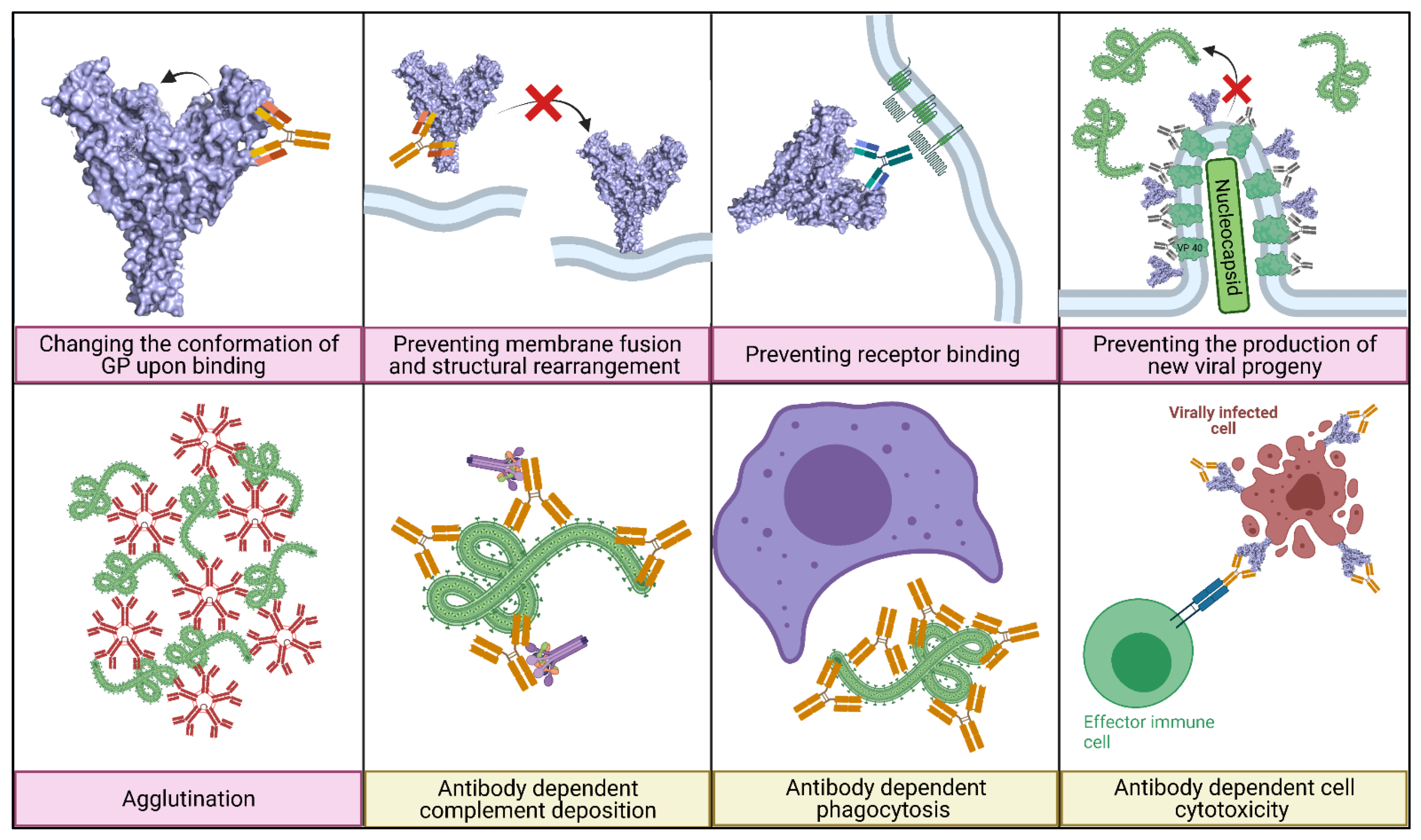
5.1. Main Mechanisms of Neutralisation
5.2. Regions Targeted by Neutralising Antibodies
6. Strategies to Prevent Immune Escape from Neutralising Antibodies
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Towner, J.S.; Khristova, M.L.; Sealy, T.K.; Vincent, M.J.; Erickson, B.R.; Bawiec, D.A.; Hartman, A.; Comer, J.A.; Zaki, S.R.; Ströher, U.; et al. Marburgvirus Genomics and Association with a Large Hemorrhagic Fever Outbreak in Angola. J. Virol. 2006, 80, 6497–6516. [Google Scholar] [CrossRef] [PubMed]
- Timothy, J.; Hall, Y.; Akoi-Boré, J.; Diallo, B.; Tipton, T.R.W.; Bower, H.; Strecker, T.; Glynn, J.R.; Carroll, M. Early transmission and case fatality of Ebola virus at the index site of the 2013–2016 west African Ebola outbreak: A cross-sectional seroprevalence survey. Lancet Infect. Dis. 2019, 19, 429–438. [Google Scholar] [CrossRef]
- Goldstein, T.; Anthony, S.J.; Gbakima, A.; Bird, B.H.; Bangura, J.; Tremeau-Bravard, A.; Belaganahalli, M.N.; Wells, H.L.; Dhanota, J.K.; Liang, E.; et al. The discovery of Bombali virus adds further support for bats as hosts of ebolaviruses. Nat. Microbiol. 2018, 3, 1084–1089. [Google Scholar] [CrossRef]
- Zampieri, C.A.; Sullivan, N.J.; Nabel, G.J. Immunopathology of highly virulent pathogens: Insights from Ebola virus. Nat. Immunol. 2007, 8, 1159–1164. [Google Scholar] [CrossRef] [PubMed]
- Negredo, A.; Palacios, G.; Vázquez-Morón, S.; González, F.; Dopazo, H.; Molero, F.; Juste, J.; Quetglas, J.; Savji, N.; Martínez, M.D.L.C.; et al. Discovery of an Ebolavirus-Like Filovirus in Europe. PLoS Pathog. 2011, 7, e1002304. [Google Scholar] [CrossRef]
- Ramírez de Arellano, E.; Sanchez-Lockhart, M.; Perteguer, M.J.; Bartlett, M.; Ortiz, M.; Campioli, P.; Hernández, A.; Gonzalez, J.; Garcia, K.; Ramos, M.; et al. First Evidence of Antibodies against Lloviu Virus in Schreiber’s Bent-Winged Insectivorous Bats Demonstrate a Wide Circulation of the Virus in Spain. Viruses 2019, 11, 360. [Google Scholar] [CrossRef]
- Kemenesi, G.; Kurucz, K.; Dallos, B.; Zana, B.; Földes, F.; Boldogh, S.; Görföl, T.; Carroll, M.W.; Jakab, F. Re-emergence of Lloviu virus in Miniopterus schreibersii bats, Hungary, 2016. Emerg. Microbes Infect. 2018, 7, 1–4. [Google Scholar] [CrossRef]
- Yang, X.-L.; Tan, C.W.; Anderson, D.E.; Jiang, R.-D.; Li, B.; Zhang, W.; Zhu, Y.; Lim, X.F.; Zhou, P.; Liu, X.-L.; et al. Characterization of a filovirus (Měnglà virus) from Rousettus bats in China. Nat. Microbiol. 2019, 4, 390–395. [Google Scholar] [CrossRef]
- Burk, R.; Bollinger, L.; Johnson, J.; Wada, J.; Radoshitzky, S.; Palacios, G.; Bavari, S.; Jahrling, P.B.; Kuhn, J.H. Neglected filoviruses. FEMS Microbiol. Rev. 2016, 40, 494–519. [Google Scholar] [CrossRef]
- Maruyama, J.; Miyamoto, H.; Kajihara, M.; Ogawa, H.; Maeda, K.; Sakoda, Y.; Yoshida, R.; Takada, A. Characterization of the Envelope Glycoprotein of a Novel Filovirus, Lloviu Virus. J. Virol. 2014, 88, 99–109. [Google Scholar] [CrossRef]
- Hume, A.; Mühlberger, E. Distinct Genome Replication and Transcription Strategies within the Growing Filovirus Family. J. Mol. Biol. 2019, 431, 4290–4320. [Google Scholar] [CrossRef]
- Lee, J.E.; Saphire, E.O. Ebolavirus glycoprotein structure and mechanism of entry. Future Virol. 2009, 4, 621–635. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Ren, J.; Harlos, K.; Jones, D.M.; Zeltina, A.; Bowden, T.A.; Padilla-Parra, S.; Fry, E.E.; Stuart, D.I. Toremifene interacts with and destabilizes the Ebola virus glycoprotein. Nature 2016, 535, 169–172. [Google Scholar] [CrossRef] [PubMed]
- Mühlberger, E. Filovirus replication and transcription. Future Virol. 2007, 2, 205–215. [Google Scholar] [CrossRef] [PubMed]
- Cantoni, D.; Rossman, J.S. Ebolaviruses: New roles for old proteins. PLoS Negl. Trop. Dis. 2018, 12, e0006349. [Google Scholar] [CrossRef]
- Volchkov, V.E.; Feldmann, H.; Volchkova, V.A.; Klenk, H.-D. Processing of the Ebola virus glycoprotein by the proprotein convertase furin. Proc. Natl. Acad. Sci. USA 1998, 95, 5762–5767. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.E.; Fusco, M.L.; Hessell, A.J.; Oswald, W.B.; Burton, D.R.; Saphire, E.O. Structure of the Ebola virus glycoprotein bound to an antibody from a human survivor. Nature 2008, 454, 177–182. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.; Banadyga, L.; Emeterio, K.; Wong, G.; Qiu, X. The Roles of Ebola Virus Soluble Glycoprotein in Replication, Pathogenesis, and Countermeasure Development. Viruses 2019, 11, 999. [Google Scholar] [CrossRef] [PubMed]
- Mehedi, M.; Falzarano, D.; Seebach, J.; Hu, X.; Carpenter, M.S.; Schnittler, H.-J.; Feldmann, H. A New Ebola Virus Nonstructural Glycoprotein Expressed through RNA Editing. J. Virol. 2011, 85, 5406–5414. [Google Scholar] [CrossRef]
- Escudero-Pérez, B.; Volchkova, V.A.; Dolnik, O.; Lawrence, P.; Volchkov, V.E. Shed GP of Ebola Virus Triggers Immune Activation and Increased Vascular Permeability. PLoS Pathog. 2014, 10, e1004509. [Google Scholar] [CrossRef]
- Yu, D.-S.; Weng, T.-H.; Wu, X.; Wang, F.X.; Lu, X.-Y.; Wu, H.-B.; Wu, N.-P.; Li, L.-J.; Yao, H.-P. The lifecycle of the Ebola virus in host cells. Oncotarget 2017, 8, 55750–55759. [Google Scholar] [CrossRef] [PubMed]
- Nanbo, A.; Imai, M.; Watanabe, S.; Noda, T.; Takahashi, K.; Neumann, G.; Halfmann, P.; Kawaoka, Y. Ebolavirus Is Internalized into Host Cells via Macropinocytosis in a Viral Glycoprotein-Dependent Manner. PLoS Pathog. 2010, 6, e1001121. [Google Scholar] [CrossRef] [PubMed]
- Brecher, M.; Schornberg, K.L.; Delos, S.E.; Fusco, M.L.; Saphire, E.O.; White, J.M. Cathepsin Cleavage Potentiates the Ebola Virus Glycoprotein to Undergo a Subsequent Fusion-Relevant Conformational Change. J. Virol. 2012, 86, 364–372. [Google Scholar] [CrossRef] [PubMed]
- Miller, E.H.; Obernosterer, G.; Raaben, M.; Herbert, A.S.; Deffieu, M.S.; Krishnan, A.; Ndungo, E.; Sandesara, R.G.; Carette, J.; Kuehne, A.I.; et al. Ebola virus entry requires the host-programmed recognition of an intracellular receptor. EMBO J. 2012, 31, 1947–1960. [Google Scholar] [CrossRef] [PubMed]
- Gregory, S.M.; Larsson, P.; Nelson, E.A.; Kasson, P.M.; White, J.M.; Tamm, L.K. Ebolavirus Entry Requires a Compact Hydrophobic Fist at the Tip of the Fusion Loop. J. Virol. 2014, 88, 6636–6649. [Google Scholar] [CrossRef]
- Moller-Tank, S.; Maury, W. Ebola Virus Entry: A Curious and Complex Series of Events. PLoS Pathog. 2015, 11, e1004731. [Google Scholar] [CrossRef]
- Olejnik, J.; Ryabchikova, E.; Corley, R.B.; Mühlberger, E. Intracellular Events and Cell Fate in Filovirus Infection. Viruses 2011, 3, 1501–1531. [Google Scholar] [CrossRef]
- Okumura, A.; Pitha, P.M.; Yoshimura, A.; Harty, R.N. Interaction between Ebola Virus Glycoprotein and Host Toll-Like Receptor 4 Leads to Induction of Proinflammatory Cytokines and SOCS1. J. Virol. 2010, 84, 27–33. [Google Scholar] [CrossRef]
- Olejnik, J.; Forero, A.; Deflubé, L.R.; Hume, A.; Manhart, W.A.; Nishida, A.; Marzi, A.; Katze, M.G.; Ebihara, H.; Rasmussen, A.L.; et al. Ebolaviruses Associated with Differential Pathogenicity Induce Distinct Host Responses in Human Macrophages. J. Virol. 2017, 91, e00179-17. [Google Scholar] [CrossRef]
- Gupta, M.; Mahanty, S.; Ahmed, R.; Rollin, P. Monocyte-Derived Human Macrophages and Peripheral Blood Mononuclear Cells Infected with Ebola Virus Secrete MIP-1α and TNF-α and Inhibit Poly-IC-Induced IFN-α in Vitro. Virology 2001, 284, 20–25. [Google Scholar] [CrossRef]
- Ströher, U.; West, E.; Bugany, H.; Klenk, H.-D.; Schnittler, H.-J.; Feldmann, H. Infection and Activation of Monocytes by Marburg and Ebola Viruses. J. Virol. 2001, 75, 11025–11033. [Google Scholar] [CrossRef]
- Hensley, L.E.; Young, H.A.; Jahrling, P.B.; Geisberta, T.W. Proinflammatory response during Ebola virus infection of primate models: Possible involvement of the tumor necrosis factor receptor superfamily. Immunol. Lett. 2002, 80, 169–179. [Google Scholar] [CrossRef]
- Wahl-Jensen, V.; Kurz, S.; Feldmann, F.; Buehler, L.K.; Kindrachuk, J.; De Filippis, V.; Correia, J.D.S.; Früh, K.; Kuhn, J.H.; Burton, D.R.; et al. Ebola Virion Attachment and Entry into Human Macrophages Profoundly Effects Early Cellular Gene Expression. PLoS Negl. Trop. Dis. 2011, 5, e1359. [Google Scholar] [CrossRef] [PubMed]
- McElroy, A.K.; Muhlberger, E.; Muñoz-Fontela, C. Immune barriers of Ebola virus infection. Curr. Opin. Virol. 2018, 28, 152–160. [Google Scholar] [CrossRef] [PubMed]
- Longhi, P.; Trumpfheller, C.; Idoyaga, J.; Caskey, M.; Matos, I.; Kluger, C.; Salazar, A.M.; Colonna, M.; Steinman, R.M. Dendritic cells require a systemic type I interferon response to mature and induce CD4+ Th1 immunity with poly IC as adjuvant. J. Exp. Med. 2009, 206, 1589–1602. [Google Scholar] [CrossRef] [PubMed]
- Rhein, B.A.; Powers, L.S.; Rogers, K.; Anantpadma, M.; Singh, B.; Sakurai, Y.; Bair, T.; Miller-Hunt, C.; Sinn, P.; Davey, R.; et al. Interferon-γ Inhibits Ebola Virus Infection. PLoS Pathog. 2015, 11, e1005263. [Google Scholar] [CrossRef]
- Dyall, J.; Hart, B.J.; Postnikova, E.; Cong, Y.; Zhou, H.; Gerhardt, D.M.; Freeburger, D.; Michelotti, J.; Honko, A.N.; Dewald, L.E.; et al. Interferon-β and Interferon-γ Are Weak Inhibitors of Ebola Virus in Cell-Based Assays. J. Infect. Dis. 2017, 215, 1416–1420. [Google Scholar] [CrossRef] [PubMed]
- Jahrling, P.B.; Geisbert, T.W.; Geisbert, J.B.; Swearengen, J.R.; Bray, M.; Jaax, N.K.; Huggins, J.W.; LeDuc, J.W.; Peters, C.J. Evaluation of Immune Globulin and Recombinant Interferon-α2b for Treatment of Experimental Ebola Virus Infections. J. Infect. Dis. 1999, 179, S224–S234. [Google Scholar] [CrossRef]
- Smith, L.M.; Hensley, L.; Geisbert, T.W.; Johnson, J.; Stossel, A.; Honko, A.; Yen, J.Y.; Geisbert, J.; Paragas, J.; Fritz, E.; et al. Interferon-β Therapy Prolongs Survival in Rhesus Macaque Models of Ebola and Marburg Hemorrhagic Fever. J. Infect. Dis. 2013, 208, 310–318. [Google Scholar] [CrossRef] [PubMed]
- Qiu, X.; Wong, G.; Fernando, L.; Audet, J.; Bello, A.; Strong, J.; Alimonti, J.B.; Kobinger, G.P. mAbs and Ad-Vectored IFN- Therapy Rescue Ebola-Infected Nonhuman Primates When Administered After the Detection of Viremia and Symptoms. Sci. Transl. Med. 2013, 5, 207ra143. [Google Scholar] [CrossRef]
- Villinger, F.; Rollin, P.; Brar, S.S.; Chikkala, N.F.; Winter, J.; Sundstrom, J.B.; Zaki, S.R.; Swanepoel, R.; Ansari, A.A.; Peters, C.J. Markedly Elevated Levels of Interferon (IFN)-σ, IFN-α, Interleukin (IL)-2, IL-10, and Tumor Necrosis Factor-α Associated with Fatal Ebola Virus Infection. J. Infect. Dis. 1999, 179, S188–S191. [Google Scholar] [CrossRef] [PubMed]
- Olejnik, J.; Hume, A.J.; Leung, D.W.; Amarasinghe, G.K.; Basler, C.F.; Mühlberger, E. Filovirus Strategies to Escape Antiviral Responses. Curr. Top. Microbiol. Immunol. 2017, 411, 293–322. [Google Scholar] [CrossRef]
- McElroy, A.; Akondy, R.; Davis, C.W.; Ellebedy, A.H.; Mehta, A.; Kraft, C.S.; Lyon, G.M.; Ribner, B.S.; Varkey, J.; Sidney, J.; et al. Human Ebola virus infection results in substantial immune activation. Proc. Natl. Acad. Sci. USA 2015, 112, 4719–4724. [Google Scholar] [CrossRef]
- Ruibal, P.; Oestereich, L.; Lüdtke, A.; Becker-Ziaja, B.; Wozniak, D.; Kerber, R.; Korva, M.; Cabeza-Cabrerizo, M.; Bore, J.A.; Koundouno, F.R.; et al. Unique human immune signature of Ebola virus disease in Guinea. Nature 2016, 533, 100–104. [Google Scholar] [CrossRef] [PubMed]
- Baize, S.; Leroy, E.; Georges-Courbot, M.-C.; Capron, M.; Lansoud-Soukate, J.; Debré, P.; Fisher-Hoch, S.P.; McCormick, J.B.; Georges, A.J. Defective humoral responses and extensive intravascular apoptosis are associated with fatal outcome in Ebola virus-infected patients. Nat. Med. 1999, 5, 423–426. [Google Scholar] [CrossRef]
- Muñoz-Fontela, C.; McElroy, A.K. Ebola Virus Disease in Humans: Pathophysiology and Immunity. Curr. Top. Microbiol. Immunol. 2017, 411, 141–169. [Google Scholar] [CrossRef]
- Gupta, M.; Mahanty, S.; Greer, P.; Towner, J.S.; Shieh, W.-J.; Zaki, S.R.; Ahmed, R.; Rollin, P.E. Persistent Infection with Ebola Virus under Conditions of Partial Immunity. J. Virol. 2004, 78, 958–967. [Google Scholar] [CrossRef] [PubMed]
- Thom, R.; Tipton, T.; Strecker, T.; Hall, Y.; Bore, J.A.; Maes, P.; Koundouno, F.R.; Fehling, S.K.; Krähling, V.; Steeds, K.; et al. Longitudinal antibody and T cell responses in Ebola virus disease survivors and contacts: An observational cohort study. Lancet Infect. Dis. 2020, 21, 507–516. [Google Scholar] [CrossRef]
- Tipton, T.R.W.; Hall, Y.; Bore, J.A.; White, A.; Sibley, L.S.; Sarfas, C.; Yuki, Y.; Martin, M.; Longet, S.; Mellors, J.; et al. Characterisation of the T-cell response to Ebola virus glycoprotein amongst survivors of the 2013–16 West Africa epidemic. Nat. Commun. 2021, 12, 1153. [Google Scholar] [CrossRef] [PubMed]
- Sakabe, S.; Sullivan, B.; Hartnett, J.N.; Robles-Sikisaka, R.; Gangavarapu, K.; Cubitt, B.; Ware, B.C.; Kotliar, D.; Branco, L.M.; Goba, A.; et al. Analysis of CD8+ T cell response during the 2013–2016 Ebola epidemic in West Africa. Proc. Natl. Acad. Sci. USA 2018, 115, E7578–E7586. [Google Scholar] [CrossRef] [PubMed]
- Longet, S.; Mellors, J.; Carroll, M.W.; Tipton, T. Ebolavirus: Comparison of Survivor Immunology and Animal Models in the Search for a Correlate of Protection. Front. Immunol. 2021, 11, 599568. [Google Scholar] [CrossRef]
- Ksiazek, T.G.; Rollin, P.; Williams, A.J.; Bressler, D.S.; Martin, M.L.; Swanepoel, R.; Burt, F.J.; Leman, P.A.; Khan, A.S.; Rowe, A.K.; et al. Clinical Virology of Ebola Hemorrhagic Fever (EHF): Virus, Virus Antigen, and IgG and IgM Antibody Findings among EHF Patients in Kikwit, Democratic Republic of the Congo, 1995. J. Infect. Dis. 1999, 179, S177–S187. [Google Scholar] [CrossRef]
- Song, P.; Zheng, N.; Liu, Y.; Tian, C.; Wu, X.; Ma, X.; Chen, D.; Zou, X.; Wang, G.; Wang, H.; et al. Deficient humoral responses and disrupted B-cell immunity are associated with fatal SFTSV infection. Nat. Commun. 2018, 9, 3328. [Google Scholar] [CrossRef]
- Corti, D.; Misasi, J.; Mulangu, S.; Stanley, D.A.; Kanekiyo, M.; Wollen, S.; Ploquin, A.; Doria-Rose, N.A.; Staupe, R.P.; Bailey, M.; et al. Protective monotherapy against lethal Ebola virus infection by a potently neutralizing antibody. Science 2016, 351, 1339–1342. [Google Scholar] [CrossRef] [PubMed]
- Maruyama, T.; Rodriguez, L.L.; Jahrling, P.B.; Sanchez, A.; Khan, A.S.; Nichol, S.T.; Peters, C.J.; Parren, P.W.H.I.; Burton, D.R. Ebola Virus Can Be Effectively Neutralized by Antibody Produced in Natural Human Infection. J. Virol. 1999, 73, 6024–6030. [Google Scholar] [CrossRef]
- Parren, P.; Geisbert, T.W.; Maruyama, T.; Jahrling, P.B.; Burton, D.R. Pre- and Postexposure Prophylaxis of Ebola Virus Infection in an Animal Model by Passive Transfer of a Neutralizing Human Antibody. J. Virol. 2002, 76, 6408–6412. [Google Scholar] [CrossRef] [PubMed]
- Flyak, A.; Shen, X.; Murin, C.D.; Turner, H.L.; David, J.; Fusco, M.L.; Lampley, R.; Kose, N.; Ilinykh, P.A.; Kuzmina, N.; et al. Cross-Reactive and Potent Neutralizing Antibody Responses in Human Survivors of Natural Ebolavirus Infection. Cell 2016, 164, 392–405. [Google Scholar] [CrossRef] [PubMed]
- Wec, A.Z.; Herbert, A.S.; Murin, C.D.; Nyakatura, E.K.; Abelson, D.M.; Fels, J.M.; He, S.; James, R.M.; de La Vega, M.-A.; Zhu, W.; et al. Antibodies from a Human Survivor Define Sites of Vulnerability for Broad Protection against Ebolaviruses. Cell 2017, 169, 878–890.e15. [Google Scholar] [CrossRef] [PubMed]
- Bornholdt, Z.; Turner, H.L.; Murin, C.D.; Li, W.; Sok, D.; Souders, C.A.; Piper, A.E.; Goff, A.; Shamblin, J.D.; Wollen-Roberts, S.; et al. Isolation of potent neutralizing antibodies from a survivor of the 2014 Ebola virus outbreak. Science 2016, 351, 1078–1083. [Google Scholar] [CrossRef] [PubMed]
- Saphire, E.O.; Schendel, S.; Gunn, B.M.; Milligan, J.C.; Alter, G. Antibody-mediated protection against Ebola virus. Nat. Immunol. 2018, 19, 1169–1178. [Google Scholar] [CrossRef] [PubMed]
- Klingler, J.; Weiss, S.; Itri, V.; Liu, X.; Oguntuyo, K.Y.; Stevens, C.; Ikegame, S.; Hung, C.-T.; Enyindah-Asonye, G.; Amanat, F.; et al. Role of IgM and IgA Antibodies in the Neutralization of SARS-CoV-2. medRxiv 2020. [Google Scholar] [CrossRef]
- Luczkowiak, J.; Arribas, J.R.; Gómez, S.; Jiménez-Yuste, V.; de la Calle-Prieto, F.; Viejo, A.; Delgado, R. Specific neutralizing response in plasma from convalescent patients of Ebola Virus Disease against the West Africa Makona variant of Ebola virus. Virus Res. 2016, 213, 224–229. [Google Scholar] [CrossRef]
- Williamson, L.E.; Flyak, A.I.; Kose, N.; Bombardi, R.; Branchizio, A.; Reddy, S.; Davidson, E.; Doranz, B.J.; Fusco, M.L.; Saphire, E.O.; et al. Early Human B Cell Response to Ebola Virus in Four U.S. Survivors of Infection. J. Virol. 2019, 93. [Google Scholar] [CrossRef] [PubMed]
- Rimoin, A.W.; Lu, K.; Bramble, M.S.; Steffen, I.; Doshi, R.H.; Hoff, N.A.; Mukadi, P.; Nicholson, B.P.; Alfonso, V.H.; Olinger, G.; et al. Ebola Virus Neutralizing Antibodies Detectable in Survivors of theYambuku, Zaire Outbreak 40 Years after Infection. J. Infect. Dis. 2018, 217, 223–231. [Google Scholar] [CrossRef] [PubMed]
- Halfmann, P.J.; Eisfeld, A.J.; Watanabe, T.; Maemura, T.; Yamashita, M.; Fukuyama, S.; Armbrust, T.; Rozich, I.; N’Jai, A.; Neumann, G.; et al. Serological analysis of Ebola virus survivors and close contacts in Sierra Leone: A cross-sectional study. PLoS Negl. Trop. Dis. 2019, 13, e0007654. [Google Scholar] [CrossRef]
- Davis, C.W.; Jackson, K.; McElroy, A.K.; Halfmann, P.; Huang, J.; Chennareddy, C.; Piper, A.E.; Leung, Y.; Albariño, C.G.; Crozier, I.; et al. Longitudinal Analysis of the Human B Cell Response to Ebola Virus Infection. Cell 2019, 177, 1566–1582.e17. [Google Scholar] [CrossRef] [PubMed]
- Khurana, S.; Ravichandran, S.; Hahn, M.; Coyle, E.M.; Stonier, S.W.; Zak, S.E.; Kindrachuk, J.; Davey, R.T.; Dye, J.M.; Chertow, D.S. Longitudinal Human Antibody Repertoire against Complete Viral Proteome from Ebola Virus Survivor Reveals Protective Sites for Vaccine Design. Cell Host Microbe 2020, 27, 262–276.e4. [Google Scholar] [CrossRef]
- Gunn, B.M.; Roy, V.; Karim, M.M.; Hartnett, J.N.; Suscovich, T.J.; Goba, A.; Momoh, M.; Sandi, J.D.; Kanneh, L.; Andersen, K.G.; et al. Survivors of Ebola Virus Disease Develop Polyfunctional Antibody Responses. J. Infect. Dis. 2020, 221, 156–161. [Google Scholar] [CrossRef]
- Ullah, I.; Prévost, J.; Ladinsky, M.S.; Stone, H.; Lu, M.; Anand, S.P.; Beaudoin-Bussières, G.; Symmes, K.; Benlarbi, M.; Ding, S.; et al. Live Imaging of SARS-CoV-2 Infection in Mice Reveals that Neutralizing Antibodies Require Fc Function for Optimal Efficacy. Immunity 2021. [Google Scholar] [CrossRef] [PubMed]
- Sobarzo, A.; Groseth, A.; Dolnik, O.; Becker, S.; Lutwama, J.J.; Perelman, E.; Yavelsky, V.; Muhammad, M.; Kuehne, A.I.; Marks, R.S.; et al. Profile and Persistence of the Virus-Specific Neutralizing Humoral Immune Response in Human Survivors of Sudan Ebolavirus (Gulu). J. Infect. Dis. 2013, 208, 299–309. [Google Scholar] [CrossRef] [PubMed]
- Sobarzo, A.; Eskira, Y.; Herbert, A.S.; Kuehne, A.I.; Stonier, S.W.; Ochayon, D.E.; Fedida-Metula, S.; Balinandi, S.; Kislev, Y.; Tali, N.; et al. Immune Memory to Sudan Virus: Comparison between Two Separate Disease Outbreaks. Viruses 2015, 7, 37–51. [Google Scholar] [CrossRef]
- Stonier, S.W.; Herbert, A.S.; Kuehne, A.I.; Sobarzo, A.; Habibulin, P.; Dahan, C.V.A.; James, R.M.; Egesa, M.; Cose, S.; Lutwama, J.J.; et al. Marburg virus survivor immune responses are Th1 skewed with limited neutralizing antibody responses. J. Exp. Med. 2017, 214, 2563–2572. [Google Scholar] [CrossRef]
- Lipman, N.S.; Jackson, L.R.; Trudel, L.J.; Weis-Garcia, F. Monoclonal Versus Polyclonal Antibodies: Distinguishing Characteristics, Applications, and Information Resources. ILAR J. 2005, 46, 258–268. [Google Scholar] [CrossRef]
- Gaudinski, M.; Coates, E.E.; Novik, L.; Widge, A.; Houser, K.V.; Burch, E.; Holman, L.A.; Gordon, I.J.; Chen, G.L.; Carter, C.; et al. Safety, tolerability, pharmacokinetics, and immunogenicity of the therapeutic monoclonal antibody mAb114 targeting Ebola virus glycoprotein (VRC 608): An open-label phase 1 study. Lancet 2019, 393, 889–898. [Google Scholar] [CrossRef]
- Misasi, J.; Gilman, M.S.A.; Kanekiyo, M.; Gui, M.; Cagigi, A.; Mulangu, S.; Corti, D.; Ledgerwood, J.E.; Lanzavecchia, A.; Cunningham, J.; et al. Structural and molecular basis for Ebola virus neutralization by protective human antibodies. Science 2016, 351, 1343–1346. [Google Scholar] [CrossRef] [PubMed]
- Mulangu, S.; Dodd, L.E.; Davey, R.T.; Mbaya, O.T.; Proschan, M.; Mukadi, D.; Manzo, M.L.; Nzolo, D.; Oloma, A.T.; Ibanda, A.; et al. A Randomized, Controlled Trial of Ebola Virus Disease Therapeutics. N. Engl. J. Med. 2019, 381, 2293–2303. [Google Scholar] [CrossRef]
- Pascal, K.E.; Dudgeon, D.; Trefry, J.C.; Anantpadma, M.; Sakurai, Y.; Murin, C.D.; Turner, H.L.; Fairhurst, J.; Torres, M.; Rafique, A.; et al. Development of Clinical-Stage Human Monoclonal Antibodies That Treat Advanced Ebola Virus Disease in Nonhuman Primates. J. Infect. Dis. 2018, 218, S612–S626. [Google Scholar] [CrossRef] [PubMed]
- Howell, K.; Qiu, X.; Brannan, J.M.; Bryan, C.; Davidson, E.; Holtsberg, F.W.; Wec, A.Z.; Shulenin, S.; Biggins, J.E.; Douglas, R.; et al. Antibody Treatment of Ebola and Sudan Virus Infection via a Uniquely Exposed Epitope within the Glycoprotein Receptor-Binding Site. Cell Rep. 2016, 15, 1514–1526. [Google Scholar] [CrossRef]
- Murin, C.D.; Fusco, M.L.; Bornholdt, Z.; Qiu, X.; Olinger, G.; Zeitlin, L.; Kobinger, G.P.; Ward, A.B.; Saphire, E.O. Structures of protective antibodies reveal sites of vulnerability on Ebola virus. Proc. Natl. Acad. Sci. USA 2014, 111, 17182–17187. [Google Scholar] [CrossRef] [PubMed]
- Davidson, E.; Bryan, C.; Fong, R.H.; Barnes, T.; Pfaff, J.M.; Mabila, M.; Rucker, J.B.; Doranz, B.J. Mechanism of Binding to Ebola Virus Glycoprotein by the ZMapp, ZMAb, and MB-003 Cocktail Antibodies. J. Virol. 2015, 89, 10982–10992. [Google Scholar] [CrossRef]
- Saphire, E.O.; Schendel, S.; Fusco, M.L.; Gangavarapu, K.; Gunn, B.M.; Wec, A.Z.; Halfmann, P.J.; Brannan, J.M.; Herbert, A.S.; Qiu, X.; et al. Systematic Analysis of Monoclonal Antibodies against Ebola Virus GP Defines Features that Contribute to Protection. Cell 2018, 174, 938–952.e13. [Google Scholar] [CrossRef] [PubMed]
- Gilchuk, P.; Murin, C.D.; Milligan, J.C.; Cross, R.W.; Mire, C.E.; Ilinykh, P.A.; Huang, K.; Kuzmina, N.; Altman, P.X.; Hui, S.; et al. Analysis of a Therapeutic Antibody Cocktail Reveals Determinants for Cooperative and Broad Ebolavirus Neutralization. Immunity 2020, 52, 388–403.e12. [Google Scholar] [CrossRef]
- Gilchuk, P.; Kuzmina, N.; Ilinykh, P.A.; Huang, K.; Gunn, B.M.; Bryan, A.; Davidson, E.; Doranz, B.J.; Turner, H.L.; Fusco, M.L.; et al. Multifunctional Pan-ebolavirus Antibody Recognizes a Site of Broad Vulnerability on the Ebolavirus Glycoprotein. Immunity 2018, 49, 363–374.e10. [Google Scholar] [CrossRef]
- Murin, C.D.; Gilchuk, P.; Ilinykh, P.A.; Huang, K.; Kuzmina, N.; Shen, X.; Bruhn, J.F.; Bryan, A.L.; Davidson, E.; Doranz, B.J.; et al. Convergence of a common solution for broad ebolavirus neutralization by glycan cap-directed human antibodies. Cell Rep. 2021, 35, 108984. [Google Scholar] [CrossRef]
- Murin, C.D.; Bruhn, J.F.; Bornholdt, Z.A.; Copps, J.; Stanfield, R.; Ward, A.B. Structural Basis of Pan-Ebolavirus Neutralization by an Antibody Targeting the Glycoprotein Fusion Loop. Cell Rep. 2018, 24, 2723–2732. [Google Scholar] [CrossRef]
- Wec, A.Z.; Bornholdt, Z.A.; He, S.; Herbert, A.S.; Goodwin, E.; Wirchnianski, A.S.; Gunn, B.M.; Zhang, Z.; Zhu, W.; Liu, G.; et al. Development of a Human Antibody Cocktail that Deploys Multiple Functions to Confer Pan-Ebolavirus Protection. Cell Host Microbe 2019, 25, 39–48.e5. [Google Scholar] [CrossRef]
- Brannan, J.M.; He, S.; Howell, K.A.; Prugar, L.I.; Zhu, W.; Vu, H.; Shulenin, S.; Kailasan, S.; Raina, H.; Wong, G.; et al. Post-exposure immunotherapy for two ebolaviruses and Marburg virus in nonhuman primates. Nat. Commun. 2019, 10, 105. [Google Scholar] [CrossRef]
- Mire, C.E.; Geisbert, J.B.; Borisevich, V.; Fenton, K.A.; Agans, K.N.; Flyak, A.I.; Deer, D.J.; Steinkellner, H.; Bohorov, O.; Bohorova, N.; et al. Therapeutic treatment of Marburg and Ravn virus infection in nonhuman primates with a human monoclonal antibody. Sci. Transl. Med. 2017, 9, eaai8711. [Google Scholar] [CrossRef]
- King, L.B.; Fusco, M.L.; Flyak, A.I.; Ilinykh, P.A.; Huang, K.; Gunn, B.; Kirchdoerfer, R.N.; Hastie, K.M.; Sangha, A.K.; Meiler, J.; et al. The Marburgvirus-Neutralizing Human Monoclonal Antibody MR191 Targets a Conserved Site to Block Virus Receptor Binding. Cell Host Microbe 2018, 23, 101–109.e4. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Shi, Y.; Song, J.; Qi, J.; Lu, G.; Yan, J.; Gao, G.F. Ebola Viral Glycoprotein Bound to Its Endosomal Receptor Niemann-Pick C1. Cell 2016, 164, 258–268. [Google Scholar] [CrossRef] [PubMed]
- Ridgeback Biotherapeutics. Drug Label: EBANGA (Ansuvimab-Zykl) for Injection; Ridgeback Biotherapeutics: Miami, FL, USA, 2020. [Google Scholar]
- FDA. Multi-Discipline Review; Inmazeb (Atoltivimab, Maftivimab, and Odesivimab-Ebgn) Application Number 761169Orig1s000; FDA: Silver Spring, MD, USA, 2018.
- Zhao, X.; Howell, K.A.; He, S.; Brannan, J.M.; Wec, A.Z.; Davidson, E.; Turner, H.L.; Chiang, C.-I.; Lei, L.; Fels, J.M.; et al. Immunization-Elicited Broadly Protective Antibody Reveals Ebolavirus Fusion Loop as a Site of Vulnerability. Cell 2017, 169, 891–904.e15. [Google Scholar] [CrossRef]
- Janus, B.M.; Van Dyk, N.; Zhao, X.; Howell, K.A.; Soto, C.; Aman, M.J.; Li, Y.; Fuerst, T.R.; Ofek, G. Structural basis for broad neutralization of ebolaviruses by an antibody targeting the glycoprotein fusion loop. Nat. Commun. 2018, 9, 3934. [Google Scholar] [CrossRef] [PubMed]
- Marano, G.; Vaglio, S.; Pupella, S.; Facco, G.; Catalano, L.; Liumbruno, G.M.; Grazzini, G. Convalescent plasma: New evidence for an old therapeutic tool? Blood Transfus. 2016, 14, 1–6. [Google Scholar] [CrossRef][Green Version]
- Simon, J. Emil Behring’s Medical Culture: From Disinfection to Serotherapy. Med. Hist. 2007, 51, 201–218. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Dean, C.L.; Hooper, J.W.; Dye, J.M.; Zak, S.E.; Koepsell, S.A.; Corash, L.; Benjamin, R.J.; Kwilas, S.; Bonds, S.; Winkler, A.M.; et al. Characterization of Ebola convalescent plasma donor immune response and psoralen treated plasma in the United States. Transfusion 2020, 60, 1024–1031. [Google Scholar] [CrossRef] [PubMed]
- Delamou, A.; Haba, N.Y.; Saez, A.M.; Gallian, P.; Ronse, M.; Jacobs, J.; Camara, B.S.; Kadio, K.J.-J.O.; Guemou, A.; Kolie, J.P.; et al. Organizing the Donation of Convalescent Plasma for a Therapeutic Clinical Trial on Ebola Virus Disease: The Experience in Guinea. Am. J. Trop. Med. Hyg. 2016, 95, 647–653. [Google Scholar] [CrossRef] [PubMed]
- Van Griensven, J.; Edwards, T.; De Lamballerie, X.; Semple, M.; Gallian, P.; Baize, S.; Horby, P.; Raoul, H.; Magassouba, N.; Antierens, A.; et al. Evaluation of Convalescent Plasma for Ebola Virus Disease in Guinea. N. Engl. J. Med. 2016, 374, 33–42. [Google Scholar] [CrossRef]
- Brown, J.F.; Dye, J.M.; Tozay, S.; Jeh-Mulbah, G.; Wohl, D.A.; Fischer, W.A.; Cunningham, C.K.; Rowe, K.; Zacharias, P.; Van Hasselt, J.; et al. Anti–Ebola Virus Antibody Levels in Convalescent Plasma and Viral Load After Plasma Infusion in Patients with Ebola Virus Disease. J. Infect. Dis. 2018, 218, 555–562. [Google Scholar] [CrossRef]
- Tedder, R.; Samuel, D.; Dicks, S.; Scott, J.T.; Ijaz, S.; Smith, C.C.; Adaken, C.; Cole, C.; Baker, S.; Edwards, T.; et al. Detection, characterization, and enrollment of donors of Ebola convalescent plasma in Sierra Leone. Transfusion 2018, 58, 1289–1298. [Google Scholar] [CrossRef]
- Marzi, A.; Robertson, S.J.; Haddock, E.; Feldmann, F.; Hanley, P.W.; Scott, D.P.; Strong, J.E.; Kobinger, G.; Best, S.M.; Feldmann, H. VSV-EBOV rapidly protects macaques against infection with the 2014/15 Ebola virus outbreak strain. Science 2015, 349, 739–742. [Google Scholar] [CrossRef]
- Jones, S.M.; Ströher, U.; Fernando, L.; Qiu, X.; Alimonti, J.; Melito, P.; Bray, M.; Klenk, H.; Feldmann, H. Assessment of a Vesicular Stomatitis Virus–Based Vaccine by Use of the Mouse Model of Ebola Virus Hemorrhagic Fever. J. Infect. Dis. 2007, 196, S404–S412. [Google Scholar] [CrossRef] [PubMed]
- Marzi, A.; Engelmann, F.; Feldmann, F.; Haberthur, K.; Shupert, W.L.; Brining, D.; Scott, D.P.; Geisbert, T.W.; Kawaoka, Y.; Katze, M.G.; et al. Antibodies are necessary for rVSV/ZEBOV-GP-mediated protection against lethal Ebola virus challenge in nonhuman primates. Proc. Natl. Acad. Sci. USA 2013, 110, 1893–1898. [Google Scholar] [CrossRef] [PubMed]
- Wong, G.; Richardson, J.S.; Pillet, S.; Patel, A.; Qiu, X.; Alimonti, J.; Hogan, J.; Zhang, Y.; Takada, A.; Feldmann, H.; et al. Immune Parameters Correlate with Protection Against Ebola Virus Infection in Rodents and Nonhuman Primates. Sci. Transl. Med. 2012, 4, 158ra146. [Google Scholar] [CrossRef]
- Wong, G.; Audet, J.; Fernando, L.; Fausther-Bovendo, H.; Alimonti, J.B.; Kobinger, G.P.; Qiu, X. Immunization with vesicular stomatitis virus vaccine expressing the Ebola glycoprotein provides sustained long-term protection in rodents. Vaccine 2014, 32, 5722–5729. [Google Scholar] [CrossRef]
- Feldmann, H.; Jones, S.M.; Daddario-DiCaprio, K.M.; Geisbert, J.B.; Ströher, U.; Grolla, A.; Bray, M.; Fritz, E.A.; Fernando, L.; Feldmann, F.; et al. Effective Post-Exposure Treatment of Ebola Infection. PLoS Pathog. 2007, 3, e2. [Google Scholar] [CrossRef]
- Geisbert, T.W.; Daddario-DiCaprio, K.M.; Geisbert, J.B.; Reed, D.; Feldmann, F.; Grolla, A.; Ströher, U.; Fritz, E.A.; Hensley, L.; Jones, S.M.; et al. Vesicular stomatitis virus-based vaccines protect nonhuman primates against aerosol challenge with Ebola and Marburg viruses. Vaccine 2008, 26, 6894–6900. [Google Scholar] [CrossRef]
- Qiu, X.; Fernando, L.; Alimonti, J.B.; Melito, P.L.; Feldmann, F.; Dick, D.; Ströher, U.; Feldmann, H.; Jones, S.M. Mucosal Immunization of Cynomolgus Macaques with the VSVΔG/ZEBOVGP Vaccine Stimulates Strong Ebola GP-Specific Immune Responses. PLoS ONE 2009, 4, e5547. [Google Scholar] [CrossRef]
- Geisbert, T.W.; Daddario-DiCaprio, K.M.; Lewis, M.G.; Geisbert, J.B.; Grolla, A.; Leung, A.; Paragas, J.; Matthias, L.; Smith, M.A.; Jones, S.M.; et al. Vesicular Stomatitis Virus-Based Ebola Vaccine Is Well-Tolerated and Protects Immunocompromised Nonhuman Primates. PLoS Pathog. 2008, 4, e1000225. [Google Scholar] [CrossRef] [PubMed]
- Agnandji, S.T.; Huttner, A.; Zinser, M.E.; Njuguna, P.; Dahlke, C.; Fernandes, J.F.; Yerly, S.; Dayer, J.-A.; Kraehling, V.; Kasonta, R.; et al. Phase 1 Trials of rVSV Ebola Vaccine in Africa and Europe. N. Engl. J. Med. 2016, 374, 1647–1660. [Google Scholar] [CrossRef]
- Regules, J.A.; Beigel, J.; Paolino, K.M.; Voell, J.; Castellano, A.R.; Hu, Z.; Muñoz, P.; Moon, J.E.; Ruck, R.C.; Bennett, J.W.; et al. A Recombinant Vesicular Stomatitis Virus Ebola Vaccine. N. Engl. J. Med. 2017, 376, 330–341. [Google Scholar] [CrossRef]
- Huttner, A.; Dayer, J.-A.; Yerly, S.; Combescure, C.; Auderset, F.; Desmeules, J.; Eickmann, M.; Finckh, A.; Goncalves, A.R.; Hooper, J.; et al. The effect of dose on the safety and immunogenicity of the VSV Ebola candidate vaccine: A randomised double-blind, placebo-controlled phase 1/2 trial. Lancet Infect. Dis. 2015, 15, 1156–1166. [Google Scholar] [CrossRef]
- ElSherif, M.S.; Brown, C.; MacKinnon-Cameron, D.; Li, L.; Racine, T.; Alimonti, J.; Rudge, T.L.; Sabourin, C.; Silvera, P.; Hooper, J.; et al. Assessing the safety and immunogenicity of recombinant vesicular stomatitis virus Ebola vaccine in healthy adults: A randomized clinical trial. Can. Med. Assoc. J. 2017, 189, E819–E827. [Google Scholar] [CrossRef]
- Henao-Restrepo, A.M.; Camacho, A.; Longini, I.M.; Watson, C.; Edmunds, W.J.; Egger, M.; Carroll, M.; Dean, N.E.; Diatta, I.D.; Doumbia, M.; et al. Efficacy and effectiveness of an rVSV-vectored vaccine in preventing Ebola virus disease: Final results from the Guinea ring vaccination, open-label, cluster-randomised trial (Ebola Ça Suffit!). Lancet 2017, 389, 505–518. [Google Scholar] [CrossRef]
- Metzger, W.G.; Vivas-Martínez, S. Questionable efficacy of the rVSV-ZEBOV Ebola vaccine. Lancet 2018, 391, 1021. [Google Scholar] [CrossRef]
- Halperin, S.A.; Das, R.; Onorato, M.T.; Liu, K.; Martin, J.; Grant-Klein, R.J.; Nichols, R.; Coller, B.-A.; Helmond, F.A.; Simon, J.K. Immunogenicity, Lot Consistency, and Extended Safety of rVSVΔG-ZEBOV-GP Vaccine: A Phase 3 Randomized, Double-Blind, Placebo-Controlled Study in Healthy Adults. J. Infect. Dis. 2019, 220, 1127–1135. [Google Scholar] [CrossRef] [PubMed]
- Khurana, S.; Fuentes, S.; Coyle, E.M.; Ravichandran, S.; Davey, R.T., Jr.; Beigel, J. Human antibody repertoire after VSV-Ebola vaccination identifies novel targets and virus-neutralizing IgM antibodies. Nat. Med. 2016, 22, 1439–1447. [Google Scholar] [CrossRef]
- Stephenson, K.E.; Le Gars, M.; Sadoff, J.; de Groot, A.M.; Heerwegh, D.; Truyers, C.; Atyeo, C.; Loos, C.; Chandrashekar, A.; McMahan, K.; et al. Immunogenicity of the Ad26.COV2.S Vaccine for COVID-19. JAMA 2021, 325, 1535–1544. [Google Scholar] [CrossRef] [PubMed]
- Priddy, F.H.; Brown, D.; Kublin, J.; Monahan, K.; Wright, D.P.; Lalezari, J.; Santiago, S.; Marmor, M.; Lally, M.; Novak, R.M.; et al. Safety and Immunogenicity of a Replication-Incompetent Adenovirus Type 5 HIV-1 Clade Bgag/pol/nefVaccine in Healthy Adults. Clin. Infect. Dis. 2008, 46, 1769–1781. [Google Scholar] [CrossRef] [PubMed]
- Cupovic, J.; Ring, S.S.; Onder, L.; Colston, J.M.; Lütge, M.; Cheng, H.-W.; De Martin, A.; Provine, N.M.; Flatz, L.; Oxenius, A.; et al. Adenovirus vector vaccination reprograms pulmonary fibroblastic niches to support protective inflating memory CD8+ T cells. Nat. Immunol. 2021, 22, 1042–1051. [Google Scholar] [CrossRef]
- Chen, T.; Li, D.; Song, Y.; Yang, X.; Liu, Q.; Jin, X.; Zhou, D.; Huang, Z. A heterologous prime-boost Ebola virus vaccine regimen induces durable neutralizing antibody response and prevents Ebola virus-like particle entry in mice. Antivir. Res. 2017, 145, 54–59. [Google Scholar] [CrossRef]
- Pollard, A.J.; Launay, O.; Lelievre, J.-D.; Lacabaratz, C.; Grande, S.; Goldstein, N.; Robinson, C.; Gaddah, A.; Bockstal, V.; Wiedemann, A.; et al. Safety and immunogenicity of a two-dose heterologous Ad26.ZEBOV and MVA-BN-Filo Ebola vaccine regimen in adults in Europe (EBOVAC2): A randomised, observer-blind, participant-blind, placebo-controlled, phase 2 trial. Lancet Infect. Dis. 2020, 21, 493–506. [Google Scholar] [CrossRef]
- Anywaine, Z.; Whitworth, H.; Kaleebu, P.; PrayGod, G.; Shukarev, G.; Manno, D.; Kapiga, S.; Grosskurth, H.; Kalluvya, S.; Bockstal, V.; et al. Safety and Immunogenicity of a 2-Dose Heterologous Vaccination Regimen With Ad26.ZEBOV and MVA-BN-Filo Ebola Vaccines: 12-Month Data From a Phase 1 Randomized Clinical Trial in Uganda and Tanzania. J. Infect. Dis. 2019, 220, 46–56. [Google Scholar] [CrossRef] [PubMed]
- Mutua, G.; Anzala, O.; Luhn, K.; Robinson, C.; Bockstal, V.; Anumendem, D.; Douoguih, M. Safety and Immunogenicity of a 2-Dose Heterologous Vaccine Regimen with Ad26.ZEBOV and MVA-BN-Filo Ebola Vaccines: 12-Month Data from a Phase 1 Randomized Clinical Trial in Nairobi, Kenya. J. Infect. Dis. 2019, 220, 57–67. [Google Scholar] [CrossRef]
- Cross, R.W.; Bornholdt, Z.A.; Prasad, A.N.; Geisbert, J.B.; Borisevich, V.; Agans, K.N.; Deer, D.J.; Melody, K.; Fenton, K.A.; Feldmann, H.; et al. Prior vaccination with rVSV-ZEBOV does not interfere with but improves efficacy of postexposure antibody treatment. Nat. Commun. 2020, 11, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Suder, E.; Furuyama, W.; Feldmann, H.; Marzi, A.; de Wit, E. The vesicular stomatitis virus-based Ebola virus vaccine: From concept to clinical trials. Hum. Vaccines Immunother. 2018, 14, 2107–2113. [Google Scholar] [CrossRef]
- Jones, S.M.; Feldmann, H.; Ströher, U.; Geisbert, J.B.; Fernando, L.; Grolla, A.; Klenk, H.-D.; Sullivan, N.J.; Volchkov, V.; Fritz, E.A.; et al. Live attenuated recombinant vaccine protects nonhuman primates against Ebola and Marburg viruses. Nat. Med. 2005, 11, 786–790. [Google Scholar] [CrossRef] [PubMed]
- Daddario-DiCaprio, K.M.; Geisbert, T.W.; Geisbert, J.B.; Ströher, U.; Hensley, L.E.; Grolla, A.; Fritz, E.A.; Feldmann, F.; Feldmann, H.; Jones, S.M. Cross-Protection against Marburg Virus Strains by Using a Live, Attenuated Recombinant Vaccine. J. Virol. 2006, 80, 9659–9666. [Google Scholar] [CrossRef]
- Mire, C.E.; Geisbert, J.B.; Agans, K.N.; Satterfield, B.; Versteeg, K.M.; Fritz, E.A.; Feldmann, H.; Hensley, L.; Geisbert, T.W. Durability of a Vesicular Stomatitis Virus-Based Marburg Virus Vaccine in Nonhuman Primates. PLoS ONE 2014, 9, e94355. [Google Scholar] [CrossRef]
- Daddario-DiCaprio, K.M.; Geisbert, T.W.; Ströher, U.; Geisbert, J.B.; Grolla, A.; Fritz, E.A.; Fernando, L.; Kagan, E.; Jahrling, P.B.; Hensley, L.; et al. Postexposure protection against Marburg haemorrhagic fever with recombinant vesicular stomatitis virus vectors in non-human primates: An efficacy assessment. Lancet 2006, 367, 1399–1404. [Google Scholar] [CrossRef]
- Geisbert, T.W.; Hensley, L.; Geisbert, J.B.; Leung, A.; Johnson, J.; Grolla, A.; Feldmann, H. Postexposure Treatment of Marburg Virus Infection. Emerg. Infect. Dis. 2010, 16, 1119–1122. [Google Scholar] [CrossRef]
- Shurtleff, A.C.; Biggins, J.E.; Keeney, A.E.; Zumbrun, E.E.; Bloomfield, H.A.; Kuehne, A.; Audet, J.L.; Alfson, K.J.; Griffiths, A.; Olinger, G.G.; et al. Standardization of the Filovirus Plaque Assay for Use in Preclinical Studies. Viruses 2012, 4, 3511–3530. [Google Scholar] [CrossRef] [PubMed]
- HSE. Biological Agents 137. The Principles, Design and Operation of Containment Level 4 Facilities; HSE: London, UK, 2006.
- Steeds, K. Antibody Correlates of Protection for Ebola Virus Infection: Effects of Mutations within the Viral Glycoprotein on Immune Escape. Ph.D. Thesis, University of Liverpool, Liverpool, UK, 2019. [Google Scholar]
- Steeds, K.; Hall, Y.; Slack, G.S.; Longet, S.; Strecker, T.; Fehling, S.K.; Wright, E.; Bore, J.A.; Koundouno, F.R.; Konde, M.K.; et al. Pseudotyping of VSV with Ebola virus glycoprotein is superior to HIV-1 for the assessment of neutralising antibodies. Sci. Rep. 2020, 10, 14289. [Google Scholar] [CrossRef] [PubMed]
- Konduru, K.; Shurtleff, A.C.; Bavari, S.; Kaplan, G. High degree of correlation between Ebola virus BSL-4 neutralization assays and pseudotyped VSV BSL-2 fluorescence reduction neutralization test. J. Virol. Methods 2018, 254, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, D.E.; Page, M.; Mattiuzzo, G.; Hassall, M.; Dougall, T.; Rigsby, P.; Stone, L.; Minor, P. Comparison of platform technologies for assaying antibody to Ebola virus. Vaccine 2017, 35, 1347–1352. [Google Scholar] [CrossRef]
- Ilinykh, P.A.; Shen, X.; Flyak, A.; Kuzmina, N.; Ksiazek, T.G.; Crowe, J.E.; Bukreyev, A. Chimeric Filoviruses for Identification and Characterization of Monoclonal Antibodies. J. Virol. 2016, 90, 3890–3901. [Google Scholar] [CrossRef]
- Shedlock, D.J.; Bailey, M.A.; Popernack, P.M.; Cunningham, J.M.; Burton, D.R.; Sullivan, N.J. Antibody-mediated neutralization of Ebola virus can occur by two distinct mechanisms. Virology 2010, 401, 228–235. [Google Scholar] [CrossRef]
- Kaletsky, R.L.; Simmons, G.; Bates, P. Proteolysis of the Ebola Virus Glycoproteins Enhances Virus Binding and Infectivity. J. Virol. 2007, 81, 13378–13384. [Google Scholar] [CrossRef]
- Roguin, L.; Retegui, L.A. Monoclonal Antibodies Inducing Conformational Changes on the Antigen Molecule. Scand. J. Immunol. 2003, 58, 387–394. [Google Scholar] [CrossRef] [PubMed]
- Yu, D.-S.; Weng, T.-H.; Shen, L.; Wu, X.; Hu, C.-Y.; Wang, F.X.; Wu, Z.-G.; Wu, H.-B.; Wu, N.-P.; Li, L.-J.; et al. Development and Characterization of Neutralizing Antibodies Against Zaire Ebolavirus Glycoprotein and Protein 40. Cell. Physiol. Biochem. 2018, 50, 1055–1067. [Google Scholar] [CrossRef]
- Stahelin, R.V. Could the Ebola Virus Matrix Protein VP40 be a Drug Target? Expert Opin. Ther. Targets 2013, 18, 115–120. [Google Scholar] [CrossRef]
- Brioen, P.; Dekegel, D.; Boeyé, A. Neutralization of poliovirus by antibody-mediated polymerization. Virology 1983, 127, 463–468. [Google Scholar] [CrossRef]
- Murin, C.D.; Wilson, I.A.; Ward, A.B. Antibody responses to viral infections: A structural perspective across three different enveloped viruses. Nat. Microbiol. 2019, 4, 734–747. [Google Scholar] [CrossRef]
- Qiao, J.; Li, Y.; Wei, C.; Yang, H.; Yu, J.; Wei, H. Rapid detection of viral antibodies based on multifunctional Staphylococcus aureus nanobioprobes. Enzym. Microb. Technol. 2016, 95, 94–99. [Google Scholar] [CrossRef] [PubMed]
- Pelegrin, M.; Naranjo-Gomez, M.; Piechaczyk, M. Antiviral Monoclonal Antibodies: Can They Be More Than Simple Neutralizing Agents? Trends Microbiol. 2015, 23, 653–665. [Google Scholar] [CrossRef]
- Mellors, J.; Tipton, T.; Longet, S.; Carroll, M. Viral Evasion of the Complement System and Its Importance for Vaccines and Therapeutics. Front. Immunol. 2020, 11, 1450. [Google Scholar] [CrossRef]
- Howell, K.A.; Brannan, J.M.; Bryan, C.; McNeal, A.; Davidson, E.; Turner, H.L.; Vu, H.; Shulenin, S.; He, S.; Kuehne, A.; et al. Cooperativity Enables Non-neutralizing Antibodies to Neutralize Ebolavirus. Cell Rep. 2017, 19, 413–424. [Google Scholar] [CrossRef]
- Kuzmina, N.; Younan, P.; Gilchuk, P.; Santos, R.I.; Flyak, A.I.; Ilinykh, P.A.; Huang, K.; Lubaki, N.M.; Ramanathan, P.; Crowe, J.E.; et al. Antibody-Dependent Enhancement of Ebola Virus Infection by Human Antibodies Isolated from Survivors. Cell Rep. 2018, 24, 1802–1815.e5. [Google Scholar] [CrossRef]
- Weisblum, Y.; Schmidt, F.; Zhang, F.; DaSilva, J.; Poston, D.; Lorenzi, J.C.; Muecksch, F.; Rutkowska, M.; Hoffmann, H.-H.; Michailidis, E.; et al. Escape from neutralizing antibodies by SARS-CoV-2 spike protein variants. eLife 2020, 9, e61312. [Google Scholar] [CrossRef]
- Hiatt, A.; Zeitlin, L.; Whaley, K.J. Multiantibody Strategies for HIV. Clin. Dev. Immunol. 2013, 2013, 632893. [Google Scholar] [CrossRef] [PubMed]
- Yasuhara, A.; Yamayoshi, S.; Ito, M.; Kiso, M.; Yamada, S.; Kawaoka, Y. Isolation and Characterization of Human Monoclonal Antibodies That Recognize the Influenza A(H1N1)pdm09 Virus Hemagglutinin Receptor-Binding Site and Rarely Yield Escape Mutant Viruses. Front. Microbiol. 2018, 9, 2660. [Google Scholar] [CrossRef]
- Taylor, P.C.; Adams, A.C.; Hufford, M.M.; de la Torre, I.; Winthrop, K.; Gottlieb, R.L. Neutralizing monoclonal antibodies for treatment of COVID-19. Nat. Rev. Immunol. 2021, 21, 382–393. [Google Scholar] [CrossRef] [PubMed]
- Bramble, M.S.; Hoff, N.; Gilchuk, P.; Mukadi, P.; Lu, K.; Doshi, R.H.; Steffen, I.; Nicholson, B.P.; Lipson, A.; Vashist, N.; et al. Pan-Filovirus Serum Neutralizing Antibodies in a Subset of Congolese Ebolavirus Infection Survivors. J. Infect. Dis. 2018, 218, 1929–1936. [Google Scholar] [CrossRef] [PubMed]
- Keck, Z.-Y.; Enterlein, S.G.; Howell, K.A.; Vu, H.; Shulenin, S.; Warfield, K.L.; Froude, J.W.; Araghi, N.; Douglas, R.; Biggins, J.; et al. Macaque Monoclonal Antibodies Targeting Novel Conserved Epitopes within Filovirus Glycoprotein. J. Virol. 2016, 90, 279–291. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Antibody (Cocktail) | Epitope | Brief Description—How It Was Discovered and Where It Targets | Neutralising |
|---|---|---|---|
| KZ52—WHO Research Standard Critical residues—red: 511, 550, 552, 552, 556 Putative residues—yellow: 24, 40, 43, 507-508, 513-514, 549, 551 |  | A potently neutralising antibody isolated from a survivor of the 1995 Kikwit outbreak binding to the GP1/GP2 interface to prevent insertion of the fusion loop into the membrane [80]. | ✓ |
| 13C6 (ZMapp) Critical residues—red: 270, 272 |  | A non-neutralising humanised mouse antibody that binds to the GP1 head and GC regions and can activate effector functions [80]. | X |
| 2G4 (ZMapp) Critical residues—red: 511, 550, 553, 556 |  | A neutralising humanised mouse antibody that binds to the GP1/GP2 interface to prevent insertion of the fusion loop into the endosome membrane [80]. | ✓ |
| 4G7 (ZMapp) Critical residues—red: 511, 552, 556 |  | A neutralising humanised mouse antibody that binds to the GP1/GP2 interface to prevent insertion of the fusion loop into the membrane [80]. | ✓ |
| Ansuvimab-zykl/mAb114 (Ebanga™) Known residues—red: 111-119 |  | Isolated from a survivor of the 1995 Kikwit outbreak that targets the RBD blocking access to NCP1 [74,91]. | ✓ |
| Atoltivimab/REGN3470 (Inmazeb™) Putative residues—yellow: 236-244, 264-297 and 298-308 |  | Developed by the VelcoImmune platform in which genetically engineered mice express fully human antibodies. Atoltivimab is a neutralising antibody that binds to GC, it is also able to activate effector functions [92]. | ✓ |
| Odesivimab/REGN3471 (Inmazeb™) Putative residues—yellow: 114 to 122, 139 to 151, 236 to 244, and 265 to 287 |  | A non-neutralising VelcoImmune antibody that binds to the GP1 head but further from the residues involved in receptor binding compared to mAb114. It is very capable of activating effector functions [92]. | X |
| Maftivimab/REGN3479 (Inmazeb™) Putative residues—yellow: 531 to 545 |  | A neutralising VelcoImmune antibody that binds to the GP1/GP2 interface to prevent insertion of the IFL into the endosome membrane [92]. | ✓ |
| CA45—Pan-ebolavirus Critical residues—red: 64, 517, 546, 550 Putative residues—yellow: 38, 40-41, 66, 68, 101-104, 184, 186-187, 211-213, 513-516, 518-519, 544-545, 547-549, 551-552, 554, 558 |  | Isolated from challenged NHPs, CA45 binds to the IFL and GP1/GP2 interface preventing insertion of the IFL into the endosome membrane and hence neutralises EBOV, SUDV, BDBV and RESTV. CA45 provides protection in mice, guinea pigs and ferrets. Also gives 100% protection to NHPs when given in a cocktail with FVM04 and MR191 [87,93,94]. | ✓ |
| FVM04—Pan-ebolavirus Known residues—red: 115, 117-118 |  | Targets the RBD blocking interactions with NPC1. It neutralises EBOV, SUDV and BDBV, though only protective from EBOV and SUDV challenge in mouse and Guinea pig model. Can be given in cocktail with CA45 and MR191 [78,87]. | ✓ |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hargreaves, A.; Brady, C.; Mellors, J.; Tipton, T.; Carroll, M.W.; Longet, S. Filovirus Neutralising Antibodies: Mechanisms of Action and Therapeutic Application. Pathogens 2021, 10, 1201. https://doi.org/10.3390/pathogens10091201
Hargreaves A, Brady C, Mellors J, Tipton T, Carroll MW, Longet S. Filovirus Neutralising Antibodies: Mechanisms of Action and Therapeutic Application. Pathogens. 2021; 10(9):1201. https://doi.org/10.3390/pathogens10091201
Chicago/Turabian StyleHargreaves, Alexander, Caolann Brady, Jack Mellors, Tom Tipton, Miles W. Carroll, and Stephanie Longet. 2021. "Filovirus Neutralising Antibodies: Mechanisms of Action and Therapeutic Application" Pathogens 10, no. 9: 1201. https://doi.org/10.3390/pathogens10091201
APA StyleHargreaves, A., Brady, C., Mellors, J., Tipton, T., Carroll, M. W., & Longet, S. (2021). Filovirus Neutralising Antibodies: Mechanisms of Action and Therapeutic Application. Pathogens, 10(9), 1201. https://doi.org/10.3390/pathogens10091201